

Abstract
The properties of oxidation of dichloroethene (DCE) and trichloroethylene (TCE) by three mutant strains of Pseudomonas butanovora containing single amino acid substitutions in the α-subunit of butane monooxygenase hydroxylase (BMOH-α) were compared to the properties of the wild-type strain (Rev WT). The rates of oxidation of three chloroethenes (CEs) were reduced in mutant strain G113N and corresponded with a lower maximum rate of butane oxidation. The rate of TCE degradation was reduced by one-half in mutant strain L279F, whereas the rates of DCE oxidation were the same as those in Rev WT. Evidence was obtained that the composition of products of CE oxidation differed between Rev WT and some of the mutant strains. For example, while Rev WT released nearly all available chlorine stoichiometrically during CE oxidation, strain F321Y released about 40% of the chlorine during 1,2-cis-DCE and TCE oxidation, and strain G113N released between 14 and 25% of the available chlorine during oxidation of DCE and 56% of the available chlorine during oxidation of TCE. Whereas Rev WT, strain L279F, and strain F321Y formed stoichiometric amounts of 1,2-cis-DCE epoxide during oxidation of 1,2-cis-DCE, only about 50% of the 1,2-cis-DCE oxidized by strain G113N was detected as the epoxide. Evidence was obtained that 1,2-cis-DCE epoxide was a substrate for butane monooxygenase (BMO) that was oxidized after the parent compound was consumed. Yet all of the mutant strains released less than 40% of the available 1,2-cis-DCE chlorine, suggesting that they have altered activity towards the epoxide. In addition, strain G113N was unable to degrade the epoxide. TCE epoxide was detected during exposure of Rev WT and strain F321Y to TCE but was not detected with strains L279F and G113N. Lactate-dependent O2 uptake rates were differentially affected by DCE degradation in the mutant strains, providing evidence that some products released by the altered BMOs reduced the impact of CE on cellular toxicity. The use of CEs as substrates in combination with P. butanovora BMOH-α mutants might allow insights into the catalytic mechanism of BMO to be obtained.
Pseudomonas butanovora utilizes a butane monooxygenase (BMO) to initiate oxidation of short-chain alkanes as its sole source of carbon and energy for growth (2). BMO fortuitously activates a wide variety of chemically stable compounds, including environmental contaminants such as chloroethenes (CEs) (3, 17). Other bacterial genera, such as Nocardioides and Mycobacterium, also utilize BMOs that are biochemically distinct from that characterized in P. butanovora (18). Trichloroethylene (TCE) turnover-dependent toxicities, including BMO inactivation (66% inactivation in Mycobacterium vaccae, compared to 96% inactivation in P. butanovora and Nocardioides sp. strain CF8) and reduction in cell viability (83% reduction in P. butanovora, compared to no reduction in M. vaccae and Nocardioides sp. strain CF8), varied substantially among strains (16). The possibility that there are differences among the BMOs in their catalytic attack on TCE and the possibility that product profiles from TCE degradation may vary and impact overall CE transformation capacities encouraged additional research aimed at the identification of mechanisms that would allow more sustainable CE degradation.
BMO from P. butanovora is a soluble diiron multicomponent monooxygenase with high similarity to soluble methane monooxygenase (sMMO). Genetic and biochemical characterization showed that BMO consists of a hydroxylase (BMOH) in an α2β2γ2 configuration, a reductase (BMOR) which transfers electrons from NADH to the active site in the hydroxylase α-subunit, and an effector protein (BMOB) whose function in BMO remains undefined (25). Mutants with single amino acid substitutions in the BMOH α-subunit (BMOH-α) of P. butanovora have provided a glimpse into the basis of its substrate and product specificity (15). For example, the broad substrate range of BMO, which includes aromatics, alkenes, alkynes, and CEs (8, 18), was recently shown to include methane (15). In addition, while wild-type BMO terminally oxidizes propane and butane (2), strain G113N, in which glycine 113 in BMOH-α was substituted for asparagine so that the enzyme resembles the sMMO hydroxylase α-subunit (sMMO-α) at that position, oxidized propane and butane almost exclusively at the subterminal position (Table 1) (15). Two other mutant strains, L279F and F321Y, were similarly engineered so that the sequences resemble the sMMOH-α sequence at the corresponding amino acid positions. The ratio of the rate of 2-butanol accumulation to the rate of 1-butanol accumulation during butane oxidation was 5.5-fold greater in strain L279F than in strain Rev WT, and mutant strain F321Y oxidized butane exclusively at the terminal position (Table 1) (15).
TABLE 1.
P. butanovora strains used in this study
| Strain | Targeted region for amino acid substitution in BMOH-α | Position of butane hydroxylation |
|---|---|---|
| Rev WT | None | Primarily terminal |
| F321Y | Surface of BMOH-α involved in interaction with BMOB | Exclusively terminal |
| L279F | Hydrophobic cavity 2 | Increased subterminal |
| G113N | Hydrophobic cavity 1 | Primarily subterminal |
Products of CE degradation have been quantified using whole cells of methanotrophs and using purified sMMO (9, 22, 24, 30). For example, 80 to 96% of the products of CE degradation are the result of CE epoxide hydrolysis or enzymatic turnover of CE epoxide. The outcome of epoxide breakdown is primarily liberation of chloride, with the organochlorines chloral (trichloroacetaldehyde) and dichloroacetaldehyde accounting for 6% and 5 to 17%, respectively, of the total TCE turned over (9, 24). In contrast, chloral and dichloroacetaldehyde comprise the majority of the products formed during oxidation of TCE by the distantly related liver microsomal cytochrome P-450 (23). Retention of the chlorine atoms is a result of electron abstraction followed by halide or hydride shift (NIH shift), an enzymatic mechanism that does not involve formation of a CE epoxide (14, 21, 23). These early experiments using CEs as substrates with purified sMMO and cytochrome P-450 provided the foundations for development of mechanistic models of the enzymatic catalytic cycles. For example, the atomic migration associated with the formation of chloral during oxidation of TCE was rationally explained by the formation of a carbocation intermediate and radical rebound chemistry (9, 20), as was the detection of a trace of monochloroacetic acid from the oxidation of 1,1-dichloroethene (1,1-DCE) by sMMO (14).
Some of the transient enzyme intermediates in the catalytic cycle of sMMO have unique spectroscopic characteristics, thus enabling identification of the specific intermediates responsible for substrate oxidation (5, 6, 32, 33). For example, sMMO oxidation of electron-rich alkenes, such as propylene, is initiated by a two-electron transfer step, followed by epoxidation of the substrate by the peroxodiiron(III) enzyme intermediate (Hperoxo) (6). Oxidation of alkanes, such as methane, ethane, and propane, progresses through the di(μ-oxo)diiron(IV) intermediate (Q), requiring two single electron abstraction steps and forming a carbocation product intermediate (4, 7, 10-12). In the case of CEs, subsequent rearrangement of the carbocation intermediate results in halide or hydride shift to the neighboring carbon. A similar mechanism has been demonstrated for attack of TCE by P-450 monooxygenases, resulting in accumulation of chloral in vitro (23). Although the Hperoxo and Q intermediates have not yet been identified in BMO, we were interested in determining if the combination of CEs as substrate probes and P. butanovora mutant strains would provide insights into the mechanism of catalysis in the BMO system.
In this study, mutant strains of P. butanovora containing altered BMOs were exposed to the CEs 1,1-DCE, 1,2-cis-DCE, and TCE. Differences in product formation and physiological responses were determined, and the results were applied to a model differentiating between the enzymatic oxidation of CEs by P. butanovora and the enzymatic oxidation of CEs by mutant strain G113N.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
P. butanovora strains were cultured at 30°C in sealed 160-ml vials as previously described (15). Mutant strains F321Y, L279F, and G113N contain single amino acid substitutions in BMOH-α (Table 1). Strain Rev WT contains the wild-type amino acid sequence. The construction of these strains was previously described (15). For all experiments, cells were grown on butane and harvested at the late exponential to early stationary phase (optical density at 600 nm, 0.60 to 0.80). Cells were washed three times and resuspended with 30 mM phosphate buffer (25 mM KH2PO4, 25 mM Na2HPO4 [pH 7.2]) to obtain a concentrated cell suspension (10 mg/ml total protein).
Chlorinated ethene exposure.
1,1-DCE, 1,2-cis-DCE, and TCE concentrations were monitored by gas chromatography. Teflon-faced butyl septa (Supelco, Bellefonte, PA) were used to seal 7.7-ml vials containing 5 mM sodium lactate, either 25 μM (initial liquid concentration) 1,1-DCE or 1,2-cis-DCE or 40 μM TCE, and sufficient phosphate buffer to bring the volume to 900 μl. Vials were equilibrated for at least 15 min in a reciprocating shaker at 30°C. Concentrated cell suspensions (100 μl containing 1.0 mg total protein) were added to initiate the experiments. Samples of the gas phase (10 to 40 μl) were removed using a gas-tight syringe for analysis by gas chromatography.
For chloride release measurements, following complete CE consumption, vials were placed on ice for 15 min. Cells were sedimented, and the supernatant was transferred to a fresh tube and stored at 4°C until analysis by ion chromatography. Chloride concentrations were determined using a Dionex (Sunnyvale, CA) model DX-120 ion chromatograph equipped with an autosampler, an electrical conductivity detector, and a Dionex AS14 column.
Oxygen uptake measurements.
O2 uptake measurements were obtained using a YSI (Yellow Springs, OH) Clark-style O2 electrode mounted in a glass water-jacketed reaction vessel (1.6 ml) kept at 30°C and filled with phosphate buffer. For each experiment, cells (0.3 mg total protein) were added to the reaction vessel and the vessel was capped. Sodium lactate (3 mM) was added to the vessel through the capillary inlet to determine a lactate-dependent O2 uptake rate. Chlorinated ethenes (13 μM 1,1-DCE, 10 μM 1,2-cis-DCE) were added to determine their effects on rates of lactate-dependent O2 uptake. O2 uptake rates were determined for each strain during exposure to each CE by measuring the slopes of tangent lines drawn to the resulting progress curves.
Determination of CE epoxides.
Reaction vials containing either 1,2-cis-DCE (25 μM) or TCE (80 μM) and cell suspension (1 mg total protein) were prepared as described above, and reactions were quenched by addition of 0.5 ml benzene at appropriate time points. 1,2-cis-DCE and TCE epoxide concentrations were determined as previously described (9, 23). Because mutant strains L279F and G113N had lower rates of TCE degradation and because of the relative instability of TCE epoxide (half-life, 21 to 39 s) (30), the quantity of cell suspension was increased for these mutant strains and decreased for Rev WT so that the total TCE epoxide measured would be based on comparable degradation rates and total amount of TCE degraded.
Analytical methods.
CE concentrations were monitored with a Shimadzu (Kyoto, Japan) GC-8A chromatograph equipped with a flame ionization detector and a capillary column (15 m by 0.53 mm; Alltech, Deerfield, IL) as described above. CE calibration curves were obtained by performing headspace gas analysis with vials containing known amounts of each compound. Dimensionless Henry's constants (1.3 for 1,1-DCE, 0.18 for 1,2-cis-DCE, and 0.49 for TCE) (13) were used to account for aqueous and gaseous partitioning of the total CE in the vials. Protein concentrations were determined using the biuret assay following cell solubilization in 3 M NaOH for 30 min at 65°C.
RESULTS
Degradation of chlorinated ethenes by P. butanovora mutants.
The effects of specific amino acid substitutions in BMOH-α mutant strains of P. butanovora on CE oxidation were measured. The mutant strains degraded the three CEs at initial rates that were less than or equivalent to those of the wild-type control strain (Rev WT) (Table 2). Mutant strain F321Y degraded the CEs at rates that were similar to those of Rev WT. Mutant strain G113N degraded all three substrates at lower rates than Rev WT, whereas strain L279F degraded only TCE at a lower rate than Rev WT. For all strains the rates of CE oxidation were lower than the corresponding rates of butane oxidation (15) except for the rate of oxidation of 1,1-DCE by strain G113N, which was equivalent to the rate of butane oxidation.
TABLE 2.
Initial rates of CE oxidation and percentages of chloride released following exposure to CEs for mutant strains of P. butanovoraa
| Strain | Butane oxidation ratec | 1,1-DCE (25 μM)b
|
1,2-cis-DCE (25 μM)b
|
TCE (40 μM)b
|
|||
|---|---|---|---|---|---|---|---|
| Rated | Chloride released (%)e | Rated | Chloride released (%)e | Rated | Chloride released (%)e | ||
| Rev WT | 76.7 ± 4.0 | 31.6 ± 3.5 | 94.5 ± 3.8 | 8.8 ± 1.9 | 98.0 ± 70 | 16.5 ± 2.7 | 95 ± 1.5 |
| F321Y | 102.4 ± 5.7 | 30.3 ± 3.0 | 106 ± 2.2 | 8.4 ± 2.2 | 39.2 ± 0.9 | 17.9 ± 3.0 | 43 ± 1.6 |
| L279F | 57.5 ± 7.1 | 28.5 ± 2.9 | 70.9 ± 2.3 | 7.2 ± 2.4 | 30.5 ± 1.6 | 8.0 ± 2.5 | 97 ± 1.6 |
| G113N | 21.4 ± 2.4 | 22.5 ± 2.5 | 25.1 ± 0.6 | 4.1 ± 1.8 | 14.1 ± 1.2 | 5.6 ± 2.0 | 56 ± 0.9 |
The values represent data from at least three separate experiments (means ± standard deviations).
The concentration in parentheses is the initial liquid phase chlorinated ethene concentration.
The data are modified from the data in reference 15 and are the sums of the rates of 1- and 2- butanol accumulation expressed in nmol min−1 (mg protein)−1.
Rate of CE degradation expressed in nmol min−1 (mg protein)−1.
Strains (1 mg total protein) were exposed to CEs. Following complete CE consumption, vials were placed on ice for 15 min. Cells were pelleted, and each supernatant was transferred to a fresh tube and stored at 4°C until chloride release was measured. Sufficient time elapsed during storage to allow all chlorine associated with epoxides to dissociate abiologically. The values are percentages of the total chlorine available at the start of each experiment.
Chloride release during CE degradation.
The amounts of chloride released following incubation of the P. butanovora mutant strains with the same amounts of each of the CEs were measured. All available chlorine was released during incubation of Rev WT with each CE (Table 2). In contrast, less than 25% of the available chlorine was released during incubation of strain G113N with either of the DCEs, and only 56% of the available chlorine was released from TCE. In addition, 100% of the chlorine was released when strain F321Y was incubated with 1,1-DCE, but only about 40% was released from 1,2-cis-DCE and TCE. In contrast, all of the available chlorine was released from TCE during incubation with strain L279F, which corresponded to a rate of TCE degradation lower than those of strains F321Y and Rev WT, but only partial amounts were released when strain L279F was exposed to either of the DCEs. Cells of each strain incubated without TCE released no chlorine. These results provide circumstantial evidence that the altered BMOs created different products during CE oxidation or the altered BMOs have different oxidative activities towards the products of initial CE oxidation that account for the variable percentage of chloride released.
Epoxide detection.
It is well established that the predominant pathway for 1,2-cis-DCE and TCE degradation by sMMO is via epoxide intermediates (1,1-DCE epoxide is thought to have a half-life of <2 s and has not been detected) (20, 29). We verified that exposure of the Rev WT control strain to 1,2-cis-DCE resulted in accumulation of the corresponding epoxide. After 10 min of incubation and consumption of 57 nmol of 1,2-cis-DCE, a stoichiometric amount of 1,2-cis-DCE epoxide was measured (Fig. 1). Subsequently, following consumption of the DCE, 82% of the epoxide was degraded (Fig. 1). Stoichiometric conversion of 1,2-cis-DCE to its epoxide was also detected in strains F321Y and L279F (data not shown), and the epoxide was similarly degraded following consumption of nearly 90% of the 1,2-cis-DCE (Table 3). However, only 30 to 40% of the available chlorine from 1,2-cis-DCE was released during incubation with these strains, compared to complete chloride release during incubation with Rev WT. Together, these results suggest that the mechanism of epoxide oxidation was changed in strains F321Y and L279F. Consumption of 57 nmol of 1,2-cis-DCE by mutant strain G113N resulted in production of only 30 nmol of the corresponding epoxide (Fig. 1). Furthermore, 1,2-cis-DCE epoxide was not degraded 50 min following complete DCE turnover, suggesting that the altered BMO in strain G113N did not attack 1,2-cis-DCE epoxide. Alternatively, products other than 1,2-cis-DCE epoxide that were produced during 1,2-cis-DCE oxidation inhibited or inactivated BMO, thereby preventing 1,2-cis-DCE epoxide turnover. Both of these scenarios would account for the low percentage of chloride release by strain G113N during oxidation of 1,2-cis-DCE. We also considered the possibility that the altered BMOs in the mutant strains were simply more easily inactivated by 1,2-cis-DCE epoxide than BMO in Rev WT. However, this scenario seems unlikely for the following reasons. First, since the mutant strains completely consumed the primary substrate (CE), the epoxide product did not strongly compete with the primary substrate. Therefore, it is unlikely that the affinity of the altered BMOs for the epoxide relative to the CE was significantly increased. Second, if the sensitivity of the altered BMOs to epoxide was increased but the affinity was not significantly altered, then stoichiometric production of epoxide would have occurred. Regardless of whether epoxide then inactivated BMO, subsequent abiotic degradation of the epoxides would have resulted in high levels of chloride release, as was observed for Rev WT.
FIG. 1.

1,2-cis-DCE epoxide formation (open symbols and dashed lines) during 1,2-cis-DCE degradation (solid symbols and solid lines) by Rev WT (⧫ and ⋄) and mutant strain G113N (▪ and □).
TABLE 3.
1,2-cis-DCE epoxide and TCE epoxide formation and DCE epoxide consumption by BMOa
| Strain | 1,2-cis-DCE
|
1,2-cis-DCE epoxide formed (nmol)c | 1,2-cis-DCE epoxide degraded (nmol)d | TCE
|
TCE epoxide formed (nmol)e | ||
|---|---|---|---|---|---|---|---|
| Initial concn (μM)b | Amt degraded (nmol) | Initial concn (μM)b | Amt degraded (nmol) | ||||
| Rev WT | 25 | 57 ± 6 | 57 ± 8 | 48 ± 11 | 80 | 265 ± 14 | 28 ± 6 |
| 75 | 152 ± 8 | 149 ± 13 | 61 ± 13 | ||||
| F321Y | 25 | 60 ± 13 | 60 ± 13 | 55 ± 14 | 80 | 300 ± 15 | 15 ± 5 |
| 75 | 159 ± 10 | 131 ± 9 | 54 ± 9 | ||||
The values represent data from at least three separate experiments (means ± standard deviations).
Initial liquid phase chlorinated ethene concentration.
Reaction vials were quenched with benzene following 10 min of exposure to 1,2-cis-DCE, and 1,2-cis-DCE epoxide was measured colorimetrically as described in Materials and Methods.
Reaction vials were quenched with benzene at appropriate time points to follow disappearance of 1,2-cis-DCE epoxide.
Reaction vials were quenched with benzene following 6 min of exposure to TCE, and TCE epoxide was measured colorimetrically as described in Materials and Methods.
TCE epoxide formation was measured during exposure to TCE. While 1,2-cis-DCE epoxide is relatively stable, the half-life of TCE epoxide is only 21 to 39 s (30). Therefore, rapid formation of TCE epoxide in sufficient quantity was required for detection. Strains were exposed to 80 μM TCE for 6 to 7 min. Rev WT consumed TCE at a rate of 44 nmol min−1 mg−1. After 6 min, the reaction was quenched with benzene, and 28 nmol TCE epoxide was detected (Table 3). Interestingly, the rate of TCE consumption by F321Y was at least as high as the rate of TCE consumption by Rev WT, but only 15 nmol TCE epoxide was detected. This result is corroborated by the fact that only 43% of the available chlorine was released during exposure of strain F321Y to TCE (Table 2). Because strains L279F and G113N have lower rates of TCE consumption, the total protein used in the assays was adjusted in an attempt to match the rates of these strains with that of Rev WT. Using 2 mg total protein in each assay, strains G113N and L279F consumed 275 to 325 nmol TCE at maximal rates of 32 to 36 nmol min−1. TCE epoxide was detected in these assays at levels that were about one-fourth the levels detected in strains Rev WT and F321Y per mg total protein. The lower levels of TCE epoxide detected in strains F321Y, L279F, and G113N were a result of either less epoxide formed during TCE turnover or differences in the fate of the TCE epoxide. Since 1,2-cis-DCE epoxide consumption commences only after ≥85% of the 1,2-cis-DCE is consumed (Fig. 1), it is unlikely that BMO-dependent TCE epoxide consumption would occur in these assays since the reactions are quenched with benzene with approximately 40 μM TCE remaining. This is the first study in which TCE epoxide was detected by BMO-dependent turnover of TCE.
Effects of CE degradation on general cellular respiration.
Degradation of chlorinated ethenes results in the release of products that may cause cellular toxicity, including inactivation of the monooxygenase and attack by nucleophilic groups on macromolecules (1, 8, 16, 26, 31). Because the ratios of the rate of 2-butanol accumulation to the rate of 1-butanol accumulation for mutant strains L279F and G113N were 5.5- and 278-fold higher than the ratio for Rev WT (15), it was plausible that differences in regiospecific oxidation of CEs by the mutant strains would result in the production of different product profiles, leading to changes in toxic effects relative to the effects for Rev WT. The effects of DCE turnover on general cellular respiration were determined by measuring lactate-dependent O2 consumption during exposure to 1,1- or 1,2-cis-DCE. The DCE concentrations used in the assays were well below the transformation capacities to ensure that all available DCE would be consumed (8). For all strains, prior to addition of the DCE, the lactate-dependent O2 uptake rates were 28 to 32 nmol min−1. Upon addition of 1,1-DCE, the O2 uptake rates increased 52% for Rev WT and strain F321Y, 33% for strain L279F, and 30% for strain G113N. Although 1,1-DCE was completely consumed by all strains within 3 min, the impact on O2 uptake rates was remarkably different among strains (Fig. 2A). Lactate-dependent respiration by mutant strain G113N appeared to be insensitive to 1,1-DCE turnover. However, the rate of O2 uptake was reduced similarly in mutant strain F321Y and strain Rev WT. In contrast, the O2 uptake rate in strain L279F was intermediate between the rate in G113N and the rate in Rev WT. Since the chloride release value was similarly intermediate for strain L279F compared to the other strains, the data imply that different products produced during CE oxidation are less toxic.
FIG. 2.
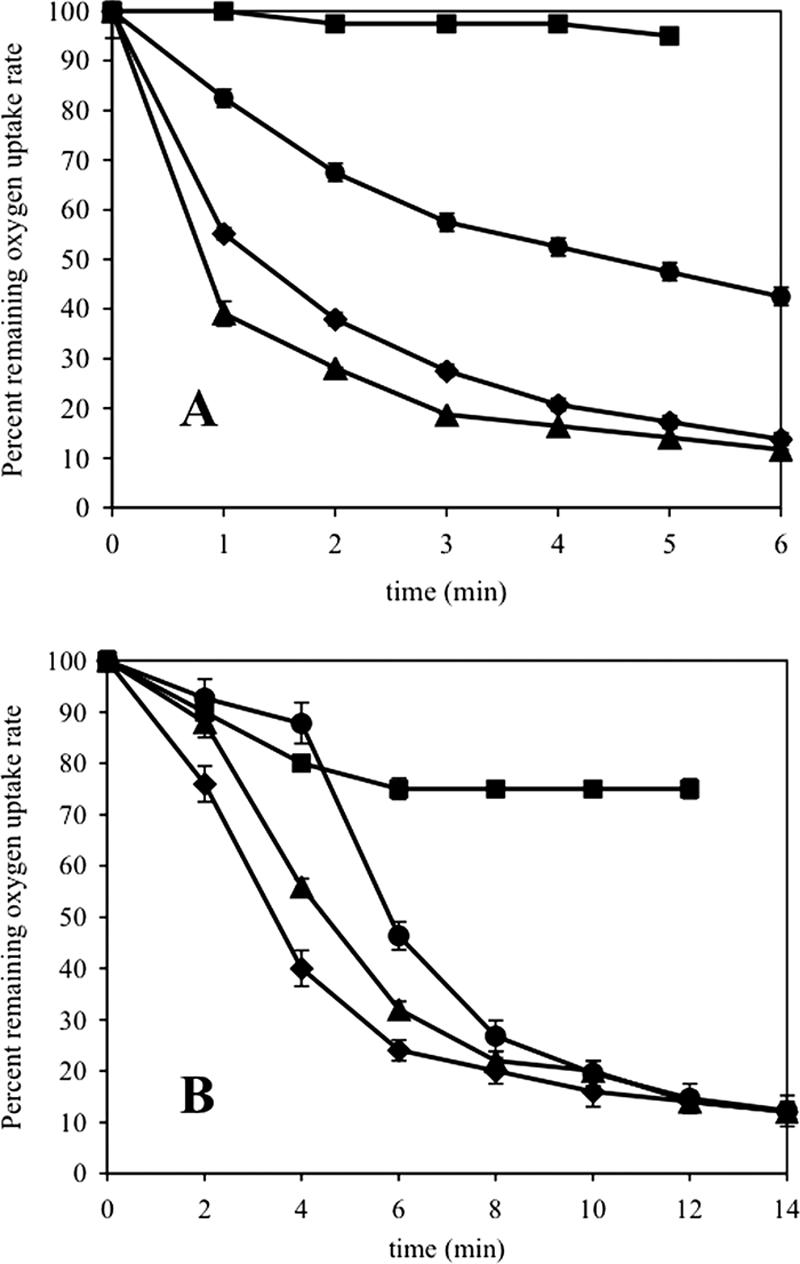
Differential effects of 1,1-DCE (A) and 1,2-cis-DCE (B) degradation on general cellular respiration in mutant strains of P. butanovora. Butane-grown cells were provided 3 mM lactate and 13 μM 1,1-DCE or 10 μM 1,2-cis-DCE. The slopes of tangents drawn to progress curves obtained during the assays were measured at the indicated time points to determine rates of O2 uptake. The results are expressed as percentages of the initial rate of respiration immediately following addition of DCE. Symbols: ▪, mutant strain G113N; •, L279F; ⧫, Rev WT; ▴, F321Y.
Differential effects on lactate-dependent O2 uptake rates were also measured during 1,2-cis-DCE oxidation (Fig. 2B). Addition of 1,2-cis-DCE immediately increased lactate-dependent O2 uptake rates 50% in Rev WT and strain F321Y, 30% in strain L279F, and 20% in strain G113N. The O2 uptake rate of Rev WT was reduced by 50% (to a rate equivalent to the original lactate-dependent O2 uptake rate) within 3.5 min, which is about the same time that the substrate was completely consumed. Mutant strain F321Y had a similar response to the control. However, the rate of O2 uptake in mutant strain L279F did not decline significantly until all available 1,2-cis-DCE was consumed (at about 4 min). Although mutant strain G113N consumed the available 1,2-cis-DCE in 8 min, its O2 uptake rate was reduced only 25% after 12 min. The differential sensitivities of general respiration to DCE turnover in strains G113N and L279F suggest that specific amino acid substitutions in BMO affected CE turnover-product distribution such that the mutant strains experienced strikingly different toxic impacts.
DISCUSSION
Mutant strains of P. butanovora containing single amino acid substitutions in BMOH-α have facilitated structure-function studies that are expanding our understanding of BMO-dependent oxidation reactions (15). To further probe the oxidative reactions of BMO, we investigated both product formation and physiological responses of butane-grown mutant and wild-type (Rev WT) strains of P. butanovora during exposure to chlorinated ethenes.
CE turnover-dependent toxicities are well documented in BMO and sMMO systems (1, 8, 16, 28). Although TCE degradation causes inactivation of sMMO (19) and BMO (16), the short half-life of TCE epoxide has prevented detailed information concerning the mechanism of inactivation from being obtained. Online gas chromatography detection of the formation and consumption of 1,2-cis-DCE epoxide in sMMO-expressing cells pinpointed turnover of the epoxide as the causative agent of the inactivation (30). A preepoxide intermediate has also been implicated in the covalent binding of the CE to the activated oxygen species of sMMO (14). Although the mechanism by which monooxygenases oxidize CE epoxides has not been elucidated, it is plausible that altered BMOs varying in epoxide affinity, turnover-dependent inactivation, and partitioning ratios will be useful tools for determining the key catalytic details that influence epoxide turnover products. For example, strain G113N was engineered to more closely resemble sMMO, and some of its phenotypes reflect an sMMO-like character, including subterminal oxidation of propane and butane and 3.5-fold less inhibition by methanol of methane oxidation (15), yet strain G113N appears to be unable to oxidize 1,2-cis-DCE epoxide.
CE epoxide breakdown products include highly reactive acyl chlorides that likely contribute to losses in cell viability by nonspecific binding to nucleic acids or other essential macromolecules. In strains Rev WT, F321Y, and L279F, 100% of the 1,2-cis-DCE degraded was accounted for as the corresponding epoxide. In contrast, only 48% of the 1,2-cis-DCE degraded by strain G113N was detected as epoxide. The differential sensitivities of the mutant strains to DCE turnover as measured by lactate-dependent O2 uptake suggest that DCE oxidation by G113N yields products that are less toxic. Furthermore, the steep decline in the O2 uptake rate of strain L279F began after commencement of 1,2-cis-DCE epoxide turnover. However, this strain liberated less chloride than Rev WT, but its rate of O2 uptake declined as fast as that of Rev WT. Taken together, these results suggest that the different products produced in strain L279F (e.g., glyoxyl chloride and/or formyl chloride) are very toxic but need to reach a critical concentration before causing cellular injury. Alternatively, the parent compound provides protection until it is depleted.
During exposure to CEs, the strain carrying the G→N-substituted BMO released at most 54% of the available chlorine, formed 48% 1,2-cis-DCE epoxide, and maintained greater than 75% of the lactate-dependent respiration. These results support the idea that strain G113N utilizes a CE oxidative pathway other than the pathway used by either wild-type BMO or sMMO during CE degradation. Because the loss of cellular respiration during CE degradation by wild-type P. butanovora is most severe during 1,1-DCE turnover (8), it was quite remarkable that respiration was unaffected by oxidation of 1,1-DCE in strain G113N. Interestingly, butane-grown M. vaccae also shows resistance to oxidation of 1,1-DCE (unpublished data), suggesting that M. vaccae probably does not form the corresponding epoxide during CE degradation. Furthermore, organochlorines accounted for 25% of the total products formed during TCE degradation by propane-grown M. vaccae, and only 53% of the available chlorine was released (26, 27).
The dramatic phenotypes described above were measured for mutant strains L279F and G113N. The ratios of the rate of 2-butanol accumulation to the rate of 1-butanol accumulation in these strains were previously shown to be 5.5- and 279-fold greater, respectively, than the ratio in Rev WT (15). Mutant strain F321Y oxidizes butane exclusively at the terminal position (15), and it formed stoichiometric amounts of 1,2-cis-DCE epoxide and was hypersensitive to 1,1-DCE degradation as measured by lactate-dependent O2 uptake. It appears that strain F321Y skews oxidation of CEs even more specifically towards epoxide formation than Rev WT.
Although the transient enzyme intermediates characterized in the sMMO system have not yet been identified in BMO, we believe that using a combination of CEs as substrate probes and the P. butanovora mutant strains may provide unprecedented insights into the mechanism of catalysis in the BMO system. The results obtained in this study indicate that wild-type BMO, like sMMO, primarily oxidizes CEs via an Hperoxo intermediate, forming unstable epoxides (Fig. 3). The epoxides are either attacked by BMO or spontaneously degrade to reactive compounds, leading to severe reductions in general cellular respiration and limiting CE transformation capacities. Because strain G113N formed less epoxide, released less chloride, and was less sensitive to CE degradation as measured by lactate-dependent O2 uptake, we propose that its reaction with CEs utilizes the Q state of the enzyme and radical rebound chemistry to a greater extent than the wild-type BMO (Fig. 3). Further work is needed to identify the specific products of CE degradation by BMO and to confirm the hypothesis in vitro with purified enzyme.
FIG. 3.

Proposed oxidative pathway of 1,1-DCE or TCE with BMO. X represents either Cl or H. Oxidation by the peroxodiiron(III) intermediate (Hperoxo) occurs by a two-electron transfer step and results in epoxide formation. Oxidation by the di(μ-oxo)diiron(IV) intermediate (Q) initiates at either carbon, with electron transfer forming a cationic intermediate. The cationic intermediate may alkylate the active site, resulting in enzyme inactivation, or leads to a chloride or hydride shift. Wild-type BMO primarily utilizes the Hperoxo enzyme intermediate for CE oxidation, and mutant strain G113N primarily utilizes the Q enzyme intermediate.
We used CEs with varying substituent positions and numbers to probe the enzymatic mechanism of P. butanovora BMOH-α mutants. The results are rationally explained by the presence of different oxidative pathways initiated by different enzymatic intermediates. Alteration of a single amino acid in BMOH-α appears to have created an enzymatic mechanism that oxidizes CEs primarily via the Q enzymatic intermediate. The resulting product profiles of CE degradation also have significant physiological consequences. Since biodegradation is limited by product toxicity in the form of enzyme inactivation or loss of cellular viability, the results obtained in this study indicate that the oxidative pathway favored by mutant strain G113N would promote more sustainable biodegradation of CEs.
Acknowledgments
This research was supported through grant 5R01GM56128-06 from the National Institutes of Health. This work was also supported by the office of Research and Development, U.S. Environmental Protection Agency, under agreement R-828772 through the Western Region Hazardous Substance Research Center.
Footnotes
Published ahead of print on 11 May 2007.
REFERENCES
- 1.Alvarez-Cohen, L., and P. L. McCarty. 1991. Product toxicity and cometabolic competitive inhibition modeling of chloroform and trichloroethylene transformation by methanotrophic resting cells. Appl. Environ. Microbiol. 57:1031-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arp, D. J. 1999. Butane metabolism by butane-grown Pseudomonas butanovora. Microbiology 145:1173-1180. [DOI] [PubMed] [Google Scholar]
- 3.Arp, D. J., C. M. Yeager, and M. R. Hyman. 2001. Molecular and cellular fundamentals of aerobic cometabolism of trichloroethylene. Biodegradation 12:81-103. [DOI] [PubMed] [Google Scholar]
- 4.Baik, M. H., M. Newcomb, R. A. Friesner, and S. J. Lippard. 2003. Mechanistic studies on the hydroxylation of methane by methane monooxygenase. Chem. Rev. 103:2385-2419. [DOI] [PubMed] [Google Scholar]
- 5.Beauvais, L. G., and S. J. Lippard. 2005. Reactions of the diiron(IV) intermediate Q. in soluble methane monooxygenase with fluoromethanes. Biochem. Biophys. Res. Commun. 338:262-266. [DOI] [PubMed] [Google Scholar]
- 6.Beauvais, L. G., and S. J. Lippard. 2005. Reactions of the peroxo intermediate of soluble methane monooxygenase hydroxylase with ethers. J Am. Chem. Soc. 127:7370-7378. [DOI] [PubMed] [Google Scholar]
- 7.Brazeau, B. J., and J. D. Lipscomb. 2000. Kinetics and activation thermodynamics of methane monooxygenase compound Q. Formation and reaction with substrates. Biochemistry 39:13503-13515. [DOI] [PubMed] [Google Scholar]
- 8.Doughty, D. M., L. A. Sayavedra-Soto, D. J. Arp, and P. J. Bottomley. 2005. Effects of dichloroethene isomers on the induction and activity of butane monooxygenase in the alkane-oxidizing bacterium “Pseudomonas butanovora.” Appl. Environ. Microbiol. 71:6054-6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fox, B. G., J. G. Borneman, L. P. Wackett, and J. D. Lipscomb. 1990. Haloalkene oxidation by soluble methane monooxygenase from Methylosinus trichosporium Ob3b: mechanistic and environmental implications. Biochemistry 29:6419-6427. [DOI] [PubMed] [Google Scholar]
- 10.Gherman, B. F., M. H. Baik, S. J. Lippard, and R. A. Friesner. 2004. Dioxygen activation in methane monooxygenase: a theoretical study. J Am. Chem. Soc. 126:2978-2990. [DOI] [PubMed] [Google Scholar]
- 11.Gherman, B. F., B. D. Dunietz, D. A. Whittington, S. J. Lippard, and R. A. Friesner. 2001. Activation of the C-H bond of methane by intermediate Q of methane monooxygenase: a theoretical study. J Am. Chem. Soc. 123:3836-3837. [DOI] [PubMed] [Google Scholar]
- 12.Gherman, B. F., S. J. Lippard, and R. A. Friesner. 2005. Substrate hydroxylation in methane monooxygenase: quantitative modeling via mixed quantum mechanics/molecular mechanics techniques. J Am. Chem. Soc. 127:1025-1037. [DOI] [PubMed] [Google Scholar]
- 13.Gossett, J. M. 1987. Measurement of Henry's law constants for C1 and C2 chlorinated hydrocarbons. Environ. Sci. Technol. 21:202-208. [Google Scholar]
- 14.Green, J., and H. Dalton. 1989. Substrate specificity of soluble methane monooxygenase. Mechanistic implications. J. Biol. Chem. 264:17698-17703. [PubMed] [Google Scholar]
- 15.Halsey, K. H., L. A. Sayavedra-Soto, P. J. Bottomley, and D. J. Arp. 2006. Site-directed amino acid substitutions in the hydroxylase alpha subunit of butane monooxygenase from Pseudomonas butanovora: implications for substrates knocking at the gate. J. Bacteriol. 188:4962-4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halsey, K. H., L. A. Sayavedra-Soto, P. J. Bottomley, and D. J. Arp. 2005. Trichloroethylene degradation by butane-oxidizing bacteria causes a spectrum of toxic effects. Appl. Microbiol. Biotechnol. 68:794-801. [DOI] [PubMed] [Google Scholar]
- 17.Hamamura, N., C. Page, T. Long, L. Semprini, and D. J. Arp. 1997. Chloroform cometabolism by butane-grown CF8, Pseudomonas butanovora, and Mycobacterium vaccae JOB5 and methane-grown Methylosinus trichosporium OB3b. Appl. Environ. Microbiol. 63:3607-3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamamura, N., R. T. Storfa, L. Semprini, and D. J. Arp. 1999. Diversity in butane monooxygenases among butane-grown bacteria. Appl. Environ. Microbiol. 65:4586-4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanson, R. S., and T. E. Hanson. 1996. Methanotrophic bacteria. Microbiol. Rev. 60:439-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liebler, D. C., and F. P. Guengerich. 1983. Olefin oxidation by cytochrome P-450: evidence for group migration in catalytic intermediates formed with vinylidene chloride and trans-1-phenyl-1-butene. Biochemistry 22:5482-5489. [DOI] [PubMed] [Google Scholar]
- 21.Lipscomb, J. D. 1994. Biochemistry of the soluble methane monooxygenase. Annu. Rev. Microbiol. 48:371-399. [DOI] [PubMed] [Google Scholar]
- 22.Lontoh, S., J. A. Zahn, A. A. DiSpirito, and J. D. Semrau. 2000. Identification of intermediates of in vivo trichloroethylene oxidation by the membrane-associated methane monooxygenase. FEMS Microbiol. Lett. 186:109-113. [DOI] [PubMed] [Google Scholar]
- 23.Miller, R. E., and F. P. Guengerich. 1982. Oxidation of trichloroethylene by liver microsomal cytochrome P-450: evidence for chlorine migration in a transition state not involving trichloroethylene oxide. Biochemistry 21:1090-1097. [DOI] [PubMed] [Google Scholar]
- 24.Shinohara, Y., H. Uchiyama, and I. Kusakabe. 1998. Oxidation of some alkanes and trichloroethylene by H2O2/hydroxylase system of soluble methane monooxygenase from Methylocystis sp. M. J. Ferment. Bioeng. 85:266-270. [Google Scholar]
- 25.Sluis, M. K., L. A. Sayavedra-Soto, and D. J. Arp. 2002. Molecular analysis of the soluble butane monooxygenase from ‘Pseudomonas butanovora.’ Microbiology 148:3617-3629. [DOI] [PubMed] [Google Scholar]
- 26.Vanderberg, L. A., B. L. Burback, and J. J. Perry. 1995. Biodegradation of trichloroethylene by Mycobacterium vaccae. Can. J. Microbiol. 41:298-301. [DOI] [PubMed] [Google Scholar]
- 27.Vanderberg, L. A., and J. J. Perry. 1994. Dehalogenation by Mycobacterium vaccae JOB-5: role of the propane monooxygenase. Can. J. Microbiol. 40:169-172. [DOI] [PubMed] [Google Scholar]
- 28.van Hylckama Vlieg, J., W. De Koning, and D. Janssen. 1997. Effect of chlorinated ethene conversion on viability and activity of Methylosinus trichosporium OB3b. Appl. Environ. Microbiol. 63:4961-4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Hylckama Vlieg, J. E., and D. B. Janssen. 2001. Formation and detoxification of reactive intermediates in the metabolism of chlorinated ethenes. J. Biotechnol. 85:81-102. [DOI] [PubMed] [Google Scholar]
- 30.van Hylckama Vlieg, J. E. T., W. deKoning, and D. B. Janssen. 1996. Transformation kinetics of chlorinated ethenes by Methylosinus trichosporium OB3b and detection of unstable epooxides by on-line gas chromatography. Appl. Environ. Microbiol. 62:3304-3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeager, C. M., P. J. Bottomley, and D. J. Arp. 2001. Cytotoxicity associated with trichloroethylene oxidation in Burkholderia cepacia G4. Appl. Environ. Microbiol. 67:2107-2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang, J., and J. D. Lipscomb. 2006. Role of the C-terminal region of the B component of Methylosinus trichosporium OB3b methane monooxygenase in the regulation of oxygen activation. Biochemistry 45:1459-1469. [DOI] [PubMed] [Google Scholar]
- 33.Zheng, H., and J. D. Lipscomb. 2006. Regulation of methane monooxygenase catalysis based on size exclusion and quantum tunneling. Biochemistry 45:1685-1692. [DOI] [PubMed] [Google Scholar]