

Abstract
The G protein Go is highly expressed in neurons and mediates effects of a group of rhodopsin-like receptors that includes the opioid, α2-adrenergic, M2 muscarinic, and somatostatin receptors. In vitro, Go is also activated by growth cone-associated protein of Mr 43,000 (GAP43) and the Alzheimer amyloid precursor protein, but it is not known whether this occurs in intact cells. To learn about the roles that Go may play in intact cells and whole body homeostasis, we disrupted the gene encoding the α subunits of Go in embryonic stem cells and derived Go-deficient mice. Mice with a disrupted αo gene (αo−/− mice) lived but had an average half-life of only about 7 weeks. No Goα was detectable in homogenates of αo−/− mice by ADP-ribosylation with pertussis toxin. At the cellular level, inhibition of cardiac adenylyl cyclase by carbachol (50–55% at saturation) was unaffected, but inhibition of Ca2+ channel currents by opioid receptor agonist in dorsal root ganglion cells was decreased by 30%, and in 25% of the αo−/− cells examined, the Ca2+ channel was activated at voltages that were 13.3 ± 1.7 mV lower than in their counterparts. Loss of αo was not accompanied by appearance of significant amounts of active free βγ dimers (prepulse test). At the level of the living animal, Go-deficient mice are hyperalgesic (hot-plate test) and display a severe motor control impairment (falling from rotarods and 1-inch wide beams). In spite of this deficiency, αo−/− mice are hyperactive and exhibit a turning behavior that has them running in circles for hours on end, both in cages and in open-field tests. Except for one, all αo−/− mice turned only counterclockwise. These findings indicate that Go plays a major role in motor control, in motor behavior, and in pain perception and also predict involvement of Go in Ca2+ channel regulation by an unknown mechanism.
Go is an αβγ heterotrimeric G protein discovered in 1984 in brain by Neer and collaborators (1, 2) and by Sternweis and Robishaw (3), who all characterized it as a substrate for the ADP-ribosyltransferase activity of pertussis toxin. Go has received special attention for the following reasons: (i) It is the most abundant G protein in neurons, where it can constitute up to 2% of membrane protein (3). (ii) In addition to neurons, it appears to be expressed only on endocrine cells and heart, albeit at much lower levels comparable to those of the other heterotrimeric G proteins. (iii) Go is activated not only by the same class of seven-transmembrane receptors that activate the inhibitory G proteins Gi1–Gi3 (4–9) but also by at least two proteins that do not belong to the rhodopsin-like family of G protein-coupled receptors, GAP43, an intracellular growth cone-associated protein active in neurite outgrowth (10), and the Alzheimer amyloid protein precursor protein responsible for familial forms of this disease (11). (iv) Except for inhibition of neuronal Ca2+ channels, for which the mechanism of action of Go has been elucidated at the molecular level and shown to be due to the interaction of its βγ moiety with the α1 subunit of the channel (6, 12–16), the mode of action of Go is essentially unknown. Tests for an αi-like function for αo have failed (17, 18). Activated αo has transformed NIH-3T3 cells (19) and activated mitogen-activated protein kinase activity in Chinese hamster ovary cells (20), phospholipase C in Xenopus oocytes, and K+ channels in neurons (21, 22), but how these effects come about has not been established. In fact, there is scant knowledge of the gamut of effector systems that may be the target(s) of activated Go.
Gene ablation in mice is a powerful yet technically complex approach to identify as yet unknown functions of proteins that become manifest in mutated animals and/or in cell lines derived from them. It has been applied to several G proteins with the following interesting results. Gi2-deficient mice were found to develop ulcerative colitis and adenocarcinomas, revealing an unexpected and as yet unexplained role of Gi2 in the development of a chronic inflammatory response and very likely in lymphocyte homing to enteric epithelia (23, 24). Ablation of Gq revealed an essential role for this G protein in platelet activation, because Gq-deficient mice bleed and their platelets fail to be activated by physiologic activators such as collagen, thrombin, thromboxane, and ADP (25). Also the ablation of Go has been reported (26). Live mice, homozygous for loss of αo, were obtained showing that αo is not essential for life in spite of the features that set it apart from other G proteins. Mice lacking Go were not like wild-type mice, however. They had a generalized tremor of unknown etiology and died at a very early postnatal ages. At the cellular level, Go-deficient mice displayed a loss of muscarinic inhibition of isoproterenol-stimulated cardiac L type Ca2+ currents. The latter finding was unexpected, given that the present view is that this channel in particular is insensitive to inhibition by a G protein-coupled pathway; specifically, it does not interact with G protein βγ dimers that inhibit the neuronal non-L type Ca2+ channels (16, 27, 28).
We have also used homologous recombination in embryonic stem cells to abolish its expression. The most prominent but not necessarily the most important of our findings is that Go-deficient mice develop a turning behavior as if afflicted by unilateral lesions of the central nervous system.
MATERIALS AND METHODS
Vector and Cell Development.
The intron exon structure of the gene encoding αo has been elucidated (refs. 29 and 30 and Fig. 1). The αo gene was disrupted in embryonic stem cells with previously described procedures and selection markers [Rudolph et al. (23, 33)]. 129Sv genomic DNA was obtained from a P1 cosmid selected from a commercial library (Genome Systems, St. Louis) by PCR screening with oligonucleotides A and B (where A is 5′-GGACAGCCTGGATCGGATTGG, a fragment of the sense strand of αo exon 5, and B is 5′-ACCTGGTCATAGCCGCTGAGT, a fragment of the antisense strand of αo exon 6) as primers (amplified fragment, 940 bp). A 10.8-kb HindIII–SalI fragment containing exons 5, 6, 7.2, and part of 8.2 was used to construct a targeting vector. The final vector had a pBSII backbone (Stratagene) and contained a 4.2-kb HindIII–BamHI fragment separated from 2.1-kb BamHI fragment of the αo gene by a Pol I-neomycin selection cassette (ref. 34 and Fig. 1). The MC1-tk counter- selection marker was placed at the 5′ end (35). This vector was transfected by electroporation into AB2.2 embryonic stem cells (from Allan Bradley, Baylor College of Medicine), and candidate recombinant clones were selected by growth on leukemia inhibitory factor-producing SNL feeder layers (also from Allan Bradley) in the presence of G418 (active ingredient; GIBCO/BRL; 180 μg/ml) and 0.2 μM 1-(2-deoxy-2-fluoro-β-d-arabinofuranosyl)-5-iodouracil (Moravek Biochemicals, Brea, CA). All cells were grown in DMEM containing glucose (4.5 g/liter), 2 mM glutamine, 15% heat-inactivated fetal bovine serum (GIBCO/BRL), 1% 2-mercaptoethanol, penicillin (100 units/ml), and streptomycin (100 μg/ml) (GIBCO/BRL). Recombinant clones were identified by Southern blot analysis using a 1.2-kb BamHI–XhoI 3′ external probe. After confirming the insertion of the Pol II-neomycin cassette into the BamHI site of the αo gene and absence of secondary insertions, the targeted 129Sv embryonic stem cells were injected into C57BL/6J blastocysts from which chimeric mice were derived. Two (129Sv × C57BL/6J) F1 cross-bred mouse strains heterozygous for loss of Goα were obtained from two chimeric (F0) mice and these were bred to homozygosity (F2 mice) for loss of Goα (αo−/− mice, Fig. 2A).
Figure 1.
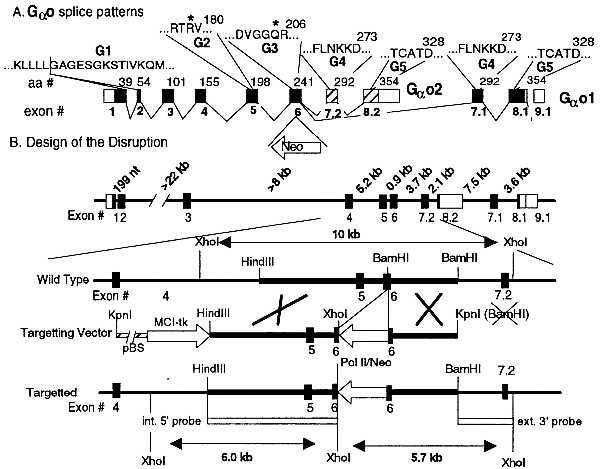
Genomic organization of the gene encoding Go α subunits and design of the disruption. (A) Intron–exon boundaries of the cDNA coding for Goα and location of key amino acid sequences. Boxes, exons; open boxes, untranslated sequences; solid or hatched boxes, translated sequences. Exon numbering is shown below and the number of the last amino acid of each exon is shown above the cDNA. Amino acids in G1 through G5 regions are responsible for binding and hydrolysis of GTP (31). Mutations in R* and Q* of G2 and G3 reduce GTPase activity (32). Pertussis toxin (PTX) ADP ribosylates a Cys at position −4 from C terminus. (B) Genomic structure of the Goα gene (29, 30), structure of the targeting vector, 3′ and 5′ probes for Southern blot analysis, and expected restriction fragment sizes of the wild-type and the mutated alleles.
Figure 2.
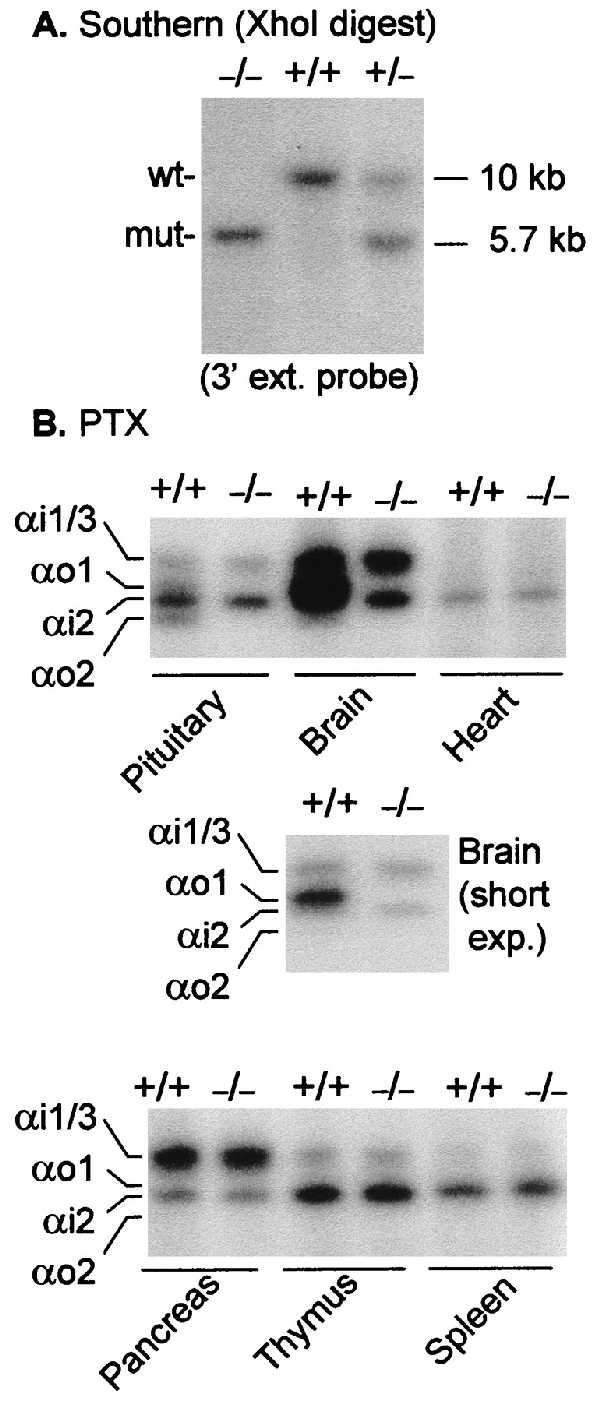
Genotype and ADP-ribosylation patterns of wild-type and αo−/− mice. (A) Southern blot analysis of XhoI-digested DNA from tail biopsies of −/− mice and +/− and +/+ littermates; the probe was the 3′ external probe shown in Fig. 1. (B) Selected tissues from wild-type mice and mice predicted by Southern blot analysis to be Go-deficient. Homogenizations were all in 27% sucrose/1 mM EDTA/10 mM Tris⋅HCl, pH 7.5. Homogenates were ADP-ribosylated without further processing in the presence of guanosine 5′-[β-thio]diphosphate, adenosine 5′-[β,γ-imido]triphosphate, 0.1% SDS, and 2 mM DTT for 30 min at 32°C in a final volume of 15 μl as described (36). NAD was then added to give 4 mM, mixed with 30 μl of 2× Laemmli’s sample buffer, and separated by urea-gradient SDS/PAGE in 9% gels (36). The gels were stained and destained over night and autoradiographed. Except for brain, the predominant bands are αi2. Note that in the pituitary the intensities of αo1 and αo2 (above and below αi2) are about equivalent; in brain, αo1 is the predominant band with αi1/3 and αi2 being similar in intensity. In heart ventricle homogenates, only the αi2 band is evident [absent in αi2-deficient hearts (37)].
Genotyping.
Southern blot analysis was performed according to Sambrook et al. (38) with DNA isolated from tail biopsies. The 3′ external probe was a 1.8-kb BamHI–XhoI fragment, and the 5′ probe was the 4.2-kb HindIII–BamHI fragment used to construct the vector. The plasmid pBluescript KSII, the PolII-Neo cassette and the MC1-tk cassette were used to probe for absence of these sequences in the targeted embryonic stem cell. Probes were labeled with [32P]dCTP by using the random priming method (39). For hybridization, DNA digests were depurinated and transferred onto GeneScreenPlus membranes (NEN/Dupont). The filters were baked at 80°C under vacuum, washed for 1 h at 65°C with 2× SSC (1× SSC = 150 mM NaCl/15 mM sodium citrate, pH 7.0), and prehybridized in 0.5 M sodium phosphate/7% SDS/1% BSA/0.5 mM EDTA, pH 7.2, for 4–6 h at 65°C. Hybridization was at the same temperature in the same solution containing the heat-denatured probes at 2–10 × 106 cpm/ml. After 15 h, the filters were washed for four 5-min periods with 0.5× SSC/0.1% SDS at room temperature and for two 15-min periods at 65°C. The membranes were autoradiographed by using Kodak BioMax MR films. The PCR was also used to genotype DNA samples. Amplification conditions were 1 min at 94°C, 2 min at 64°C, and 3 min at 72°C for 35 cycles with oligonucleotides A and B for wild-type DNA (940 bp) and C and B for mutant DNA (where C is 5′-CAATGGCCGATCCCATATTGGCTGC, an antisense fragment located in the coding region of the neomycin gene; 700 bp) as primers.
Isolation of Dorsal Root Ganglion (DRG) Cells and Recording of Voltage-Gated Ca2+ Channel Currents.
Sensory neurons were obtained from the lumbar and thoracic dorsal root ganglia of adult mice. Ganglia were enzymatically treated and mechanically dispersed as described (40), except that the ganglia were not desheathed and that collagenase treatment was 90 min. DRG neurons were plated onto laminin/ornithine-coated glass coverslips and incubated overnight in MEM containing 10% fetal bovine serum and mouse nerve growth factor (2.5 S; 10–20 ng/ml) at 37°C, 90% humidity, and 3% CO2 before recording.
Voltage-clamp recordings were performed within 30 h of plating by using an Axopatch 200A amplifier (Axon Instruments, Foster City, CA) in the whole-cell patch configuration as described (39). Data were filtered with a 4-pole Bessel filter and digitized. Series resistance, 0.3–5 MΩ, was estimated from the settling rate of the voltage clamp and the membrane capacitance and was compensated (>80%) by using amplifier circuitry. Only data obtained from neurons in which uncompensated series resistance resulted in voltage-clamp errors of less than 5 mV were used. A P/4 protocol was used for leak subtraction. To ensure the linearity of leakage currents around the holding potential for the P/4 protocol, the resting conductance for each cell was determined at potentials between −100 and −60 mV. Data were fit with a nonlinear least-square method.
Ba2+ was used as the charge carrier. The bath solution contained 130 mM choline chloride, 2.5 mM BaCl2, 0.6 mM MgCl2, 10 mM Hepes, and 10 mM glucose (pH was adjusted to 7.4 with Tris base and osmolarity was adjusted with sucrose to 325 milliosmolar). The electrode solution contained 140 mM CsCl, 0.1 mM CaCl2, 2 mM MgCl2, 11 mM EGTA, 10 mM Hepes, 2 mM Mg-ATP, and 1 mM Li-GTP (pH was adjusted to 7.2 with Tris base and osmolarity was adjusted with sucrose to 310 milliosmolar). Patch pipettes filled with electrode solution had resistances of 2–6 MΩ.
Whole-cell impedance and capacitance of each cell were estimated by stepping the membrane potential to −90 mV for four 10-ms periods and measuring the area under the transient associated with the voltage step. Ba2+ currents were evoked from a holding potential of −70 mV by 40-ms steps to 0 mV every 10 s. Currents were sampled at 50 kHz and filtered at 5 kHz. [d-Ala,N-MePhe4,Gly-ol]Enkephalin (DAMGO at 1 μM, dissolved in bath solution; from Research Biochemicals) was applied for 1–2 min, and its effect was determined as a percent change in peak evoked current (39) or, with a prepulse potentiation protocol, as an increase in peak current after a depolarizing step to +80 mV for 100 ms (prepulse) preceding a 30-ms test depolarization to −5 mV initiated 10 ms after the end of the prepulse. Conductance–voltage curves were constructed for each neuron from instantaneous tail currents evoked at −70 mV after 40-ms voltage steps between −80 and +50 mV taken at every 5 mV. Current density was estimated by dividing the peak current evoked at the start of the experiment by the cell capacitance and did not differ in neurons from αo+/+ and αo−/− mice (data not shown).
ADP Ribosylation of Membrane Particles from Murine Tissues.
Except for heart, all other homogenates were made in Dounce homogenizers and 27% (wt/wt) sucrose/1 mM EDTA/10 mM Tris⋅HCl, pH 7.5. Homogenates were centrifuged in glass tubes for 5 min at 1,000 × g, the loose pellets were discarded, and membranes to be ADP-ribosylated were collected by centrifugation at 12,000 × g for 20 min. Cardiac tissue was homogenized in the same medium but with a Polytron at a setting of 8 for 60 s. A pellet, obtained by centrifuging at 10,000 × g for 20 min, was discarded, and membranes to be ADP-ribosylated were collected by centrifugation at 100,000 × g for 30 min in a Beckman SW 50.1 rotor. Other procedures used to ADP-ribosylate and separate the ADP-ribosylated proteins by urea and polyacrylamide gradient gel electrophoresis (urea-gradient SDS/PAGE) were as described (40).
Adenylyl cyclase activity was assayed in murine ventricular heart muscle homogenates as described (41).
RESULTS AND DISCUSSION
Disruption of the Gene Encoding αo Variants.
Two Go α subunit cDNAs, derived from alternatively spliced mRNAs, have been identified by molecular cloning: αo1, encoded in exons 1 through 6 plus 7.1, 8.1, and 9 (42–44); and αo2, encoded in exons 1 through 6 plus 7.2 and 8.2 (45, 46). αo1 and αo2 differ in 26 amino acids dispersed along their last 111 amino acids (total length, 354 amino acids; Fig. 1A). In urea-gradient SDS/PAGE gels, ADP-ribosylated αo1 migrates very close but slightly behind ADP-ribosylated αi2, which it often occludes, and ADP-ribosylated αo2 migrates slightly ahead of ADP-ribosylated αi2 (47). A posttranslational modification of unknown nature generates a third αo (αoC) that comigrates in SDS/PAGE gels with αo2 (47, 48). Fig. 1 shows the intron–exon organization of the Goα gene (29, 30) and the structural features of the insertion vector constructed to disrupt this gene. The neomycin selection cassette was inserted into exon 6, which is common to both splice variants, and thus prevents synthesis of all forms of Go.
Intercrosses of +/− mice yielded Go-deficient mice (αo −/− mice). αo−/− mice had DNA with the expected restriction endonuclease pattern (e.g., Fig. 2A) and, by ADP ribosylation of brain, pituitary, pancreatic islet, and cardiac membranes, showed the expected loss of Go proteins (Fig. 2B). No difference in ADP-ribosylation pattern was seen in tissues not expressing Goα (e.g., thymus, spleen, liver, lung, and exocrine pancreas). A comparison of the pituitary and brain ADP-ribosylation patterns clearly shows that, relative to αo1, αo2 is more abundant in the pituitary.
αo−/− Mice: Inheritance of the Disruption and Poor Survival.
Thirty-eight embryos resulting from four +/− × +/− crosses were genotyped at embryonic days 18–20. We found 8 −/−, 14 +/−, and 15 +/+ mice (22, 38, and 40%, respectively), indicating an under-representation of embryos with one or two disrupted alleles (χ2 test, P > 0.05). Loss of −/− mice became very noticeable after birth. Thus, at weaning we had 348 live mice from 46 litters: 21 −/−, 177 +/−, and 105 +/+ mice, for a distribution of 7, 58, and 35%, respectively. This distribution indicates early death of −/− mice. Also, 3-week-old −/− animals are small, with a mean body weight that is only 45% the weight of their littermates. Weaned −/− animals are unable to feed well by themselves in standard cages either because of the small size or because of general weakness. However, they eat food supplied as a paste on a dish and, those that survive, grow well so that by 8 weeks they have a body weight that does not differ from that of their littermates (Fig. 3A). Weight and growth characteristics of αo+/− mice are indistinguishable from those of wild-type littermates. Fig. 3B shows the survival curve of 31 αo−/− mice starting after the first postnatal week; by 7 weeks 50% had died of unknown reasons. Two survived for more than 50 weeks. When subjected to hot-plate tests, αo−/− animals were found to be hyperalgesic (Fig. 3C). This suggests a role for a Go-coupled pathway in normal pain perception.
Figure 3.
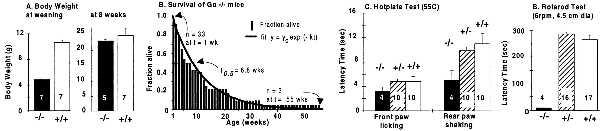
Characteristics of Go-deficient mice. (A) Body weight of littermates at 3 and 8 weeks of age. (B) Survival of 33 mice αo−/− mice at 1 week old. (C) Hot-plate tests of −/−, +/−, and +/+ mice. (D) Time elapsed between start of rotation and fall off of mice from a 4.5-cm-diameter cylinder rotating at 6 rpm. For A, C, and D, data are the mean ± SEM.
Ca2+ Current in Sensory DRG Cells of αo−/− Mice: Inhibition by Opioid Receptor and Activation by Voltage.
Go has been implicated as a mediator of negative feedback regulation of presynaptic Ca2+ channels (13), and the spinal analgesic effect of opioids has been proposed to be due, at least in part, to inhibition of presynaptic Ca2+ channels in the afferent pathway. This was based on the ability of opioids to inhibit the evoked release of neurotransmitter (49) and their ability to inhibit neuronal Ca2+ channels thought to be located at presynaptic terminals (27). We thus tested whether opioid-receptor-mediated inhibition of Ca2+ currents in sensory DRGs would be affected by loss of Go. As shown in Fig. 4, lack of Go resulted in a partial (30%, P < 0.05) loss of the inhibitory effect of the μ-selective opioid receptor agonist DAMGO. This indicates that inhibition of these type of currents is not solely mediated by Go (Fig. 4). We noted further that in 7 of 31 αo−/− cells examined, the position of the conductance–voltage relationship was shifted by −13.3 ± 1.7 mV. Whether the combination of the reduced response to Gi/Go-coupled receptors and the shift in activation voltage are related to and/or responsible for the hyperalgesia seen in hot-plate tests or whether the hyperalgesia is rooted in some other change(s) in neuronal sensitivity remains to be determined.
Figure 4.
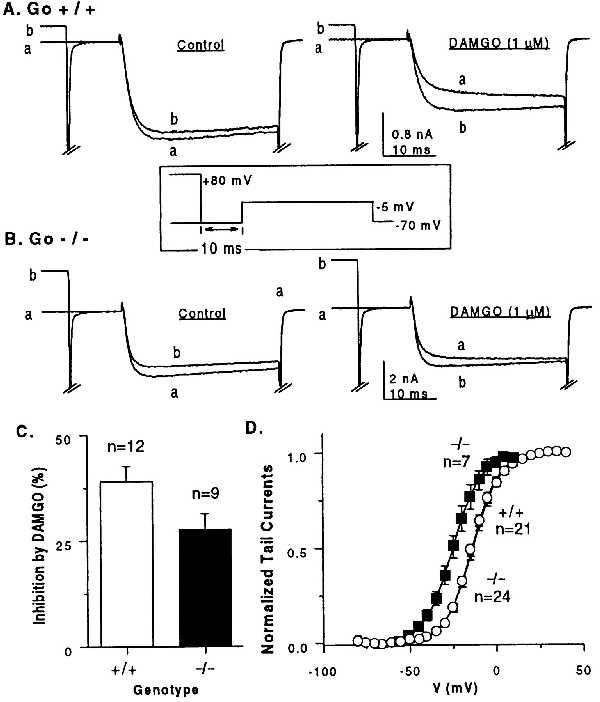
Ca2+ current regulation in DRG cells from αo−/− mice and αo+/+ littermates. (A) αo+/+ cells. (Left) Lack of a depolarizing prepulse to potentiate Ca2+ channel current elicited by a test pulse to −5 mV. (Right) Reversal by prepulse of opioid receptor-mediated inhibition of Ca2+ channel current. (B) αo−/− cells. (Left and Right) As in A. (Inset) Voltage protocol used in A and B. (C) Means of opioid inhibition of Ca2+ channel current in αo−/− and αo+/+ cells. Inhibition by DAMGO was 39 ± 3.6% for +/+ and 27.6 ± 3.8 for −/− cells. The difference, 11.4 ± 5.2%, is significant at the P < 0.05 level. (D) Current–voltage relationship of Ca2+ channel current in αo+/+ (○) and αo−/− cells (▴, occluded by circles, and ▪). Thirty-one αo−/− and 21 αo+/+ cells were analyzed. Current–voltage curves of αo−/− cells fell into two groups: one with a V1/2 of activation of −26 ± 1.5 mV and the other with V1/2 of −12.7 ± 0.8 (mean ± SEM; P < 0.001). V1/2 in +/+ cells was −14.2 ± 0.9 mV.
The opioid receptor-induced inhibition of neuronal non-L type Ca2+ currents is thought to be the direct consequence of the βγ dimer of Go interacting with the α1 Ca2+ channel subunit (for review, see ref. 50; see also ref. 16). The “prepulse facilitation” protocol was applied to test whether the inhibition seen in the absence of Go was still mediated by βγ dimers. This test is based on the observation that inhibition of Ca2+ current by the δ-opioid receptor in NG108–15 cells is very pronounced at low test potentials, −20 to 0 mV, but wanes at higher test potentials (12). This phenomenon is common for the effects of Gi/Go-coupled receptors including the type B γ-aminobutryic acid receptor in chicken DRG cells (51) and the α2-adrenergic receptor in PC12 cells (52). In the “prepulse” protocol, receptor agonist is added to the bath, time is given to allow for installment of channel inhibition, and channel activity is recorded. First, the inhibited state is measured in response to a test pulse, and then it is remeasured after a strong depolarizing pulse to +80 mV or higher (prepulse), which relieves the inhibition exerted by receptor stimulation. Later studies have shown that inhibition is mediated by Gβγ and that loss of inhibition after the prepulse is simply because of dissociation of Gβγ from the channel’s α1 subunit. Repolarization allows for rebinding of Gβγ and reinstallment of the inhibition (e.g., refs. 28, 53, and 54). As shown in Fig. 4C, the Ca2+ current recorded in the presence of the μ-opioid receptor agonist DAMGO was “potentiated,” i.e., uninhibited, by a prepulse. This proved the effective installment of channel inhibition through the βγ arm of the G protein-coupled pathway.
Lack of Increase in Functionally Active (Free) βγ Dimers in αo−/− Cells.
It has been shown that free βγ dimers lead to development of prepulse potentiation in the absence of receptor agonist, so that prepulse potentiation becomes a sensitive test for the existence of functionally active βγ dimers within the intact cell (14, 15). Fig. 4C shows the absence of prepulse potentiation before agonist addition in both αo+/+ and αo−/− cells. This indicates that loss of the Go α subunit did not lead to accumulation of an unbalanced excess of functionally active βγ dimers.
Unimpaired Inhibition of Cardiac Adenylyl Cyclase Activity by a Gi/Go-Coupled Receptor.
In agreement with predictions from studies with partially reconstituted systems (55), with homogenates and membranes (56, 57), and with intact cells (58), it is now recognized that Gi proteins inhibit adenylyl cyclase activities through their activated αi subunits. However, when compared with αi subunits, the very similar αo subunits do not appear to inhibit adenylyl cylcases (17, 18). The report of Valenzuela et al. (26), who in αo−/− mice showed loss of carbachol-induced inhibition of isoproterenol-stimulated cardiac Ca2+ currents, raised the possibility that the M2 muscarinic receptor in ventricle cells might be acting by reducing the levels of cAMP, which stimulates this Gβγ-insensitive channel, and that this reduction of cAMP levels might come about by activation of Go. The possibility existed that loss of the carbachol action was caused by a loss in its ability to reduce adenylyl cyclase activty and thus that Go might be acting as a Gi and inhibit adenylyl cyclase in cardiac ventriculocytes to the exclusion of Gi, perhaps because of a colocalization phenomenon. However, we found that carbachol (100 μM) inhibits cardiac adenylyl cyclase equally well in αo+/+ and αo−/− homogenates [55 ± 3% inhibition for αo+/+ (n = 4) vs. 52 ± 4% (n = 3) for αo−/−; mean ± SD; data not shown). This indicated that whatever the mechanism is by which Go-deficient mice lose muscarinic regulation of Ca2+ currents, it is not because of loss of regulation of cardiac adenylyl cyclase and points to yet another form of regulation of this effector system that is controlled by Go by an unknown mechanism.
Motor Control.
As reported (26), we also noted that αo−/− mice have a generalized tremor. This tremor is accompanied by impairment of motor control. Thus, when placed on a rotating rod (rotarod) 4.5 cm in diameter, αo−/− mice showed a marked deficiency in coordinated motor movement and a tendency to fall off, even from the stationary rod. None stayed on the rotating rod for more than 5 s (Fig. 3D). Likewise, when placed on a 1-m beam that was 1 inch wide (1 inch = 2.54 cm), αo−/− mice generally did not walk along it or, when they did, they did so hesitantly and fell off after just a few paces.
Hyperactivity and Turning Behavior.
Because of the impaired motor control, it was surprising to observe a higher level of activity in cages with αo−/− mice. On closer observation, we noticed that −/− mice appeared to be moving continuously. We thus placed −/− mice individually into a box with the bottom covered with an approximately 1-inch-thick layer of washed and sterilized sand from the beach of Santa Monica, CA. Wild-type and +/− mice behaved the same: walked for the most part along the sides of the box, stopped, reared, groomed, and inspected each corner. Goα −/− mice, in contrast, after a short delay during which they moved tentatively and slowly, gradually accelerated their walk, ignored corners, and were soon (within 1–2 min) running in circles. Depending on the individual, these circles were large and close to the periphery or, for 80% of the mice, changed continuously in diameter covering the complete surface of the box in a nonsystematic way. Thus, −/− mice display a turning behavior that was not seen in wild-type or +/− mice. We videotaped the movements of the mice, replayed the tape on a 9-inch television set, and traced the paths onto transparencies. Fig. 5 shows the paths of one +/+, one +/−, and one −/− mouse and the superposition of the paths of five animals of each group. Once they had started turning, −/− mice continued to do so at varying speeds. In a circular field 18 inches in diameter, the fastest (2155M) run 162.7 ± 20 circles (or ovals) (mean ± SD, n = 3) in 6 min (one every 2–3 s); the slowest made 25.5 ± 3.3 large circles in 6 min. Running is not totally continuous. The mice make short stops to defecate, resume running, stop shortly to inspect, and smell their droppings the next time by, and also stop for short grooming events—licking their coats and stroking their whiskers with their front paws. Otherwise αo−/− mice run for hours on end. Short records of their turning behavior can be seen at http://rfh.anes.ucla.edu/~lutzb/realmice.htm. (requires realplay software, which is down-loadable free of cost). The αo−/− mice see: they avoid objects and other mice placed in their paths. Interactions with littermates (−/−, also running, +/−, or +/+) do occur and are friendly but hasty. −/− animals eat and drink but do so also with haste. Neither −/− females nor −/− males have mated successfully. A histological examination of serial coronal sections of the brains of αo−/− mice (fixed in 4% formalin, 5 μm thick at 100-μm intervals, stained with Luxol fast blue and counterstained with cresyl violet) revealed no obvious anatomical left–right differences.
Figure 5.

Turning behavior of αo−/− mice. Results from an open-field test in which a mouse is placed in a 13 × 17 inch box with a flat bottom covered with a 1-inch layer of sand (for details, see text). The lower superpositions show the movements of five αo−/− mice (2065M, 2093F, 2155M, 2370M, and 2371F, where M is male and F is female). 2065M and 2371F walked almost exclusively along the sides of the box.
In summary, Go-deficient mice show an array of defects that span from the molecular (e.g., unexpected defects and alterations in the regulation of Ca2+ currents) to the integrative (e.g., impaired motor control and extreme turning behavior). Elucidation of the biochemical and cellular mechanisms affected by loss of Go is expected to lead to insights into cell signaling and homeostatic controls.
Acknowledgments
This work was supported in part by National Institutes of Health Grant DK-19318 to L.B.; a postdoctoral fellowship from the Medical Research Council, Ontario, Canada, to G.B.; a postdoctoral Fellowship of the Institute Nationale pour la Recherche Médicale to P.B.; and a postdoctoral fellowship from the Deutsche Forschungsgesellschaft to K.S. M.S.G. was the recipient of a Giannini Foundation Fellowship.
ABBREVIATIONS
- DRG
dorsal root ganglion
- DAMGO
[d-Ala,N-MePhe4,Gly-ol]enkephalin
References
- 1.Neer E J, Lok J M, Wolf L G. J Biol Chem. 1984;259:14222–14229. [PubMed] [Google Scholar]
- 2.Huff R M, Axton J M, Neer E J. J Biol Chem. 1985;260:10864–10871. [PubMed] [Google Scholar]
- 3.Sternweis P C, Robishaw J D. J Biol Chem. 1984;259:13806–13813. [PubMed] [Google Scholar]
- 4.Cerione R A, Regan J W, Nakata H, Codina J, Benovic J L, Gierschick P, Somers R L, Spiegel A M, Birnbaumer L, Lefkowitz R J, Caron M G. J Biol Chem. 1986;261:3901–3909. [PubMed] [Google Scholar]
- 5.Florio V A, Sternweis P C. J Biol Chem. 1985;260:3477–3483. [PubMed] [Google Scholar]
- 6.Hescheler J, Rosenthal W, Trautwein W, Schultz G. Nature (London) 1987;325:445–447. doi: 10.1038/325445a0. [DOI] [PubMed] [Google Scholar]
- 7.Kleuss C, Hescheler J, Ewel C, Rosenthal W, Schultz G, Wittig B. Nature (London) 1991;353:43–48. doi: 10.1038/353043a0. [DOI] [PubMed] [Google Scholar]
- 8.Taussig R, Sanchez S, Rifo M, Gilman A G, Belardetti F. Neuron. 1992;8:799–809. doi: 10.1016/0896-6273(92)90100-r. [DOI] [PubMed] [Google Scholar]
- 9.Chen C, Clarke I J. J Physiol. 1996;491:21–29. doi: 10.1113/jphysiol.1996.sp021193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strittmatter S M, Valenzuela D, Kennedy T E, Neer E J, Fishman M C. Nature (London) 1990;344:836–841. doi: 10.1038/344836a0. [DOI] [PubMed] [Google Scholar]
- 11.Nishimoto I, Okamoto T, Matsuura Y, Takahashi S, Okamoto T, Murayama Y, Ogata E. Nature (London) 1993;362:75–79. doi: 10.1038/362075a0. [DOI] [PubMed] [Google Scholar]
- 12.Tsunoo A, Yoshi M, Narahashi T. Proc Natl Acad Sci USA. 1986;83:9832–9836. doi: 10.1073/pnas.83.24.9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell V, Berrow N, Dolphin A C. J Physiol. 1993;470:1–11. doi: 10.1113/jphysiol.1993.sp019842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikeda S R. Nature (London) 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 15.Herlitze S, Garcia D E, Mackie K, Hille B, Scheuer T, Catterall W A. Nature (London) 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 16.Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Proc Natl Acad Sci USA. 1997;94:8866–8871. doi: 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong Y H, Conklin B R, Bourne H R. Science. 1992;255:339–342. doi: 10.1126/science.1347957. [DOI] [PubMed] [Google Scholar]
- 18.Taussig R, Iñiguez-Lluhi J A, Gilman A G. Science. 1993;261:218–221. doi: 10.1126/science.8327893. [DOI] [PubMed] [Google Scholar]
- 19.Kroll S D, Chen J, De Vivo M, Carthy D J, Buku A, Premont R T, Iyengar R. J Biol Chem. 1992;267:23183–23188. [PubMed] [Google Scholar]
- 20.Van Biesen T, Hawes B E, Raymond J R, Luttrell L M, Koch W J, Lefkowitz R J. J Biol Chem. 1996;272:1266–1269. doi: 10.1074/jbc.271.3.1266. [DOI] [PubMed] [Google Scholar]
- 21.Moriarty T M, Padrell E, Corby D J, Omri G, Landau E M, Iyengar R. Nature (London) 1990;343:79–82. doi: 10.1038/343079a0. [DOI] [PubMed] [Google Scholar]
- 22.VanDongen A, Codina J, Olate J, Mattera R, Joho R, Birnbaumer L, Brown A M. Science. 1988;242:1433–1437. doi: 10.1126/science.3144040. [DOI] [PubMed] [Google Scholar]
- 23.Rudolph U, Brabet P, Kaplan J, Hasty P, Bradley A, Birnbaumer L. J Recept Res. 1993;13:619–637. doi: 10.3109/10799899309073683. [DOI] [PubMed] [Google Scholar]
- 24.Rudolph U, Finegold M J, Rich S S, Harriman G R, Srinivasan Y, Brabet P, Boulay G, Bradley A, Birnbaumer L. Nat Genet. 1995;10:143–150. doi: 10.1038/ng0695-143. [DOI] [PubMed] [Google Scholar]
- 25.Offermans S, Toombs C F, Hu Y H, Simon M I. Nature (London) 1997;389:183–186. doi: 10.1038/38284. [DOI] [PubMed] [Google Scholar]
- 26.Valenzuela D, Han X, Mende U, Fankhauser C, Mashimo H, Huang H, Pfeffer J, Neer E J, Fishman M C. Proc Natl Acad Sci USA. 1997;94:1727–1732. doi: 10.1073/pnas.94.5.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bourinet E, Soong T W, Stea A, Snutch T P. Proc Natl Acad Sci USA. 1996;93:1486–1491. doi: 10.1073/pnas.93.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J F, Ellinor P T, Aldrich R W, Tsien R W. Neuron. 1996;17:991–1003. doi: 10.1016/s0896-6273(00)80229-9. [DOI] [PubMed] [Google Scholar]
- 29.Tsukamoto T, Toyama R, Itoh H, Kozasa T, Matsuoka M, Kaziro Y. Proc Natl Acad Sci USA. 1991;88:2974–2978. doi: 10.1073/pnas.88.8.2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bertrand P, Sanford J, Rudolph U, Codina J, Birnbaumer L. J Biol Chem. 1990;265:18576–18580. [PubMed] [Google Scholar]
- 31.Noel J P, Hamm H E, Sigler P B. Nature (London) 1993;366:654–663. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- 32.Graziano M P, Gilman A G. J Biol Chem. 1989;264:15475–15482. [PubMed] [Google Scholar]
- 33.Rudolph U, Brabet P, Hasty P, Bradley A, Birnbaumer L. Transgenic Res. 1993;2:345–355. doi: 10.1007/BF01976176. [DOI] [PubMed] [Google Scholar]
- 34.Soriano P, Montgomery C, Geske R, Bradley A. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 35.Mansour S L, Thomas K R, Capecchi M R. Nature (London) 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 36.Codina J, Grenet D, Chang K-J, Birnbaumer L. J Receptor Res. 1991;11:587–601. doi: 10.3109/10799899109066429. [DOI] [PubMed] [Google Scholar]
- 37.Rudolph U, Spicher K, Birnbaumer L. Proc Natl Acad Sci USA. 1996;93:3209–3214. doi: 10.1073/pnas.93.8.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 39.Feinberg A P, Vogelstein B. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 40.Gold M S, Dastmalchi S, Levine J D. Neuroscience. 1996;71:265–275. doi: 10.1016/0306-4522(95)00433-5. [DOI] [PubMed] [Google Scholar]
- 41.Khasar S G, Gold M S, Dastmalchi S, Levine J D. Neurosci Lett. 1996;218:17–20. doi: 10.1016/0304-3940(96)13111-6. [DOI] [PubMed] [Google Scholar]
- 42.Itoh H, Kozasa T, Nagata S, Nakamura S, Katada T, Ui M, Iwai S, Ohtsuka E, Kawasaki H, Suzuki K, Kaziro Y. Proc Natl Acad Sci USA. 1986;83:3776–3780. doi: 10.1073/pnas.83.11.3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones D T, Reed R R. J Biol Chem. 1987;262:14241–14249. [PubMed] [Google Scholar]
- 44.VanMeurs K P, Angus W, Lavu S, Kung H F, Czarnecki S K, Moss J, Vaughan M. Proc Natl Acad Sci USA. 1987;84:3107–3111. doi: 10.1073/pnas.84.10.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsu W H, Rudolph U, Sanford J, Bertrand P, Olate J, Nelson C, Moss L G, Boyd A E, III, Codina J, Birnbaumer L. J Biol Chem. 1990;265:11220–11226. [PubMed] [Google Scholar]
- 46.Strathmann M, Wilkie T, Simon M I. Proc Natl Acad Sci USA. 1990;87:6477–6481. doi: 10.1073/pnas.87.17.6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Codina J, Yatani A, VonDongen A M J, Padrell E, Carty D, Mattera R, Brown A M, Iyengar R, Birnbaumer L. In: G Proteins. Iyengar R, Birnbaumer L, editors. San Diego: Academic; 1990. pp. 267–294. [Google Scholar]
- 48.Wilcox M D, Dingus J, Balcueva E A, McIntire W E, Mehta N D, Schey K L, Robishaw J D, Hildebrandt J D. J Biol Chem. 1995;270:4189–4192. doi: 10.1074/jbc.270.9.4189. [DOI] [PubMed] [Google Scholar]
- 49.Arbilla S, Langer S Z. Nature (London) 1978;271:559–560. doi: 10.1038/271559a0. [DOI] [PubMed] [Google Scholar]
- 50.Dolphin A C. Trends Neurosci. 1996;19:35–43. doi: 10.1016/0166-2236(96)81865-0. [DOI] [PubMed] [Google Scholar]
- 51.Marchetti C, Carbone E, Lux H D. Pflüegers Arch. 1986;406:104–111. doi: 10.1007/BF00586670. [DOI] [PubMed] [Google Scholar]
- 52.Bean B P. Nature (London) 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- 53.Lipscombe D, Kongsamut S, Tsien R W. Nature (London) 1989;340:639–642. doi: 10.1038/340639a0. [DOI] [PubMed] [Google Scholar]
- 54.Lopez H S, Brown A M. Neuron. 1991;7:1061–1068. doi: 10.1016/0896-6273(91)90350-9. [DOI] [PubMed] [Google Scholar]
- 55.Hildebrandt J D, Codina J, Birnbaumer L. J Biol Chem. 1984;259:13178–13185. [PubMed] [Google Scholar]
- 56.Toro M-J, Montoya E, Birnbaumer L. Mol Endocrinol. 1987;1:669–676. doi: 10.1210/mend-1-10-669. [DOI] [PubMed] [Google Scholar]
- 57.Hildebrandt J D, Kohnken R E. J Biol Chem. 1990;265:9825–9830. [PubMed] [Google Scholar]
- 58.Wong Y H, Federman A, Pace A M, Zachary I, Evans T, Pouysségur, Bourne H R. Nature (London) 1991;351:63–65. doi: 10.1038/351063a0. [DOI] [PubMed] [Google Scholar]