

Abstract
Transient overexpression of either DsbA or DsbC can double the yield of periplasmic insulin-like growth factor (IGF)-I in Escherichia coli to 8.5 g/liter. Strikingly, most of the overexpressed DsbA or DsbC is found in the reduced form, implying that enhanced disulfide isomerization is responsible for the substantial increase in IGF-I yield. All of the accumulated IGF-I has had the signal sequence removed, underscoring the secretion capacity of this organism as well as its utility for efficient production of polypeptide with the correct amino terminus. The overexpressed IGF-I constitutes approximately 30% of the total cell protein. Overproduction of active site mutants of DsbA instead of the wild-type gene do not produce this increase in yield. With wild-type levels of DsbA and DsbC, most of the secreted IGF-I is found in disulfide-linked aggregates, although 10% is soluble and about 5% is correctly folded. Contrary to expectations, overexpression of the disulfide oxidoreductases decreased the soluble fraction. Because the aggregated protein can be efficiently solubilized and refolded, the increased yield is a significant benefit for the production of IGF-I.
Secretion of recombinant proteins to the periplasm of Escherichia coli confers two advantages over cytoplasmic production. First, an authentic amino terminus can be generated after cleavage by leader peptidase as opposed to cytoplasmic accumulation, which often results in an amino-terminal methionine. Second, the periplasm is relatively oxidizing compared with the cytoplasm, favoring the formation of disulfide bonds (1–4). Several proteins with relatively simple disulfide bond patterns are correctly folded after secretion to the periplasm (5–7). Many of the commercially important proteins for the biotechnology industry are secreted proteins and contain several disulfide bonds. The elucidation of the pathways responsible for disulfide bond formation and isomerization has allowed the creation of strains that have altered levels of the proteins involved in these processes. The creation of null strains has allowed investigators to rapidly assess which proteins are critical for the disulfide bond formation and isomerization (8–11). DsbA is the primary thiol oxidant of newly secreted periplasmic proteins. Although dsbA is not an essential gene, dsbA knockout strains have a slow growth rate, poor final growth yield, and also cannot accumulate recombinant protein directed to the periplasm, even one without any cysteinyl residues (12). The protein acts as both an oxidant and disulfide isomerase in vitro, depending on the redox state of the two cysteines in the protein (13). However, in rich media, DsbA is found exclusively in the oxidized form (12, 14). DsbA is reoxidized in vivo by DsbB, a cytoplasmic membrane protein that contains six cysteines, five of which are located in periplasmic loops based on alkaline phosphatase fusions (15–17).
DsbC is another soluble periplasmic protein and seems to be responsible for isomerizing disulfide bonds (10–12, 18, 19). The protein has four cysteine residues, two of which are paired in a CXXC pattern, a hallmark of the thiol:disulfide oxidoreductases (2, 3). It is the redox state of these two cysteines that determines the activity of the protein as it can act as either an oxidant or isomerase in vitro (12, 18). In vivo, the protein is predominantly found in the singly reduced form, indicating that normally the protein acts as an isomerase (12, 19). To maintain the protein in this singly reduced form, a functional dsbD allele is required. When dsbD is disrupted, the DsbC protein is found mostly in the oxidized form (9, 12). DsbD is also required for biogenesis of cytochrome c proteins. Without DsbD, the heme groups are not attached to the cysteine residues in the apocytochrome c proteins (20).
The overexpression of DsbA in conjunction with recombinant protein production has led to mixed results. Initially Knappik et al. (5) found that overexpression of DsbA did not decrease the amount of aggregated antibody fragments in vivo. Additionally, Wulfing and Pluckthun (21) found that the yield of only one out of three pairs of T-cell receptor fragments tested were improved by overexpression of DsbA. To see this benefit, the heat-shock sigma factor σ32 also had to be overexpressed. On the other hand, Wunderlich and Glockshuber (6) reported seeing a 14-fold increase in the folding of a bifunctional α-amylase/trypsin inhibitor when DsbA was overexpressed, but only when reduced glutathione was added. The yield of aggregated protein did not change when DsbA and glutathione were added. The approaches to date for increasing putative catalysts of protein folding have all employed either strongly inducible promoters, which when induced were not subsequently turned off, or have used promoters that were constitutively turned on. These approaches result in the simultaneous expression of both the catalyst and the recombinant protein. We have employed a different strategy involving the use of an inducible promoter where the inducer is consumed by the cells. After consumption of the added inducer, catalyst expression ceases, and the expression of the recombinant protein is begun. This experimental strategy results in only one protein being overproduced at a time. Using this approach, a large increase in the total yield of the recombinant protein, human insulin-like growth factor-1 (IGF-I*), was observed after increasing the concentration of periplasmic thiol:disulfide oxidoreductase, DsbA, or DsbC. Although nearly all of the IGF-I accumulates in aggregates, there exists efficient methodology for the isolation and folding of these deposits (22, 23).
EXPERIMENTAL PROCEDURES
Vectors and Strains.
The host strain used for all fermentation work was a kanamycin-sensitive version of ATCC no. 55244. Additionally, the host contains ilvG2096 to avoid valine toxicity. The final host genotype is W3110fhuAΔ phoAΔE15 Δ(argF-lac)169 ptrA3 degP41 ΔompTΔ(nmpc-fepE) ilvG2096. The construction of the IGF-I production plasmid pBKIGF2B is described (24). The dsbA and dsbC genes were amplified by PCR and cloned into pBAD22 (25). The ClaI–HindIII fragment containing araC-PBAD-dsbA was made blunt ended and cloned into the EcoRV site in pBKIGF2B. This plasmid (pBKIGF2B-A) (see Fig. 1A) produces IGF-I in low phosphate media and produces DsbA in the presence of l-arabinose. The analogous plasmid expressing DsbC was constructed identically. For chromosomal knockouts of the dsbC or dsbD gene, the procedure of Metcalf et al. (26) as modified by Bass et al. (27) was employed. The upstream and downstream regions of the genes were amplified by PCR and ligated together in a vector allowing positive (ampicillin resistance) and negative selection (sucrose resistance). Null strains were confirmed by PCR for either dsbC or dsbD. Null strains for dsbC were also confirmed by the lack of DsbC protein in immunoblots of whole cell extracts.
Figure 1.
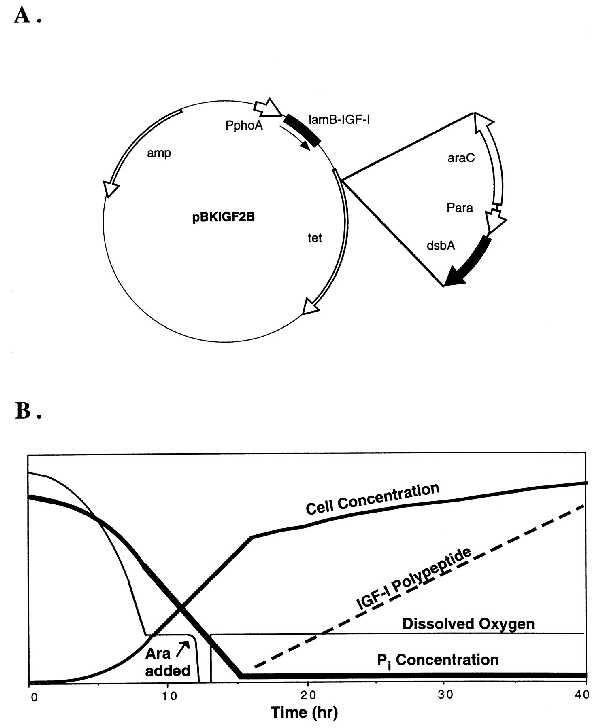
Plasmid schematic and fermentation profiles. The plasmid construction for pBKIGF2B and the modification are depicted (A). The idealized fermentation protocol for the production of rhIGF-I is shown (B). The arrow denotes the approximate time of l-arabinose addition (10–12 h). The thick solid line represents the inorganic phosphate concentration. The medium solid line represents the cell concentration, and the thin solid line represents the dissolved oxygen concentration. The dashed line represents the IGF-I polypeptide concentration. The y axis is in arbitrary units.
10-Liter Fermentations.
A 0.5-liter L-broth inoculum flask grown at 37°C for 12 h was added to the fermenter (Biolafite, 15 liters). The initial fermentation medium was composed of 55.7 mM ammonium sulfate, 13.9 mM sodium monobasic phosphate, 21.9 mM potassium dibasic phosphate, 5 mM sodium citrate, 29.6 mM potassium chloride, 14.7 mM magnesium sulfate, 1.11% NZ amine AS, 1.11% yeast extract, 0.11% glucose, 0.002% ferric chloride, 30 mg/liter ampicillin. A trace element solution (5 ml/6.8 liters) was added containing 100 mM ferric chloride and 30 mM of each of the following: zinc sulfate, cobalt chloride, sodium molybdate, copper sulfate, boric acid, and manganese sulfate. The fermenter was operated at 37°C, 1000 rpm, 10 standard liter/min aeration with 0.3 bar back-pressure, which delivers approximately 3.0 mmol/liter/min for the oxygen transfer rate. When the initial glucose was consumed, a concentrated glucose solution was fed until the dissolved oxygen concentration (dO2) reached 30% of air saturation as measured by an on-line oxygen electrode. Glucose was then fed at a rate to maintain 30% dO2. Typically, the dissolved oxygen levels fluctuate only plus or minus 2% from the set point. When the optical density (550 nm) reached 40 OD, a solution of 20% NZ amine AS, 20% yeast extract was fed at 0.2 ml/min for the rest of the fermentation. The total fermentation time was 40 h. Ammonium hydroxide was added automatically to maintain the pH at 7.3. Sulfuric acid was added manually when needed. To induce transient DsbA or DsbC expression, 0.2 liter of 40% l-arabinose (Pfanstiehl) was rapidly added at approximately 85 OD.
IGF-I Assays.
Samples were taken at 2-h intervals from the fermenter and quickly frozen in dry ice. Non-native IGF-I yields were measured as previously described (22) except the time of extraction was 30 min instead of 5 min. To measure the IGF-I that was not disulfide cross-linked, DTT was omitted from the extraction solution. The final volume analyzed by HPLC was increased 4-fold to increase the signal measured.
Redox State Assay.
Samples corresponding to 0.5 OD/ml were taken and treated immediately with 10% trichloroacetic acid. Samples were then stored at 4°C until processed. The acid precipitates were centrifuged for 10 min at 4°C, 16,000 × g, and the supernatants were discarded. The pellets were then washed with cold acetone to remove trace acid and recentrifuged as before. The supernatants were discarded, and the pellets were air dried. The precipitates were resuspended in 0.1 ml of 1% SDS, 50 mM Tris⋅Cl, pH 8.0, 1 mM EDTA containing 10 mM 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonate (Molecular Probes). The samples were mixed for 30 min at room temperature, and then 10 volumes of cold acetone was added to precipitate the proteins. The precipitates were centrifuged as described above, and the pellets were air dried. The pellets were resuspended in 0.1 ml of 2× Laemmli sample buffer (Novex) and mixed for 30 min at room temperature. The samples were heated for 5 min at 100°C and then analyzed by non-reducing SDS-PAGE on 16% Tris–glycine polyacrylamide gels (Novex). The gels were blotted to nitrocellulose and then probed with polyclonal anti-DsbA or anti-DsbC sera followed by development with chemiluminescent detection. Standards were generated with purified DsbA or DsbC in vitro and then diluted as previously reported (12).
RESULTS
Overexpression of DsbA or DsbC.
To increase the concentration of protein folding catalysts in vivo, we chose to employ the arabinose-inducible PBAD promoter. This well studied promoter is efficiently repressed in the absence of l-arabinose and is strongly induced in the presence of l-arabinose (25). Strains have been constructed and are widely available that can catabolize the inducer (wild type for arabinose) or have a genetic lesion that prevents catabolism of the inducer, resulting in constitutive expression. We used strains that were wild type with respect to arabinose catabolism for these studies. Plasmids were constructed so that either dsbA or dsbC was expressed from the PBAD promoter. The entire arabinose-regulated element was then transferred to the plasmid that expressed human IGF-I from the phoA promoter (Fig. 1A). The signal sequence from lamB was used to direct IGF-I secretion to the periplasm.
The strains were tested in 10-liter fermentations as described in Experimental Procedures. A fixed concentration of inorganic phosphate was present at the start of the fermentation. After several generations of growth, the inorganic phosphate in the culture was depleted, thereby inducing the phoA promoter and turning on IGF-I expression (Fig. 1B). For the fermentations involving overexpression of either DsbA or DsbC, l-arabinose was added prior to phosphate depletion. The large quantity of inducer added caused the dissolved oxygen to decrease to zero for approximately 45 min as the cells consumed the arabinose and expressed the Dsb protein. The arabinose was depleted as indicated by a sudden increase in the dissolved oxygen. Phosphate depletion and IGF-I expression followed.
The total yield of IGF-I polypeptide was measured with an HPLC method after extraction with chemical denaturant and reducing agent. This assay quantitates the IGF-I accumulated within the periplasm as well as the IGF-I that is located in the external media. We observed that the yield of IGF-I was increased from 4.3 g/liter to 8.5 g/liter just from increasing the DsbA concentration (Fig. 2). Similarly, when DsbC was overexpressed the total IGF-I yield was increased to 7.3 g/liter. Adding arabinose to fermentations without the arabinose-regulated element on the plasmid provided no increase in the IGF-I yield (data not shown). To determine whether active periplasmic enzyme was required, we mutated the active site cysteines of DsbA and expressed the mutant enzymes in exactly the same fermentation protocol. When both cysteines were changed to serines and the mutant DsbA induced, the yield of IGF-I was unchanged from the control yield (Fig. 2). When only the N-terminal cysteine was changed to serine, an increase of 30% was seen in the IGF-I yield compared with the control.
Figure 2.
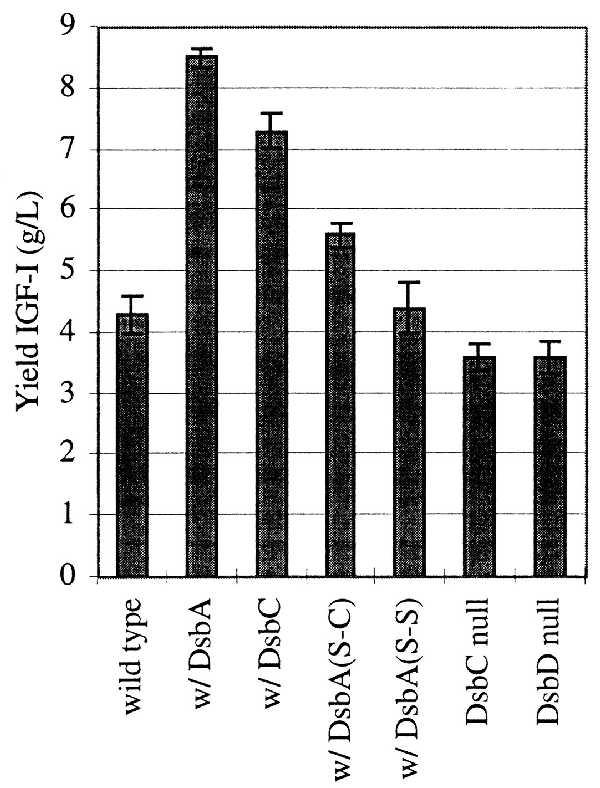
Yields of total IGF-I in 10-liter fermentations. The peak titers of IGF-I were measured by reversed phase HPLC after guanidine hydrochloride and DTT extraction. Fermentations containing overexpressed Dsb protein are denoted by w/Dsb. Results from at least three (n = 3–5) separate fermentations were averaged. The bars correspond to the standard error of the mean.
The dry cell weight yield for the control fermentation, without any DsbA or DsbC overexpression, was 46 g/liter. When DsbA was overexpressed, the dry cell weight yield increased to 50 g/liter. Assuming that protein corresponds to 50–55% of the dry cell weight, the yield of IGF-I polypeptide seen with DsbA overexpressed corresponds to 30% of the total cell protein. For the control fermentations, the yield of IGF-I corresponds to 17% of the total cell protein.
We have examined the IGF-I yield in strains that lack components of the Dsb system. Knockout strains of dsbA grow poorly even in rich media and cannot accumulate a recombinant protein devoid of cysteines, much less IGF-I which has six cysteines (12). Null strains of dsbA did not accumulate any periplasmic IGF-I in our fermentation protocol. If the null strain was transformed with the plasmid overexpressing DsbA and the culture transiently induced with arabinose, the yield of IGF-I was equal to that of wild type overexpressing DsbA (data not shown). DsbC knockout strains exhibit a much milder phenotype and grow quite well in rich media and in our fermentation protocol. Strains where dsbC or dsbD have been disrupted have a small (16%) but reproducible decrease in IGF-I yield (see Fig. 2).
Disposition of the IGF-I Polypeptide.
Most of the IGF-I accumulated in control fermentations was found in disulfide cross-linked aggregates. A small percentage (approximately 5–10%) was found in the external media with even a smaller percentage found in the correctly folded form (22, 28). If either DsbA or DsbC was overexpressed, the percentages found in the external media and in the correctly folded form decreased. The percentage of IGF-I found in the external media dropped from approximately 10% to below reliable detection (approximately 1–2%, data not shown). To test whether the IGF-I was still in disulfide cross-linked aggregates, we extracted the culture broth with guanidine hydrochloride alone with no reducing agent. This assay measured only the IGF-I that was not disulfide cross-linked. Guanidine hydrochloride is an effective agent to release soluble periplasmic contents (29). No additional monomeric IGF-I could be extracted in the guanidine hydrochloride insoluble pellet even after boiling in 1% SDS for 5 min. With normal levels of DsbA and DsbC, approximately 17% of the total IGF-I was extracted with chemical denaturant alone (Fig. 3). This agrees well with shake flask experiments and osmotic shock treatment of the cells (23). A similar proportion of denaturant-extractable polypeptide was measured in dsbC null strains (15%, Fig. 3) as well as dsbD null strains (data not shown). When the fermentations were induced for either DsbA or DsbC, the amount of denaturant-extractable polypeptide decreased (Fig. 3), indicating that a greater percentage of the IGF-I was disulfide cross-linked.
Figure 3.
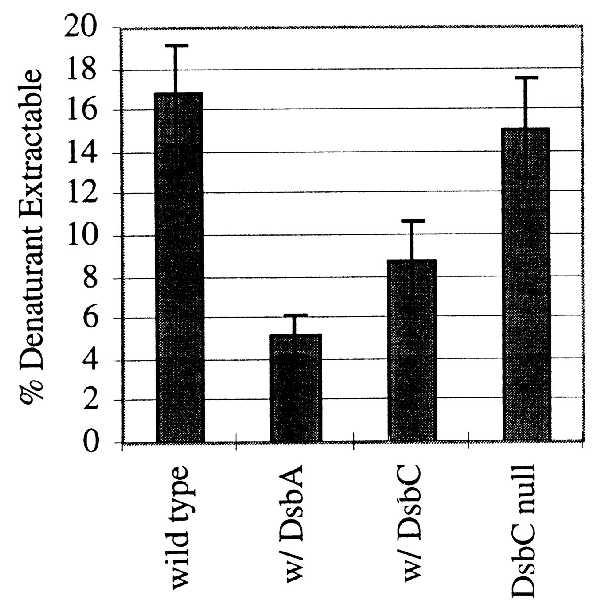
Percentage of denaturant only extractable IGF-I. Whole fermentation broth samples were extracted for IGF-I, omitting the reducing agent to detect only the IGF-I polypeptide not found in intermolecular disulfide cross-links. Fermentations where Dsb proteins were overexpressed are denoted by w/Dsb. The mean of three separate fermentations is reported, and the bars represent the standard error of the mean. The differences between the means of wild type and w/DsbA and wild type and w/DsbC are P < 0.005 and P < 0.025, respectively.
Redox States of DsbA and DsbC.
In shake flask experiments in rich media, the redox states of DsbA and DsbC are opposite (12). DsbA is in the oxidized form, and DsbC is found with one disulfide formed and one pair of free cysteines. Because overexpression of either protein aided IGF-I accumulation, the redox state of these proteins in wild-type and overexpressed conditions was addressed. Samples were taken from the fermentors at the time points indicated in Fig. 4 and processed for determining the in vivo redox state. The redox state of wild-type levels of DsbA seemed to be a mixture of oxidized and reduced forms. The protein was mostly in the oxidized form early in the fermentation and then shifted to roughly equal amounts of both species for the rest of the time course (Fig. 4). For DsbC, the protein was almost solely in the singly reduced form until the point of IGF-I production. The last two time points taken showed mixtures of oxidized and singly reduced forms of DsbC with the trend of more oxidized DsbC in the last time point relative to the penultimate time point.
Figure 4.
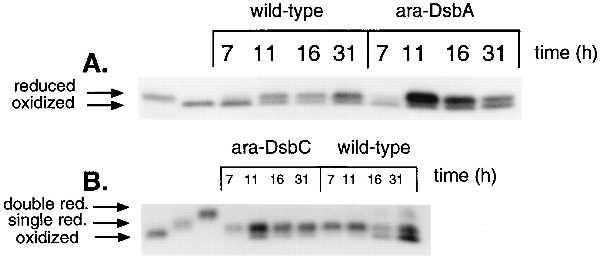
Redox state analysis of DsbA and DsbC in fermentations. Samples were taken at the hours indicated and processed as described in Experimental Procedures. When the dsb gene was present on the plasmid, half the volume of whole cell extract was loaded relative to wild-type conditions prior to addition of l-arabinose (7 h time point only). After arabinose addition, a 15-fold dilution of the extract was loaded (remaining time points). Standards were generated in vitro and correspond to approximately 3 ng of each. IGF-I expression starts at approximately 14 h.
With overexpression of DsbA, most of the DsbA was in the reduced form in the first time point taken after arabinose addition. The ratio of reduced to oxidized protein decreases with time and seems to be approximately one at the last time point taken. Prior to arabinose addition, the DsbA is identical with respect to redox states as the wild-type levels. If no inducer is added, there is no difference in the ratio of oxidized to reduced DsbA from the wild-type levels of protein (data not shown). When DsbC was overexpressed, most of the protein was found in the singly reduced form with some being completely oxidized in the first time point after induction. Most of the protein stayed in the singly reduced form as the fermentation proceeded. This contrasts with the wild-type fermentation, where some of the protein was oxidized in the last two time points. When DsbA was overexpressed, the redox state of DsbC did not change compared with the wild-type fermentation. The converse was true as overexpressing DsbC did not change the pattern for DsbA (data not shown). The peak level of induction of DsbA or DsbC was quantitated as approximately 40-fold as judged by an HPLC assay and immunoblots (data not shown). Overexpressed DsbA is very soluble, as almost all of the protein is found in supernatant fractions after cell breakage and centrifugation or in osmotic shock fractions.
DISCUSSION
The ability of E. coli to overexpress and correctly fold recombinant proteins containing multiple disulfide bonds is limited. Most E. coli periplasmic and outer membrane proteins contain two or fewer disulfides per monomer. To boost the endogenous capacity of the wild-type cells to accomplish this task, overexpression of the Dsb proteins was performed. We have found that the transient overexpression of either DsbA or DsbC resulted in a large yield increase of IGF-I. The fact that E. coli can accumulate a mammalian protein in the periplasm at up to 30% of the total cell protein is striking. The secretion capacity of this organism obviously can be very robust. With high levels of IGF-I secretion, we hypothesized that elevated concentrations of disulfide bond-forming enzymes would improve IGF-I folding. It was then surprising to find that overexpression of these enzymes led to increased aggregation and higher yields of IGF-I.
We were also surprised to find mixtures of oxidized and reduced forms of the Dsb proteins in fermentations when either Dsb protein was overexpressed or only expressed from the chromosomal locus. Normally, DsbA is found only in the oxidized form and DsbC in the singly reduced form in low cell density shake flask experiments (12, 14). Because some DsbA is found in the reduced form in our fermentations, it seems that there is a limitation in the system for maintaining DsbA in the oxidized state. The source of the limitation is currently unknown. The finding that DsbA is in both the oxidized and reduced forms in our fermentations may explain why the dsbC null strain showed only a 16% decrease in IGF-I yield. Disulfide isomerase activity may not be as compromised in the dsbC null strain due to the presence of some reduced DsbA. On the other hand, DsbA null strains are not complemented by the presence of oxidized DsbC in terms of IGF-I production even when DsbC was overexpressed (data not shown). However, it is difficult to draw any inference from this as dsbA null strains exhibit many pleiotropic effects (8).
There are at least two possible reasons for increased IGF-I yields due to overexpression of the Dsb proteins. First, elevated concentrations of the Dsb proteins may decrease the amount of IGF-I protein that is proteolyzed. If the rate of aggregation is increased, the levels of protease-sensitive folding intermediates could be decreased. Although the host strain used in these experiments has three known protease genes disrupted (degP, ptrA, ompT), there may still exist other proteases in the periplasm and outer membrane that could degrade IGF-I. Additionally, E. coli contains the rpoE regulon, which is an inducible response system for the presence of unfolded proteins and periplasmic stress. Although the known rpoE-induced protease degP is disrupted in these strains, other proteases may exist and may be induced by unfolded IGF-I in the periplasm. Disruption of either degP or ptrA contributes to increased IGF-I yields. When IGF-I is produced in a host that contains wild-type degP, ptrA, and ompT, the yield is 0.3 g/liter. When DsbA is overexpressed in this host, the yield of IGF-I increases to 2.5 g/liter consistent with the idea that DsbA is decreasing proteolysis of the polypeptide. The second possibility for increased IGF-I yields is that the Dsb proteins may assist in translocation, either indirectly by increasing the activity of the preprotein translocase enzyme or directly by pulling the preprotein through the site of translocation more efficiently. Folding of the nascent secretory protein has been suggested to aid in translocation across membranes (30). Pulse chase experiments in shake flask conditions do not show increased signal peptide cleavage with increased DsbA concentrations (data not shown). Not all recombinant proteins may show this yield increase when DsbA or DsbC is overexpressed. Proteins with complex disulfide bond patterns such as IGF-I seem to be more affected by null mutations in dsbC than proteins such as human growth hormone that have consecutive cysteines paired (12). Overexpression of disulfide isomerases may only benefit those proteins having nonconsecutive cysteines paired.
The fact that elevated concentrations of either DsbA or DsbC proteins increase the fraction of aggregated IGF-I is an unexpected finding. If the yield increase is simply due to more preprotein translocation, the increased concentration of protein folding intermediates could explain why more IGF-I is found in the refractile bodies. Conversely, increased DsbA or DsbC may affect the distribution and concentration of IGF-I folding intermediates that are more likely to aggregate. Ironically, we must also consider the possibility that increased disulfide isomerization may actually increase intermolecular disulfide bond formation and aggregate stabilization, especially at the high substrate concentrations produced in these experiments.
The reason that overexpression of IGF-I results in the formation of inclusion bodies may be due to the sequence of the protein that is produced. The active form of IGF-I is truncated at the carboxyl terminus relative to the pro-protein form that is normally secreted into the mammalian endoplasmic reticulum. Due to differential mRNA splicing, two forms of IGF-I can be produced that differ at the C terminus (31). IGF-Ia contains 35 additional amino acids including one potential N-linked glycosylation site. Overexpression of this isoform in E. coli still results in extensive aggregation and an insoluble product (32). IGF-Ib contains an additional 77 amino acid residues, many of which are charged. This IGF-Ib extension also has one cysteinyl residue six amino acids from the carboxyl terminus that may influence the disulfide bond pattern of IGF-I folding intermediates. These carboxyl-terminal extensions are presumably cleaved during subsequent trafficking steps as the mature form of IGF-I is found in the plasma. Experiments testing the production of IGF-Ib are ongoing.
Acknowledgments
We are grateful for the assistance of Nancy McFarland in strain constructions and members of the E. coli community at Genentech for fruitful discussions.
ABBREVIATION
- IGF-I
insulin-like growth factor-1
References
- 1.Pollit S, Zalkin H. J Bacteriol. 1983;153:27–32. doi: 10.1128/jb.153.1.27-32.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bardwell J C A. Mol Microbiol. 1994;14:199–205. doi: 10.1111/j.1365-2958.1994.tb01281.x. [DOI] [PubMed] [Google Scholar]
- 3.Missiakas D, Raina S. J Bacteriol. 1997;179:2465–2471. doi: 10.1128/jb.179.8.2465-2471.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wulfing C, Pluckthun A. Mol Microbiol. 1994;12:685–692. doi: 10.1111/j.1365-2958.1994.tb01056.x. [DOI] [PubMed] [Google Scholar]
- 5.Knappik A, Krebber C, Pluckthun A. Bio/Technology. 1993;11:77–83. doi: 10.1038/nbt0193-77. [DOI] [PubMed] [Google Scholar]
- 6.Wunderlich M, Glockshuber R. J Biol Chem. 1993;268:24547–24550. [PubMed] [Google Scholar]
- 7.Ostermeier M, Georgiou G. J Biol Chem. 1994;269:21072–21077. [PubMed] [Google Scholar]
- 8.Bardwell J C A, McGovern K, Beckwith J. Cell. 1991;67:581–589. doi: 10.1016/0092-8674(91)90532-4. [DOI] [PubMed] [Google Scholar]
- 9.Missiakas D, Schwager F, Raina S. EMBO J. 1995;14:3415–3424. doi: 10.1002/j.1460-2075.1995.tb07347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rietsch A, Belin D, Martin N, Beckwith J. Proc Natl Acad Sci USA. 1996;93:13048–13053. doi: 10.1073/pnas.93.23.13048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sone M, Akiyama Y, Ito K. J Biol Chem. 1997;272:10349–10352. doi: 10.1074/jbc.272.16.10349. [DOI] [PubMed] [Google Scholar]
- 12.Joly J C, Swartz J R. Biochemistry. 1997;36:10067–10072. doi: 10.1021/bi9707739. [DOI] [PubMed] [Google Scholar]
- 13.Joly J C, Swartz J R. Biochemistry. 1994;33:4231–4236. doi: 10.1021/bi00180a017. [DOI] [PubMed] [Google Scholar]
- 14.Kishigami S, Akiyama Y, Ito K. FEBS Lett. 1995;364:55–58. doi: 10.1016/0014-5793(95)00354-c. [DOI] [PubMed] [Google Scholar]
- 15.Bardwell J C A, Lee J O, Jander G, Martin N, Beckwith J. Proc Natl Acad Sci USA. 1993;90:1038–1042. doi: 10.1073/pnas.90.3.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jander G, Martin N, Beckwith J. EMBO J. 1994;13:5121–5127. doi: 10.1002/j.1460-2075.1994.tb06841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kishigami S, Kanaya E, Kikuchi M, Ito K. J Biol Chem. 1995;270:17072–17074. doi: 10.1074/jbc.270.29.17072. [DOI] [PubMed] [Google Scholar]
- 18.Zapun A, Missiakas D, Raina S, Creighton T E. Biochemistry. 1995;34:5075–5089. doi: 10.1021/bi00015a019. [DOI] [PubMed] [Google Scholar]
- 19.Rietsch A, Bessette P, Georgiou G, Beckwith J. J Bacteriol. 1997;179:6602–6608. doi: 10.1128/jb.179.21.6602-6608.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crooke H, Cole J. Mol Microbiol. 1995;15:1139–1150. doi: 10.1111/j.1365-2958.1995.tb02287.x. [DOI] [PubMed] [Google Scholar]
- 21.Wulfing C, Pluckthun A. J Mol Biol. 1994;242:655–669. doi: 10.1006/jmbi.1994.1615. [DOI] [PubMed] [Google Scholar]
- 22.Chang J Y, Swartz J R. In: Protein Folding In Vivo and In Vitro. Cleland J L, editor. American Chemical Society, Washington, DC: A.C.S. Symposium Series; 1993. pp. 178–188. [Google Scholar]
- 23.Hart R A, Lester P M, Reifsnyder D H, Ogez J R, Builder S E. Bio/Technology. 1994;12:1113–1117. doi: 10.1038/nbt1194-1113. [DOI] [PubMed] [Google Scholar]
- 24.Swartz, J. R. (1994) Method for Producing Polypeptide via Bacterial Fermentation. U.S. Patent No. 5,342,763.
- 25.Guzman L M, Belin D, Carson M J, Beckwith J. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metcalf W W, Jiang W, Wanner B L. Gene. 1994;138:1–7. doi: 10.1016/0378-1119(94)90776-5. [DOI] [PubMed] [Google Scholar]
- 27.Bass S, Gu Q, Christen A. J Bacteriol. 1996;178:1154–1161. doi: 10.1128/jb.178.4.1154-1161.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong E Y, Seetharam R, Kotts C, Heeren R A, Klein B K, Braford S R, Mathis K J, Bishop B F, Siegel N R, Smith C E, Tacon W C. Gene. 1988;68:193–203. doi: 10.1016/0378-1119(88)90021-2. [DOI] [PubMed] [Google Scholar]
- 29.Naglak T J, Wang H Y. Enzyme Microb Technol. 1990;12:603–611. doi: 10.1016/0141-0229(90)90134-c. [DOI] [PubMed] [Google Scholar]
- 30.Neupert W, Hartl F U, Craig E A, Pfanner N. Cell. 1990;63:447–450. doi: 10.1016/0092-8674(90)90437-j. [DOI] [PubMed] [Google Scholar]
- 31.Rotwein P, Pollock K M, Didier D K, Krivi G G. J Biol Chem. 1986;261:4828–4832. [PubMed] [Google Scholar]
- 32.Hober S, Hansson A, Uhlen M, Nilsson B. Biochemistry. 1994;33:6758–6761. doi: 10.1021/bi00188a003. [DOI] [PubMed] [Google Scholar]