

An unannotated protein reported from B. subtilis has been expressed in E. coli and identified as possessing penicillin V acylase activity. The crystallization and preliminary crystallographic analysis of this penicillin V acylase is presented.
Keywords: Ntn hydrolase, penicillin V acylase, conjugated bile-salt hydrolase
Abstract
Penicillin acylase proteins are amidohydrolase enzymes that cleave penicillins at the amide bond connecting the side chain to their β-lactam nucleus. An unannotated protein from Bacillus subtilis has been expressed in Escherichia coli, purified and confirmed to possess penicillin V acylase activity. The protein was crystallized using the hanging-drop vapour-diffusion method from a solution containing 4 M sodium formate in 100 mM Tris–HCl buffer pH 8.2. Diffraction data were collected under cryogenic conditions to a spacing of 2.5 Å. The crystals belonged to the orthorhombic space group C2221, with unit-cell parameters a = 111.0, b = 308.0, c = 56.0 Å. The estimated Matthews coefficient was 3.23 Å3 Da−1, corresponding to 62% solvent content. The structure has been solved using molecular-replacement methods with B. sphaericus penicillin V acylase (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=2pva) as the search model.
1. Introduction
Penicillin acylases (also called amidases or amidohydrolases; EC 3.5.1.11) are enzymes that cleave the amide bond between the side chain and the β-lactam nucleus of penicillins without affecting the amide bond within the β-lactam ring. They are pharmaceutically very important enzymes and are used to manufacture 6-aminopenicillanic acid (6-APA), the precursor for the industrial production of various semi-synthetic penicillins (Shewale & SivaRaman, 1989 ▶; Bruggink et al., 1998 ▶).
Penicillin acylases (PAs) are classified based on their substrate specificities. Penicillin V acylase (PVA), for example, cleaves the phenoxyacetic acid side chain of penicillin V (PenV), yielding 6-APA. The advantage of using PenV over penicillin G (PenG) for the production of semi-synthetic penicillins is its higher stability in aqueous solutions and its tolerance to the conditions of extraction. Moreover, PVA offers the advantage of being more specific and tolerates higher concentrations of substrate compared with penicillin G acylase (PGA; Shewale & Sudhakaran, 1997 ▶). PAs are produced by a range of organisms, with PVA proteins mostly found in fungi. The physiological role of PAs in the organisms that produce them is not clear.
PVA has been shown to belong to the family of N-terminal nucleophile (Ntn) hydrolases. These proteins undergo a post-translational autocatalytic cleavage that exposes the N-terminal amino group of the nucleophile residue and this free terminal amino group in turn acts as the base in catalysis (Suresh et al., 1999 ▶). The members of the Ntn hydrolase superfamily share a common αββα fold (Brannigan et al., 1995 ▶). PVA exhibits sequence similarity with other penicillin V acylases as well as with conjugated bile-salt (acid) hydrolases (CBH), enzymes that catalyse the hydrolysis of the bond conjugating bile acids with glycine or taurine.
During a homology search in protein sequence databases using Bacillus sphaericus PVA as the query sequence, we came across YxeI, a protein of unknown function, in B. subtilis. This protein shares 40% amino-acid sequence identity with B. sphaericus PVA, including conserved residues identified as crucial for its activity. This led us to test the B. subtilis YxeI protein for PVA activity. Here, we report the cloning, purification, crystallization and preliminary investigations into the structure of YxeI, which we have determined to be B. subtilis PVA.
2. Materials and methods
2.1. Cloning and overexpression
The yxeI gene was amplified by PCR using chromosomal DNA of B. subtilis strain IG-20 (NCIMB 11621) as template. Forward (5′-GGGACTGATCATATGTGCACAAGTCTTAC-3′) and reverse (5′-ATTGAGGATCCTTAATTAAGCTCATGAATACTCT-3′) oligonucleotide primers were designed to incorporate NdeI and BamHI restriction sites and were synthesized (Applied Biosystems 373). The PCR fragment was digested and cloned into the plasmid pET26b (Novagen). Recombinants were transformed into Escherichia coli BL21(DE3) cells and screened for overproduction of an ∼35 kDa protein to indicate a positive clone. An overnight culture of E. coli BL21(DE3)/pET26-yxeI cells in Luria–Bertani medium containing 30 µg ml−1 kanamycin was used as a 5% inoculum for shake-flask cultures, which were incubated at 310 K and shaken at 200 rev min−1 until the optical density reached 0.6 at 600 nm. Cell culture in the mid-logarithmic growth phase was supplied with isopropyl-β-d-thiogalactopyranoside to a final concentration of 1 mM to induce protein production. The culture was allowed to incubate at 303 K for a further 4 h with shaking. The culture broth was centrifuged at 8000g in a Sorvall refrigerated centrifuge for 30 min to sediment the cells (1.6 g wet weight per litre), which were stored frozen at 253 K until use.
2.2. Protein purification
Cells were resuspended in 10 ml 50 mM potassium phosphate buffer pH 6.0 (buffer A) and sonicated on ice using a Biosonik III sonic oscillator for 5 × 1 min at 20 kHz and 300 W, with 10 min intervals between pulses. The sonicate was centrifuged at 10 000g for 20 min to remove cell debris. The supernatant was mixed with 7 mg ml−1 streptomycin sulfate, stirred in the cold for 1 h and centrifuged for 20 min at 10 000g to remove precipitated DNA. The supernatant was mixed slowly with ammonium sulfate (AS) in the cold to a final concentration of 80% saturation. After centrifugation (10 000g for 20 min), the precipitate was dissolved in buffer A and dialysed against buffer B (buffer A + 24% AS). This sample was loaded onto a phenyl Sepharose column (CL 4B, Sigma) that had been pre-equilibrated with buffer B. The column was washed with 50 ml of the same buffer and eluted with buffer A. The fractions were checked for enzyme activity and the positive fractions were run on 12% SDS–PAGE gels. Fractions containing pure protein were pooled, dialysed against buffer A and concentrated by ultrafiltration through a YM-10 membrane (Amicon). The protein concentration was determined using the method of Lowry et al. (1951 ▶) with bovine serum albumin as calibration standard. The purified enzyme, which was >99% homogeneous as estimated by SDS–PAGE, was concentrated to 10 mg ml−1 and stored at 253 K in aliquots. Penicillin V acylase activity was measured by reacting the 6-amino group of the product 6-APA with p-dimethylaminobenzaldehyde to yield a chromogenic Schiff base (Shewale et al., 1987 ▶).
2.3. Crystallization
After screening using a wide range of different conditions and precipitants, the protein eventually crystallized with reservoir solution containing 4 M sodium formate in 100 mM Tris–HCl buffer pH 8.2 using the hanging-drop vapour-diffusion method (Ducruix & Giegé, 1992 ▶; McPherson, 1982 ▶) with 1 µl of the precipitant solution mixed with 1 µl protein solution at a concentration of 10 mg ml−1 in 0.1 M NaCl. Crystals grew over a week at 291 K.
2.4. X-ray diffraction analysis
Diffraction data were collected in-house at 120 K. The cryoprotectant used was 30% glycerol in crystal-growth buffer. A single crystal scooped up using a cryoloop was momentarily soaked in cryoprotectant solution and immediately flash-frozen in a stream of liquid nitrogen before being mounted on the goniometer. Diffraction data to a Bragg spacing of 2.5 Å were recorded on an R-AXIS IV++ image-plate system mounted on a Rigaku rotating-anode generator operating at 50 kV and 100 mA. The crystal was rotated through 0–180° in oscillation intervals of 0.5° with 5 min exposure per frame; the crystal-to-detector distance was 260 mm. The Cu Kα radiation was monochromated and focused using Osmic mirrors and the data-collection control and initial indexing were performed using the program CrystalClear (Rigaku-MSC, USA).
3. Results and discussion
The yxeI gene from B. subtilis has been cloned and the protein overexpressed in E. coli. The DNA sequence of independent recombinants suggests a number of differences to the published sequence (Yoshida et al., 1995 ▶), leading to four amino-acid substitutions: Ala68, Phe69, Met90 and Ser218 are replaced with Gly, Ile, Thr and Pro, respectively (Fig. 1 ▶). Similarity searches revealed that the most homologous sequences to YxeI are predicted proteins that are annotated as conjugated bile-salt hydrolase (CBH) enzymes from other Bacillus species, e.g. B. licheniformis. Sequence comparison with the structurally characterized B. sphaericus PVA (Suresh et al., 1999 ▶) and the recently crystallized bile-salt hydrolase from Bifidobacterium longum (Suresh Kumar et al., 2004 ▶) reveals that like CBH, the catalytic Cys is unmasked by simple processing of the preceding fMet and the protein thus does not possess an N-terminal propeptide as reported for B. sphaericus PVA (Manish Chandra et al., 2005 ▶). The sequence comparison also highlights that YxeI shares 40% identity with B. sphaericus PVA and 30% identity to the more distantly related Bifidobacterium bile-salt hydrolase.
Figure 1.

Sequence comparison of B. subtilis YxeI with B. sphaericus PVA (BspPVA) and conjugated bile-acid hydrolase from Bifidobacterium longum (BloCBH) using the program ESPript (Gouet et al., 1999 ▶). The proteins are numbered from their mature form, with the catalytic N-terminal Cys residue at position 1.
The purified protein behaves as a tetramer with an estimated molecular weight of ∼140 kDa on gel filtration (data not shown), displays PVA activity (5 µmol min−1 mg−1 specific activity) and lacks any detectable CBH activity. The full biochemical characterization of PVA from B. subtilis will be described elsewhere. The crystals of PVA from B. subtilis grew from precipitant solutions containing 4 M sodium formate buffered at pH 8.2 and reached their full size (typically 0.54 mm in the longest dimension) in a week (Fig. 2 ▶). The crystals belong to the orthorhombic space group C2221, with unit-cell parameters a = 111.0, b = 308.0, c = 56.0 Å and unit-cell volume 1 913 716 Å3. The crystals displayed a mosaicity of 0.7° during data collection. The HKL programs DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶) were used for integrating, processing, scaling and merging the reflections. Data were processed from 354 collected images. The signal-to-noise ratio was 11 (4 in the outer resolution shell). The completeness of the data was 98.8% (97.8% in the outer shell) and the R merge was 8% (20% in the outer shell). The Matthews coefficient V M (Matthews, 1968 ▶), estimated assuming two molecules in the asymmetric unit, was 3.23 Å3 Da −1 and corresponded to 62% solvent content. Data-collection and processing statistics are tabulated in Table 1 ▶.
Figure 2.

Crystal of PVA from B. subtilis.
Table 1. Data-collection statistics.
Values in parentheses refer to the highest resolution shell.
| Experimental conditions | |
| Source | Rigaku rotating-anode generator |
| Wavelength (Å) | 1.5418 |
| Detector | R-AXIS IV++ |
| Temperature (K) | 120 |
| Crystal parameters | |
| Space group | C2221 |
| Unit-cell parameters (Å) | |
| a | 110.96 |
| b | 307.95 |
| c | 56.00 |
| Data processing | |
| Resolution (Å) | 40–2.5 (2.59–2.5) |
| Data completeness (%) | 98.8 (97.8) |
| No. of measured reflections | 114421 |
| No. of unique reflections | 33573 |
| Merging R factor† | 0.084 (0.196) |
| I/σ(I) | 11.11 (4.14) |
| Mosaicity (°) | 0.7 |
| Molecules per AU | 2 |
| Matthews coefficient (Å3 Da−1) | 3.23 |
R
merge = 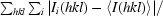
 .
.
The structure was determined using the molecular-replacement method implemented in AMoRe (Navaza, 1994 ▶) from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). B. sphaericus penicillin V acylase (PDB code http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=2pva; see Fig. 1 ▶) was used as the search model. Details of the molecular-replacement solutions are given in Table 2 ▶. As expected from the V M, there is a dimer in the asymmetric unit; the two dimers of a stable tetramer are related by a crystallographic twofold axis. Further refinement and detailed structure analysis are in progress. We hope the structure of YxeI will (i) reveal differences in post-translational processing events to B. sphaericus PVA and (ii) shed light on the relationship between PVA and CBH enzymes.
Table 2. Details of the molecular-replacement solutions.
The resolution range used for rotation function and initial translation was 10.0–3.0 Å.(a) Molecular-replacement solutions.
| Eulerian angles | Translations (fractional) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Solution | α | β | γ | CC | Tx | Ty | Tz | CC | R factor |
| 1 | 18.56 | 35.25 | 13.22 | 17.0 | 0.3602 | 0.2993 | 0.3486 | 34.0 | 55.8 |
| 2 | 51.00 | 12.07 | 336.50 | 13.8 | 0.3529 | 0.0599 | 0.2050 | 31.3 | 56.8 |
| 3 | 21.58 | 65.16 | 185.59 | 13.2 | 0.0656 | 0.4767 | 0.3324 | 24.2 | 59.9 |
| 4 | 149.33 | 21.02 | 234.00 | 11.8 | |||||
(b).
Translation function fixing one solution and searching for the second (10–3 Å).
| Solution | Tx | Ty | Tz | CC | R factor |
|---|---|---|---|---|---|
| 1 | 0.3602 | 0.2993 | 0.3486 | Fixed | |
| 2 | 0.8530 | 0.0600 | 0.7050 | 45.1 | 51.8 |
(c).
Rigid-body fit (10–3 Å).
| Solution | α | β | γ | Tx | Ty | Tz | CC | R factor |
|---|---|---|---|---|---|---|---|---|
| 1 | 18.68 | 36.17 | 13.28 | 0.3603 | 0.2993 | 0.3468 | ||
| 2 | 57.73 | 12.53 | 331.29 | 0.8529 | 0.0601 | 0.7062 | 62.2 | 46.4 |
Acknowledgments
We thank the British Council for sponsoring the Higher Education Link programme. PR is a senior research fellow of the Council of Scientific and Industrial Research, New Delhi, India. PR thanks the Commonwealth for a split-site PhD fellowship.
References
- Brannigan, J. A., Dodson, G., Duggleby, H. J., Moody, P. C. E., Smith, J. L., Tomchick, D. R. & Murzin, A. G. (1995). Nature (London), 378, 416–419. [DOI] [PubMed] [Google Scholar]
- Bruggink, A., Roos, E. C. & de Vroom, E. (1998). Org. Process Res. Dev.2, 128–133.
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Ducruix, A. & Giegé, R. (1992). Crystallization of Nucleic Acids and Proteins, pp. 130–135. Oxford University Press.
- Gouet, P., Courcelle, E., Stuart, D. I. & Metoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed] [Google Scholar]
- Lowry, O. H., Rosebrough, N. J., Farr, A. L. & Randall, R. J. (1951). J. Biol. Chem.193, 265–275. [PubMed] [Google Scholar]
- McPherson, A. (1982). Preparation and Analysis of Protein Crystals, pp. 96–97. New York: John Wiley & Sons.
- Manish Chandra, P., Brannigan, J. A., Prabhune, A., Pundle, A., Turkenburg, J. P., Dodson, G. G. & Suresh, C. G. (2005). Acta Cryst. F61, 124–127. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Navaza, J. (1994). Acta Cryst. A50, 157–163. [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Shewale, J. G., Kumar, K. K. & Ambedkar, G. R. (1987). Biotechnol. Tech.1, 69–72.
- Shewale, J. G. & SivaRaman, H. (1989). Process Biochem.24, 146–154.
- Shewale, J. G. & Sudhakaran, V. K. (1997). Enzyme Microb. Technol.20, 402–410.
- Suresh, C. G., Pundle, A. V., SivaRaman, H., Rao, K. N., Brannigan, J. A., McVey, C. E., Verma, C. S., Dauter, Z., Dodson, E. J. & Dodson, G. G. (1999). Nature Struct. Biol.6, 414–416. [DOI] [PubMed] [Google Scholar]
- Suresh Kumar, R., Brannigan, J. A., Pundle, A., Prabhune, A., Dodson, G. G. & Suresh, C. G. (2004). Acta Cryst. D60, 1665–1667. [DOI] [PubMed] [Google Scholar]
- Yoshida, K., Fujimura, M., Yanai, N. & Fujita, Y. (1995). DNA Res.2, 295–301. [DOI] [PubMed] [Google Scholar]