

Abstract
Intramolecular carbon isotope distributions reflect details of the origin of organic compounds and may record the status of complex systems, such as environmental or physiological states. A strategy is reported here for high-precision determination of 13C/12C ratios at specific positions in organic compounds separated from complex mixtures. Free radical fragmentation of methyl palmitate, a test compound, is induced by an open tube furnace. Two series of peaks corresponding to bond breaking from each end of the molecule are analyzed by isotope ratio mass spectrometry and yield precisions of SD(δ-13C) < 0.4‰. Isotope labeling in the carboxyl, terminal, and methyl positions demonstrates the absence of rearrangement during activation and fragmentation. Negligible isotopic fractionation was observed as degree of fragmentation was adjusted by changing pyrolysis temperature. [1-13C]methyl palmitate with overall δ-13C = 4.06‰, yielded values of +457‰ for the carboxyl position, in agreement with expectations from the dilution, and an average of −27.95‰ for the rest of the molecule, corresponding to −27.46‰ for the olefin series. These data demonstrate the feasibility of automated high-precision position-specific analysis of carbon for molecules contained in complex mixtures.
High-precision isotope ratio mass spectrometry (IRMS) instruments used for the analysis of bulk, chemically heterogeneous materials were developed in the 1940s and 50s (1, 2) after the remarkable advances of earlier decades with low-precision instruments (3). The classical dual-inlet approach formed the basis of IRMS for several decades thereafter, delivering precision for differential measurements of up to six significant figures and enabling measurement of variation in the stable isotopes of C, N, O, S, and H due to natural processes (4, 5). In contrast to organic mass spectrometry (MS), IRMS instruments are designed to operate optimally with a few selected gases (e.g., CO2 for C and O analysis), which must be prepared as isotopically representative of the analyte prior to introduction to the IRMS. The majority of IRMS studies have therefore focused on chemically simple systems such as natural waters or CO2, or on chemically heterogeneous materials that can be converted to the appropriate gas by a straightforward chemical process, such as combustion. Since manual preparative pretreatment steps are usually cumbersome, isotope studies of individual chemically pure compounds are limited to simple systems, such as natural waters, and microgram-to-milligram samples are required.
In the mid-1970s, Sano et al. (6) and Matthews and Hayes (7) demonstrated a means for coupling online chemical purification by chromatography to MS-based isotope measurements, adding a combustion step subsequent to the separation. Termed “isotope-ratio monitoring,” it was first applied to a high-precision multicollector IRMS in the mid-1980s (8), which has led to the capability to determine carbon isotope ratios from as little as 10 ng of purified compounds. Recently termed compound-specific isotope analysis (CSIA), its commercial introduction around 1990 precipitated a dramatic expansion to hundreds of laboratories worldwide in fields as diverse as geochemistry, ecology (9), and biomedicine (10, 11). Application of CSIA to biogeochemical topics has, for instance, yielded clues to origins of ancient sediments (12, 13) and climates (14).
In 1961, Abelson and Hoering (15) showed the first intramolecular variations in carbon isotopes in their study of amino acids, by isolating the carboxyl position manually via the ninhydrin reaction, and showing this position to be enriched relative to the rest of the molecule. The few cases where high-precision intramolecular isotope analysis has since been reported include classic work showing the origin of the low 13C content of lipids (16–20) and metabolic variability of fatty acid carboxyl 13C (21). These results are consistent with kinetic isotope effect theory and experiments, where isotopic fractionation is known to be greatest at carbons involved in bond breaking and bond making. Theoretical and experimental studies suggest that isotopic signatures within molecules record a wide range of phenomena (e.g., physiological and biochemical status in organisms, including mammals; refs. 22–24). Presently, the measurement of intramolecular isotope ratios is at a similar stage as pre-1978 CSIA, requiring manual isolation of carbon positions within a molecule prior to analysis, which is cumbersome at best and practically impossible for many internal positions (25). Low-precision intramolecular isotope analysis is routine by a variety of techniques including NMR and organic MS. The latter is generally limited to precision of no better than 0.1 atom percent, which is sufficient only for enriched tracer studies. Even at this level, experimental protocols often are limited by the quantity of initial tracer material required to produce an adequate signal after dilution with ambient levels. The goal of this work was to develop an instrument for automated position-specific isotope analysis (PSIA) of carbon at a precision of SD(δ-13C) < 1‰.
Automated fragmentation by pyrolytic means followed by GC separation was investigated in the 1960s for molecular fingerprinting and identification (26–30). These early studies showed that organic compounds of biological and commercial interest fragment in useful and characteristic ways, following predictions of simple free radical fragmentation mechanisms worked out decades earlier for hydrocarbons (31, 32). After activation by formation of a free radical, a single C—C bond breaks and the two resulting fragments ultimately stabilize and can be isolated. Because fragments related structurally to one another differ in isotope ratio by C not common to both, the isotope ratio of a position or moiety in the parent compound can be calculated from measurements of appropriate fragments if no C scrambling or severe fractionation occurs during pyrolysis.
Experimental.
An instrument for online PSIA was constructed by coupling a GC-pyrolysis-GC system to both a combustion/high-precision IRMS and an ion-trap MS for structure identification, as shown in Fig. 1. GC-I was equipped with a split/splitless injector, operated in split mode, and a 60 m × 0.32 mm Rtx-225 (0.25 μm film: 50% cyanopropylmethyl/50% phenylmethyl-polysiloxane) capillary column (Restek, Bellefonte, PA) purified the test compound, methyl palmitate (Me16:0), from the solvent, hexane. By means of an automated two-position valve (Valco Instruments, Houston), the precursor compound can be directed to either a flame ionization detector (FID) for quantitative analysis and methods development or to the pyrolysis furnace. Alternatively, a bypass injector plumbed directly to the pyrolysis furnace can be used for analysis of pure compounds. The pyrolysis chamber was 25 cm × 0.25 mm deactivated fused silica capillary surrounded by a resistively heated ceramic furnace, with temperature controlled to ±0.5°C with a CN9000A series temperature controller (Omega Engineering, Stamford, CT). Products emerging from the furnace were collected and cryofocused at −40°C in GC-II, equipped with a 50 m × 0.2 mm HP-1 (0.5 μm film: methyl silicone gum) capillary column (Hewlett–Packard). The GC-II oven temperature was increased linearly with time to 290°C to separate the fragments. With a second two-position valve, products were then directed to either a QISMS ion trap mass spectrometer (Varian) for structural analysis, or to a combustion/water-trap/open-split interface and then to a Finnigan-MAT model 252 IRMS (Bremen, Germany) for isotope ratio analysis. The fused silica combustion furnace was filled with oxidized Cu metal and held at 850°C, and the water trap was of the Nafion type described previously (5). An FID is available on the GC-II as well, which is most useful for determination of degree of fragmentation. The ion trap was controlled by Varian saturn GC/MS software, version 5.2 for dos 6.22; the Finnigan IRMS was controlled by isodat 5.2 for Concurrent DOS (Digital Equipment).
Figure 1.

Experimental system for online PSIA. Capillary GC-I separates the target compound from the mixture, passes it into a hollow, deactivated fused-silica tube pyrolysis furnace held at about 550°C. Fragments pass through the heated transfer line to GC-II with the oven cooled to −40°C for collection and focusing. After separation, fragments can be directed by valving to (i) an FID, (ii) a Varian QISMS ion trap MS for structural analysis, or (iii) a combustion/water-trap/open-split interface to a Finnigan–MAT model 252 IRMS for high-precision isotopic analysis.
Natural abundance Me16:0 was purchased from Sigma, and 13C-labeled Me16:0s were purchased from Cambridge Isotope Laboratories (Cambridge, MA). Both were used without further purification. The solvent was HPLC-grade hexane.
Organic mass spectra were acquired in positive ion electron impact (EI) and chemical ionization (CI; methane) modes. To augment spectral interpretation for structure assignment, spectra were searched with the wiley mass spectral database (Palisades, Newfield, NY) using the “purity” search parameter.
Isotope ratios were calibrated against a working standard of CO2 gas ultimately calibrated against NIST RM-22, graphite, and converted to the delta notation given by:
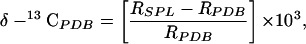 |
1 |
where Rx = [13C]/[12C], SPL refers to the sample, and PDB refers to the international standard PeeDee Belemnite, with RPDB = 0.0112372.
RESULTS AND DISCUSSION
Fig. 2 is a typical pyrogram produced with a pyrolysis furnace temperature of 550°C. Previous work (30) suggests that all peaks can be accounted for as two series of fragments with structures corresponding to single C—C bond breaking along the chain, shown in Fig. 3 and postulated to be ω-unsaturated fatty acid methyl esters (FAMEs), while the other is a series of α-olefins. For the FAME series, CI conditions produced a pseudomolecular ion of the highest molecular mass pyrolysis product at m/z 255 corresponding to a loss of 16 Da, CH2, and 2 H, from the unpyrolyzed Me16:0 pseudomolecular ion appearing at m/z 271. Pseudomolecular ions of subsequent members of the series appear with losses of 14 Da, consistent with pyrolytic losses of additional methylenes. Mass spectral peaks at [MH-32]+ and [MH-50]+ corresponding to a loss of CH3OH and CH3OH + H2O are consistent with the methyl ester structure. EI spectral searching gives first priority “hits” for the four to eight carbon members of the ω-unsaturated FAME series, with the top five priority hits for all other postulated structures of this series present in the database.
Figure 2.

Pyrogram resulting from analysis of Me16:0 with pyrolysis temperature of 550°C and detection by the ion trap operated in CI mode. Two series of peaks are observed; their overlapping retention times give rise to several doublets eluting at intermediate times as shown in the Inset.
Figure 3.

Structures of the two series of peaks observed in the pyrogram of Fig. 2. An ω-unsaturated FAME series running from C4 to C16 and an α-olefin series from C1 to C16 (including methane) account for all peaks above 2% intensity in the pyrogram.
Mass spectra of members of the postulated α-olefin series are consistent with that assignment, producing hydrocarbon ion series very similar to one another but not yielding molecular ions in either positive methane CI or EI modes, as α-olefins with structurally definitive ions are difficult to obtain in the ion trap MS. Series members of up to five carbons yield expected molecular ions. The appearance of a double bond including the terminal carbon is well known as a stabilization mechanism for free radicals and is the most likely assignment for these fragments (31).
Rearrangement Studies.
To test the extent of carbon rearrangement in the pyrolysis furnace, a series of four isotopically labeled Me16:0s, [1-13C], [Me,1-13C], [16-13C], and [Me,16-13C], were prepared and each were separately subjected to pyrolytic fragmentation. CI mass spectra of a representative ω-unsaturated FAME pyrolysis product, Me11:1, are presented in Fig. 4. The molecular ion of the unlabeled pseudomolecular ion appears at m/z 199, and prominent peaks correspond to loss of methanol (M-32) and water [M-32–18]+. Me11:1 produced from pyrolysis of the [Me,16-13C]Me16:0 parent produces the mass spectrum in Fig. 4B, with the molecular ion increased in mass by only 1 Da due to the methyl-13C and no contribution from the carbon-labeling position 16 of the parent. Also observed are a loss of 33 Da from labeled methanol, and 51 Da by an additional loss of water; no evidence of pyrolytic rearrangement is apparent in this spectra. There was no evidence of any labeled carbon in any of the α-olefin MS spectra. Table 1 presents the molecular ion masses for the unlabeled and all labeled methyl ester species. All molecular ions appear at appropriate masses corresponding to no rearrangement; the sole exception is the five carbon fragments that all appear at a mass 1 Da lower than predicted. This is best explained by a mass calibration error for these fragments, which appear at the lowest abundance in the pyrograms. These data show conclusively that no rearrangement of the Me, carboxyl, or terminal carbons of Me16:0 takes place during pyrolytic fragmentation.
Figure 4.

CI mass spectra obtained with the ion trap for the Me11:1 fragment of the natural abundance Me16:0 (A), or the [Me,16-13C] test compound (B). The labeled molecular ion is shifted by +1 Da after pyrolytic loss of the label at C-16 and retention of the methyl label. The peak m/z 167 reflects loss of one labeled C, with loss of methanol in the ion trap.
Table 1.
Masses of molecular ions for all ω-unsaturated FAME fragments for unlabeled and all labeled test Me16:0. All masses appear at the m/z values expected if there is no rearrangement during pyrolytic fragmentation.
| Methyl ester fragment | Molecular ions
|
||||
|---|---|---|---|---|---|
| Unlabeled | 1-13C | 1-13C, Me-13C | 16-13C | 16-13C, Me-13C | |
| Me3:1 | 87 | 88 | 89 | 87 | 88 |
| Me4:1 | 100 | 101 | 102 | 100 | 101 |
| Me5:1 | 115 | 116 | 117 | 115 | 116 |
| Me6:1 | 129 | 130 | 131 | 129 | 130 |
| Me7:1 | 143 | 144 | 145 | 143 | 144 |
| Me8:1 | 157 | 158 | 159 | 157 | 158 |
| Me9:1 | 171 | 172 | 173 | 171 | 172 |
| Me10:1 | 185 | 186 | 187 | 185 | 186 |
| Me11:1 | 199 | 200 | 201 | 199 | 200 |
| Me12:1 | 213 | 214 | 215 | 213 | 214 |
| Me13:1 | 227 | 228 | 229 | 227 | 228 |
| Me14:1 | 241 | 242 | 243 | 241 | 242 |
| Me15:1 | 255 | 256 | 257 | 255 | 256 |
| Me16:0 (parent) | 271 | 272 | 273 | 272 | 273 |
Pyrolysis-Induced Fractionation.
To determine whether severe isotopic fractionation occurs during pyrolysis, Me16:0 was pyrolyzed at two temperatures to induce differing degrees of fragmentation, with subsequent measurement of fragment isotope ratios. At 550°C or 600°C, FID analysis showed that Me16:0 pyrolyzed to 20% or 60% completion, respectively. The mean (± SD) isotope ratios of the fragments with at least five C atoms at 550°C and 600°C was δ-13C = −26.99 ± 0.59‰ and −25.18 ± 0.65‰, respectively, for the α-olefins (n = 11), and −29.14 ± 0.88‰ and −28.30 ± 1.05‰ for the methyl esters (n = 12). The differences in means for olefins and methyl esters are 1.81‰ and 0.84‰, respectively, both statistically significant (pairwise t test, P < 0.001). Because the magnitude of the difference is small, we conclude that pyrolysis-induced fractionation is relatively insensitive to temperature and that pyrolysis conducted at a single temperature results in fragments with isotope ratios that are readily calibrated against standards.
High-Precision PSIA of a Labeled Compound.
A sample of [1-13C]Me16:0 was diluted ≈200-fold to yield 0.5 atom percent excess 13C in the carboxyl position, and with an overall isotope ratio of δ-13C = 4.06‰. High-precision isotope ratio analysis yielded the data plotted in Fig. 5. The average precision for both methyl ester and olefin series was SD(δ-13C) < 0.4‰. The olefin series falls uniformly in the natural abundance range averaging about δ-13C = −27‰ for the fragments greater than C3, consistent with uniform loss of the labeled carboxyl carbon. The methyl ester series resembles a mixing curve with the highest isotope ratio, δ-13C = 92.6‰, for the smallest fragment, the four carbon Me3:1, and decreasing to about δ-13C = 4.21‰ for large fragments. The sole exception to this trend is the Me12:1 fragment, which is reproducibly depleted. Careful analysis of the EI mass spectrum of this fragment revealed a series of peaks not present in mass spectra of adjacent peaks, consistent with a coeluting natural abundance contaminant. We therefore excluded this fragment from further calculations.
Figure 5.

(A) Isotope ratios expressed in atom fraction for both series of fragments generated upon pyrolysis of [1-13C]Me16:0 diluted 1:200. A plot of the methyl ester series 13C atom fractions vs. carbon chain length is reminiscent of a mixing curve where the C4:1 fragment is most enriched and each sequential member is diluted with natural abundance carbon. (B) A plot of the atom fractions vs. the reciprocal chain length yields a straight line with the intercept corresponding to the mean atom fraction of all positions except C1 (i.e., at infinite dilution), while the sum of the intercept and the slope yields the atom fraction of the C1 position. All members of the olefin series correspond to natural abundance since the C1 position is removed pyrolytically.
Mass balance can be used to calculate the isotope ratio of the carboxyl position from the methyl ester data. The equation governing the isotope ratio of any particular fragment can be expressed as:
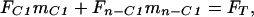 |
2 |
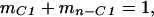 |
3 |
where F is 13C atom fraction, m is C molecular mass fraction, C1 designates the carboxyl position, and n−C1 designates all other positions in the fragment. Substituting mn−C1 = 1−mC1 into Eq. 2 and rearranging we obtain:
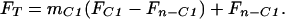 |
4 |
A plot of FT vs. mC1 yields an intercept corresponding to the average 13C atom fraction of the molecule excluding the carboxyl position, while the sum of the intercept and slope yields the atom fraction of the carboxyl position (FC1 = Fn−C1 + slope). Applying this equation to the data of Fig. 5, we find an intercept corresponding to δ-13C(n − C1) = −27.95‰ (Fn−C1 = 0.010805) in excellent correspondence to the mean isotope ratio for the α-olefin series (excluding methane and ethene) of δ-13C(n − C1) = −27.46‰ (Fn−C1 = 0.010810). Calculation using the slope yields for the carboxyl position, δ-13C(C1) = 457.5‰ (FC1 = 0.0161143), which is in good agreement with the estimated 200:1 dilution of [1-13C]Me16:0 with unlabeled Me16:0.
This calculation can be performed more conveniently by substituting for δ-13C, introducing the quantity φ-13C to represent the relative isotope fraction compared with an international standard:
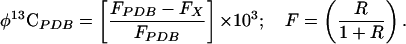 |
5 |
FPDB = 0.0111123 is readily calculated from RPDB (=0.0112372). The final form of the mass balance equation is then:
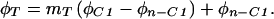 |
6 |
This form is accurate and the units are similar to the conventional δ-13C, which cannot be cast in this simple mass balance form because it is based on isotope ratios rather than isotope fractions or concentrations. The advantage of this form is manifest when calculations involve isotope fractions far removed from the international standard, although the errors introduced using mass balance based on δ-13C are insignificant around natural abundance. φ-13C for the unlabeled C can be read off of the intercept estimate, and φ-13C is the sum of the intercept and slope.
Extending this approach to a broad range of volatile compounds should be straightforward. For instance, although double bonds of unsaturated fatty acids complicate pyrolytic fragmentation considerably, hydrogenation either before analysis or online after separation, as has been demonstrated (33), reduces the problem to that of saturates as reported here; thus this approach should apply to all fatty acids. Lower molecular weight compounds should result in correspondingly simpler pyrograms. Application to nonvolatiles such as amino acids relies on fragmentation properties of volatile derivatives.
These results demonstrate that controlled pyrolysis can produce single-bond breakage, stabilization, and isotopically representative fragments for carbon. The absence of measurable rearrangement or severe fractionation show the method to be applicable to intramolecular studies of carbon isotopes at natural abundance or at very low enrichment. Prospects seem promising for position-specific analysis of other organic elements using pyrolysis to generate isotopically representative fragments. In cases where O, N, and S are structurally separated in organic molecules, fragmentation at remote bonds may be exploited to yield separate fragments containing the positions of interest. On the other hand, hydrogen isotopes are known to undergo intramolecular rearrangement in hydrocarbons via a coiling mechanism and may well undergo intermolecular exchange prior to C—C bond breakage. Thus, position-specific isotope analysis for hydrogen must be approached with caution.
Acknowledgments
We thank M. Schoell, P. Haggarty, L. Houghton, H. Tobias, and R. Caimi for helpful discussions, and are grateful to K. Goodman for the design of the fused silica combustion furnace. This work was supported by National Institutes of Health Grant GM49209 and the Chevron Petroleum Technology Corporation. T.N.C. acknowledges predoctoral support from National Institutes of Health Training Grant DK07158.
Footnotes
Abbreviations: PSIA, position-specific isotope analysis; CSIA, compound-specific isotope analysis; MS, mass spectrometry; IRMS, isotope ratio MS; EI, electron impact; CI, chemical ionization; Me16:0, methyl palmitate; FAME, fatty acid methyl ester; FID, flame ionization detector.
References
- 1.Murphey B F. Phys Rev. 1947;72:834–837. [Google Scholar]
- 2.McKinney C R, McCrea J M, Epstein S, Allen H A, Urey H C. Rev Sci Instrum. 1950;21:724–730. doi: 10.1063/1.1745698. [DOI] [PubMed] [Google Scholar]
- 3.Nier A O. Rev Sci Instrum. 1940;11:212–216. [Google Scholar]
- 4.Pillinger C T. Int J Mass Spectrom Ion Processes. 1992;118/119:477–501. [Google Scholar]
- 5.Brenna J T. Acc Chem Res. 1994;27:340–346. [Google Scholar]
- 6.Sano M, Yotsui Y, Abe H, Sasaki S. Biomed Mass Spectrom. 1976;3:1–3. doi: 10.1002/bms.1200030102. [DOI] [PubMed] [Google Scholar]
- 7.Matthews D E, Hayes J M. Anal Chem. 1978;50:1465–1473. [Google Scholar]
- 8.Barrie A, Bricout J, Koziet J. Biomed Mass Spectrom. 1984;11:583–588. [Google Scholar]
- 9.Schoell, M. & Hayes, J. M., eds. (1994) Org. Geochem. 21.
- 10.Tissot S, Normand S, Guilluy R, Pachiaudi C, Beylot M, Laville M, Cohen R, Mornex R, Riou J P. Diabetologia. 1990;33:449–456. doi: 10.1007/BF00405104. [DOI] [PubMed] [Google Scholar]
- 11.Dammelmair H, Schenck U V, Behrendt E, Sauerwald T, Koletzko B. J Pediatr Gastro Nutr. 1995;21:31–36. doi: 10.1097/00005176-199507000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Freeman K H, Hayes J M, Trendel J M, Albrecht P. Nature (London) 1989;353:254–256. doi: 10.1038/343254a0. [DOI] [PubMed] [Google Scholar]
- 13.Hayes J M, Freeman K H, Popp B N, Hoham C H. Org Geochem. 1990;16:1115–1128. doi: 10.1016/0146-6380(90)90147-r. [DOI] [PubMed] [Google Scholar]
- 14.Schoell M, Schouten S, Sinninghe-Damste J S, de Leeuw J W, Summons R-E. Science. 1994;263:1122–1125. doi: 10.1126/science.263.5150.1122. [DOI] [PubMed] [Google Scholar]
- 15.Abelson P H, Hoering T C. Proc Natl Acad Sci USA. 1961;47:623–632. doi: 10.1073/pnas.47.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeNiro M J, Epstein S. Geochim Cosmochim Acta. 1978;42:485–506. [Google Scholar]
- 17.Monson K D, Hayes J M. J Biol Chem. 1980;255:11435–11441. [PubMed] [Google Scholar]
- 18.Monson K D, Hayes J M. J Biol Chem. 1982;257:5568–5575. [PubMed] [Google Scholar]
- 19.Monson K D, Hayes J M. Geochim Cosmochim Acta. 1982;46:139–149. [Google Scholar]
- 20.Melzer E, Schmidt H L. J Biol Chem. 1987;262:8159–8164. [PubMed] [Google Scholar]
- 21.Vogler E A, Hayes J M. In: Physics and Chemistry of the Earth. Douglas A G, Maxwell J R, editors. Vol. 12. Oxford: Pergamon; 1980. pp. 697–704. [Google Scholar]
- 22.Ivlev A A. Biophysics. 1985;30:92–97. [Google Scholar]
- 23.Ivlev A A. Biokhimiya. 1986;50:1605–1615. [Google Scholar]
- 24.Galimov E M. The Biological Fractionation of Isotopes. New York: Academic; 1985. [Google Scholar]
- 25.Ivlev A A. Biophysics. 1991;36:1078–1087. [Google Scholar]
- 26.Dhont J H. Nature (London) 1961;192:747–748. [Google Scholar]
- 27.Dhont J H. Nature (London) 1963;200:882. doi: 10.1038/200882a0. [DOI] [PubMed] [Google Scholar]
- 28.Walker J Q, Wolf C J. Anal Chem. 1968;40:711–714. [Google Scholar]
- 29.Fanter D L, Walker J Q, Wolf C J. Anal Chem. 1968;40:2168–2175. [Google Scholar]
- 30.Levy E J, Paul D J. J Gas Chromatogr. 1967;5:136–145. [Google Scholar]
- 31.Kossiakoff A, Rice F O. J Am Chem Soc. 1943;65:590–595. [Google Scholar]
- 32.Fabuss B M, Smith J O, Satterfield C N. In: Advances in Petroleum Chemistry and Refining. McKetta J J, editor. Vol. 9. New York: Wiley; 1964. pp. 157–201. [Google Scholar]
- 33.Schomberg G, Hubinger E, Husmann H, Weete F. Chromatographia. 1982;16:228–232. [Google Scholar]