

The three-dimensional structure of the APE2540 protein from A. pernix K1 has been determined by the multiple anomalous dispersion method at 1.7 Å resolution. The structure includes two monomers in the asymmetric unit and shares structural similarity with the YbaK protein or cysteinyl-tRNAPro deacylase from H. influenzae.
Keywords: trans-editing enzymes, APE2540
Abstract
The crystal structure of APE2540, the putative trans-editing enzyme ProX from Aeropyrum pernix K1, was determined in a high-throughput manner. The crystal belongs to the monoclinic space group P21, with unit-cell parameters a = 47.4, b = 58.9, c = 53.6 Å, β = 106.8°. The structure was solved by the multiwavelength anomalous dispersion method at 1.7 Å and refined to an R factor of 16.8% (R free = 20.5%). The crystal structure includes two protein molecules in the asymmetric unit. Each monomer consists of eight β-strands and seven α-helices. A structure-homology search revealed similarity between the trans-editing enzyme YbaK (or cysteinyl-tRNAPro deacylase) from Haemophilus influenzae (HI1434; 22% sequence identity) and putative ProX proteins from Caulobacter crescentus (16%) and Agrobacterium tumefaciens (21%).
1. Introduction
The aerobic hyperthermophilic crenarchaeon Aeropyrum pernix K1, which is evolutionarily separated from the anaerobic hyperthermophilic euryarchaeota including the genus Pyroccocus, is a good target for structural genomics; the genome size is small and the proteins are highly thermostable. The A. pernix K1 genome has been completely sequenced (Kawarabayasi et al., 1999 ▶). We are particularly interested in proteins involved in archaeal protein synthesis in comparison with those involved in protein synthesis in bacteria and eukaryotes. In the present study we chose the APE2540 protein as a target. The APE2540 protein consists of 152 amino-acid residues, with a molecular weight of 16.3 kDa. A Pfam analysis (Bateman et al., 2004 ▶) predicted that the APE2540 protein could be a prolyl-tRNA synthetase-associated domain. A sequence-homology search using BLAST (Altschul et al., 1997 ▶) for the APE2540 protein revealed 22% sequence identity to the YbaK protein, which deacylates cysteinyl-tRNAPro misformed by prolyl-tRNA synthetase, from the bacterium Haemophilus influenzae (An & Musier-Forsyth, 2004 ▶). Here, we report the three-dimensional structure of the APE2540 protein determined by the multiple anomalous dispersion method (MAD) at 1.7 Å.
2. Materials and methods
The APE2540 gene was amplified by PCR from A. pernix K1 genomic DNA. The PCR product was cloned into the pET11a expression vector (Novagen). The selenomethionine (SeMet) labelled APE2540 protein was expressed in the Escherichia coli methionine auxotroph B834(DE3). The cells were cultured at 310 K in LeMaster medium (LeMaster & Richards, 1985 ▶) containing SeMet.
After disruption of the cells by sonication, the lysate was incubated at 343 K for 30 min and centrifuged to remove the denatured protein. The supernatant was applied onto a HiTrap SP-Sepharose column (Amersham Biosciences) equilibrated with 20 mM MES buffer pH 5.5, 1 mM DTT. The APE2540 protein was eluted with a linear gradient of NaCl to 1 M. Peak fractions were pooled and ammonium sulfate was added to the pooled solution to a final concentration of 1.2 M. The mixture was loaded onto a Resource PHE column (Amersham Biosciences). The APE2540 protein was eluted with a decreasing linear gradient of 1.2–0 M ammonium sulfate. After gel filtration on a HiLoad 16/60 Superdex75 prep-grade column (Amersham Biosciences) equilibrated with 20 mM Tris–HCl buffer pH 8.0 containing 150 mM NaCl and 1 mM DTT, the protein was concentrated to a final concentration of 10 mg ml−1 using a Centricon filter unit (Millipore).
Using a screen implemented in the TERA automatic crystallization system using the microbatch method (Sugahara & Miyano, 2002 ▶), crystals of the APE2540 protein were obtained in 30 d at 291 K in 20% PEG 20 000, 0.1 M citrate buffer pH 5.2. X-ray diffraction data were collected with a Jupiter CCD detector installed on BL26B2 at the SPring-8 synchrotron facility (Harima, Japan) using flash-frozen crystals with Paratone-N at 100 K. All X-ray diffraction data were integrated and scaled using the HKL2000 package (Otwinowski & Minor, 1997 ▶). Data-collection statistics are summarized in Table 1 ▶.
Table 1. Summary of crystal parameters, data collection and refinement statistics.
(a).
Crystal characteristics.
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 47.435, b = 58.917, c = 53.603, β = 106.801 |
| Molecules per AU | 2 |
| SeMet residues AU | 6 |
(b).
MAD data. Values in parentheses refer to the highest resolution shell, which is 1.76–1.70 Å for all wavelength data.
| Edge | Peak | High-energy remote | |
|---|---|---|---|
| Wavelength (Å) | 0.97957 | 0.97920 | 0.96400 |
| Resolution range (Å) | 30.0–1.70 | 30.0–1.70 | 30.0–1.70 |
| Redundancy | 5.3 | 5.3 | 5.3 |
| Unique reflections | 31182 | 31201 | 31231 |
| Completeness (%) | 99.9 (100.0) | 99.9 (100.0) | 99.9 (100.0) |
| I/σ(I) | 22.9 (13.5) | 15.4 (12.5) | 20.9 (13.5) |
| Rsym† | 0.071 (0.116) | 0.083 (0.135) | 0.072 (0.120) |
| Figure of merit (FOM) | 0.50 |
(c).
Refinement statistics.
| Resolution range (Å) | 30.0–1.70 |
| Unique reflections | 31180 |
| R factor/free R factor‡ | 0.168/0.205 |
| No. protein atoms | 2264 |
| No. water molecules | 503 |
| R.m.s. deviations from ideal geometry | |
| Bond lengths (Å) | 0.011 |
| Bond angles (°) | 1.5 |
| Average isotropic B value (Å2) | 16.6 |
R
sym = 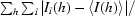
 .
.
R factor = 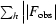 −
− 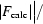
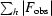 . The free R factor was calculated using 5% of reflections omitted from refinement.
. The free R factor was calculated using 5% of reflections omitted from refinement.
The crystal structure of the SeMet-labelled APE2540 protein was determined by the MAD method. SOLVE (Terwilliger & Berendzen, 1999 ▶) was used to locate the selenium sites and to calculate the phases. A total of four selenium sites were included for phase calculations. Two selenium sites at the N-termini were not determined owing to disorder. Electron-density modification was subsequently performed with the program RESOLVE (Terwilliger & Berendzen, 1999 ▶). The automatic tracing procedure in ARP/wARP (Morris et al., 2004 ▶) and manual model building by O (Jones et al., 1991 ▶) were used to complete the modelling of the protein molecules.
The structure was refined using the CNS program package (Brünger et al., 1998 ▶). All refinement steps were monitored with the free R factor based on 5% of the X-ray data. Following a simulated-annealing protocol, the structure was refined using atom-positional and temperature-factor refinement, as well as manual model building. The stereochemical quality of the final model was assessed using PROCHECK (Laskowski et al., 1993 ▶) and WHATIF (Vriend, 1990 ▶). The Ramachandran plot demonstrated that 96.8% of the residues lie in the most favoured regions and 3.2% of the residues are in the additionally allowed regions. The refinement statistics are presented in Table 1 ▶.
3. Results and discussion
The structure of the APE2540 protein was determined using highly automated systems. Crystals were produced by the crystallization robot TERA with microbatch plates; initial screening trials were for 144 conditions, containing a wide range of precipitants, buffers and salts (Sugahara & Miyano, 2002 ▶). The crystals thus obtained were flash-frozen and then stored in liquid nitrogen with a special cryo-loop for automounting (Ueno et al., 2004 ▶). Data collection was performed automatically, including the XAFS measurement, after appropriate process scheduling. For the high-resolution data, the automatic model-building algorithm worked well. Two residues in the N-terminal regions of both molecules could not be identified in the electron-density map owing to disorder. It took less than a week to complete the structure refinement after the synchrotron trip for data collection. Fig. 1 ▶(a) shows the final electron-density map.
Figure 1.

(a) Quality of the electron-density map. The refined structure is represented by a thin wire. The map is contoured at the 1.0σ level. (b) Stereo diagram of the APE2540 protein. The secondary structures are coloured blue (α-helices) and red (β-strands). (c) Comparison of the structural homologues of the APE2540 protein. The three structures are coloured and oriented in the same direction as in Fig. 1 ▶(b). Figs. 1(b) and 1(c) were produced with Molscript (Kraulis, 1991 ▶) and Raster3D (Merritt & Bacon, 1997 ▶).
The crystal structure includes two molecules, A and B, in the asymmetric unit. These molecules are related by local twofold symmetry and can be superimposed with an r.m.s.d. value of 0.9 Å. The structure shows an α/β fold, which possesses eight β-strands (β1–β8) and seven α-helices (αA–αG) (Fig. 1 ▶ b). Although all the β-strands form a continuously connected β-sheet, the sheet can be divided into two parts because of the highly twisted strand (β2). The first part is an antiparallel β-sheet with the order β1–β7–β6. The second sheet is composed of β4–β2–β3–β5–β8, with β2 arranged in an antiparallel direction. The β-sheet forms a hydrophobic core flanked by α-helices.
A structural homology search was conducted using the DALI server (Holm & Sander, 1998 ▶). The structural homologues (Z score > 10) of the APE2540 protein are the YbaK protein from H. influenzae (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1dbu; Zhang et al., 2000 ▶), with a Z score of 21.4, and a putative DNA-binding protein from Caulobacter crescentus (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1vjf), with a Z score of 13.8. In addition, the MATRAS program (Kawabata, 2003 ▶) indicated that a hypothetical protein from Agrobacterium tumefaciens (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1vki) is also a structural homologue of APE2450. Note that all the three proteins are from bacteria. Fig. 1 ▶(c) shows these structures from the same viewpoint. The overall structures of these proteins superimpose well, with an r.m.s.d. value of 1.8 Å for 145 common Cα atoms (APE2540 versus http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1dbu), 2.4 Å for 140 common Cα atoms (APE2540 versus http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1vjf) and 2.3 Å for 145 common Cα atoms (APE2540 versus http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1vki). On the other hand, the sequence of APE2540 is not highly homologous to those of these three proteins; its sequence identities with H. influenzae YbaK (http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1dbu), the C. crescentus putative DNA-binding protein (http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1vjf) and the A. tumefaciens hypothetical protein (http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1vki) are as low as 22, 16 and 21%, respectively (Fig. 2 ▶).
Figure 2.

Sequence alignment among structural homologues of APE2540. Blue bars and red arrows above the sequences show secondary-structural elements (α-helix and β-sheet). Identical amino acids are highlighted in red. The Lys residues conserved for deacylation activity are highlighted in green.
Sequence homologues of the H. influenzae YbaK (ProX proteins) are widely found in bacteria, archaea and eukaryotes. A ProX-like insertion domain also occurs in a number of prolyl-tRNA synthetases (ProRSs) from bacteria, including H. influenzae. ProRSs are known to misrecognize non-cognate amino acids such as alanine and cysteine and charge them to proline-specific tRNAs (tRNAPro). In the case of H. influenzae, alanyl-tRNAPro is deacylated (or ‘edited’) by the domain inserted in ProRS (the ‘cis-editing’ domain), while cysteinyl-tRNAPro is deacylated by YbaK or the ‘trans-editing’ enzyme ProX (An & Musier-Forsyth, 2004 ▶). In archaea, eukaryotes and some bacteria, ProRSs lack the cis-editing domain and therefore the trans-editing enzyme ProX is supposed to deacylate mischarged tRNAPro (Ahel et al., 2003 ▶). ProRS from the bacterium Clostridium sticklandii lacks the cis-editing domain, while PrdX, the trans-editing enzyme ProX from C. sticklandii, has been shown to specifically deacylate alanyl-tRNAPro (Ahel et al., 2003 ▶). A Lys residue that plays a crucial role in the deacylation activity of the C. sticklandii and H. influenzae ProXs (Ahel et al., 2003 ▶; An & Musier-Forsyth, 2004 ▶) is conserved in the ProX sequences (Lys44 for A. pernix ProX or APE2540; Wong et al., 2002 ▶; Fig. 2 ▶).
The A. pernix, C. crescentus and A. tumefaciens ProRSs lack the cis-editing domain. It is therefore possible that the A. pernix, C. crescentus and A. tumefaciens ProXs have deacylation activity for both alanyl-tRNAPro and cysteinyl-tRNAPro, while it is also possible that they deacylate only one of them. The bacterial ProXs from C. crescentus and A. tumefaciens, which lack cis-editing by ProRS, are highly homologous to each other (the identity of 46%), but far less homologous to another bacterial ProX, that from H. influenzae, with the cis-editing ProRS (both 16% identity). The C. sticklandii ProX (PrdX), which has well been characterized with respect to its cis-editing function (Ahel et al., 2003 ▶) but whose crystal structure is not yet known, is more homologous to the C. crescentus and A. tumefaciens ProXs (30 and 25% identity, respectively) than to the H. influenzae ProX (YbaK) (19% identity). On the other hand, APE2540, the putative ‘trans-editing’ enzyme ProX from an archaeon, A. pernix, shows sequence identities of 16–22% with these bacterial ProXs. The present structure is the first archaeal ProX structure. The specificities and mechanisms of trans-editing in archaeal protein synthesis could be further studied on the basis of the 1.7 Å resolution structure of the A. pernix ProX, as well as the mischarging specificities of archaeal and archaea-like ProRSs lacking the cis-editing domain on the basis of their structures (Yaremchuk et al., 2000 ▶, 2001 ▶; Kamtekar et al., 2003 ▶).
Supplementary Material
PDB reference: APE2540, 1wdv, r1wdvsf
Acknowledgments
Special thanks to the project secretarial staff for all their assistance, particularly that of Ms Tomoko Nakayama. We thank Dr R. Hirose for help with data collection at the RIKEN beamline BL26B2 at SPring-8, Dr M. Sugahara and Y. Nakamura for manipulation of the TERA robot and E. Fusatomi for technical assistance with protein purification. This work was supported by the RIKEN Structural Genomics/Proteomics Initiative (RSGI) and the National Project on Protein Structural and Functional Analyses, Ministry of Education, Culture, Sports, Science and Technology.
References
- Ahel, I., Korencic, D., Ibba, M. & Söll, D. (2003). Proc. Natl Acad. Sci. USA, 100, 15422–15427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997). Nucleic Acids Res.25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An, S. & Musier-Forsyth, K. (2004). J. Biol. Chem.279, 42359–42362. [DOI] [PubMed] [Google Scholar]
- Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E. L. L., Studholme, D. J., Yeats, C. & Eddy, S. R. (2004). Nucleic Acids Res.32, D138–D141. [DOI] [PMC free article] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Holm, L. & Sander, C. (1998). Nucleic Acids Res.26, 316–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kamtekar, S., Kennedy, W. D., Wang, J., Stathopoulos, C., Söll, D. & Steitz, T. A. (2003). Proc. Natl Acad. Sci. USA, 100, 1673–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata, T. (2003). Nucleic Acids Res.31, 3367–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayasi, Y. et al. (1999). DNA Res.6, 83–101. [DOI] [PubMed] [Google Scholar]
- Kraulis, P. J. (1991). J. Appl. Cryst.24, 946–950. [Google Scholar]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291. [Google Scholar]
- LeMaster, D. M. & Richards, F. M. (1985). Biochemistry, 24, 7263–7268. [DOI] [PubMed] [Google Scholar]
- Merritt, E. A. & Bacon, D. J. (1997). Methods Enzymol.277, 505–524. [DOI] [PubMed]
- Morris, R. J., Zwart, P. H., Cohen, S., Fernandez, F. J., Kakaris, M., Kirillova, O., Vonrhein, C., Perrakis, A. & Lamzin, V. S. (2004). J. Synchrotron Rad.11, 56–59. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Sugahara, M. & Miyano, M. (2002). Tanpakushitsu Kakusan Koso, 47, 1026–1032. [PubMed] [Google Scholar]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno, G., Hirose, R., Ida, K., Kumasaka, T. & Yamamoto, M. (2004). J. Appl. Cryst.37, 867–873. [Google Scholar]
- Vriend, G. (1990). J. Mol. Graph.8, 52–56. [DOI] [PubMed] [Google Scholar]
- Wong, F. C., Beuning, P. J., Nagan, M., Shiba, K. & Musier-Forsyth, K. (2002). Biochemistry, 41, 7108–7155. [DOI] [PubMed] [Google Scholar]
- Yaremchuk, A., Cusack, S. & Tukalo, M. (2000). EMBO J.19, 4745–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaremchuk, A., Tukalo, M., Grøtli, M. & Cusack, S. (2001). J. Mol. Biol.309, 989–1002. [DOI] [PubMed] [Google Scholar]
- Zhang, H., Huang, K., Li, Z., Banerjei, L., Fisher, K. E., Grishin, N. V., Eisenstein, E. & Herzberg, O. (2000). Proteins, 40, 86–97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: APE2540, 1wdv, r1wdvsf