

Abstract
The real-time observation of protein dynamics in living cells and organisms is of fundamental importance for understanding biological processes. Most approaches to labeling proteins exploit noncovalent interactions, unsuitable to long-term studies, or genetic fusion to naturally occurring fluorescent proteins that often have unsatisfactory optical properties. Here we used the fungal enzyme cutinase and its suicide substrate p-nitrophenyl phosphonate to covalently attach a variety of labels to the integrin lymphocyte function-associated antigen-1 (LFA-1) on the surface of living cells. Cutinase was embedded in the extracellular domain of LFA-1 with no appreciable influence on integrin function and conformational regulation. p-nitrophenyl phosphonate-conjugated fluorochromes, including the very bright and stable quantum dots, bound efficiently and specifically to LFA-1/cutinase. The availability of a genetically encoded tag that binds covalently to quantum dots could foster the development of new experimental strategies for the study of protein dynamics in vivo.
Keywords: imaging, integrin, leukocyte adhesion, protein engineering
Proteins can be labeled with fluorochromes through various techniques (1). Immunolabeling with fluorescent mAbs is a widely used approach but can significantly interfere with protein function, because of the mAb size (≥150 kDa) and multivalency, which cause cross-linking and oligomerization of their target. In vivo, the constant portion of Ig chains is detected by complement and Fc receptors and triggers a range of physiological and immunological responses in the host (2). Alternatively, monovalent Fab fragments with no constant portion are used, but these interactions typically have off-rates (koff) of 10−4-10−3·s−1 (3, 4). This translates in half-lives of minutes to few hours (half-life = ln2/koff), restricting the use of Fab fragments to short-term imaging of acute processes. Fusion of genes of interest with naturally fluorescent proteins, such as GFP, is also a popular approach (5), but typically the brightness and resistance to photobleaching of these fluorochromes are inferior to what can be achieved with nonbiological molecules. In addition, most naturally fluorescent proteins have a tendency to oligomerize, unless careful protein engineering is undertaken (6, 7). Finally, once a fusion protein is generated, expressed, and characterized, the experimental strategy is locked, and the optical properties of the fluorochrome can be changed only with a new fusion construct.
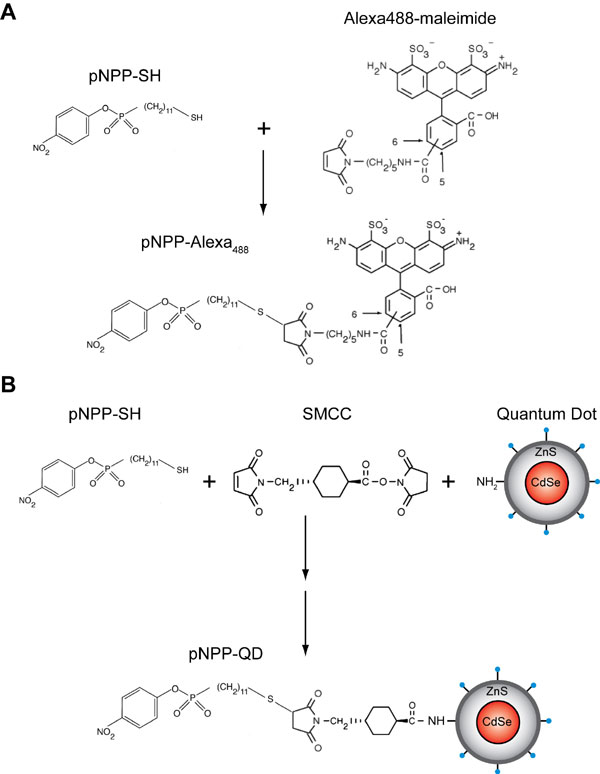
To overcome these limitations, we used cutinase, a small globular serine esterase of ≈22 kDa. The N and C termini of cutinase are close to each other (28.2 Å) and opposed to the active site (Fig. 1A), thus offering the possibility to insert the enzyme into a target protein without affecting its activity. The cutinase suicide inhibitor, p-nitrophenyl phosphonate (pNPP), can easily be synthesized with an alkyl chain carrying a reactive sulfhydryl group at its end (pNPP-SH; Fig. 1A), which in turn allows for conjugation reactions with functionalized fluorochromes, including maleimide-activated quantum dots (QDs). pNPP reacts with the active-site serine of cutinase to generate a covalent adduct, in which the alkyl chain protrudes from the catalytic pocket (8); therefore, conjugation of relatively big molecules to pNPP should not interfere with binding to cutinase.
Fig. 1.

Insertion of cutinase into integrin αL. (A) Tertiary structure of cutinase (Protein Data Bank ID code 1AGY) and 3D model of pNPP-SH. The N and C terminus and the catalytic Ser-120 are indicated. To emphasize the atomic structure, pNPP-SH is not drawn to scale. Black bars in the bottom left and right corners correspond to 5 Å in the scale used for cutinase and pNPP, respectively. (B) Model of LFA-1/cutinase in its bent (Left) and extended (Right) conformation. The structure of bent LFA-1, composed of αL (green) and β2 (cyan) chains, was obtained by homology modeling based on the crystal structure of αVβ3 (12, 13); the extended conformation was modeled as in ref. 20. The structures for cutinase and linker peptides were inserted by molecular modeling. (C) Homology model of the integrin αL β-propeller domain; the loops used for the insertion of cutinase in constructs W2, W3, and W4 are in red.
As a proof-of-concept target, we selected integrin LFA-1, an adhesion molecule of central importance to leukocyte function. The dynamic redistribution of LFA-1 during cell migration and cell–cell adhesion is a topic under intense investigation. In addition, because integrins need to undergo dramatic conformational changes, we reasoned that LFA-1 would provide a particularly stringent test for the feasibility of our labeling approach.
Results
Integrin Structure and Fusion Strategy.
LFA-1 is a heterodimer composed of a unique integrin αL associated with a β2 chain, also present in three other leukocyte integrins. Activatory stimuli induce the separation of the cytoplasmic domains of the two subunits (9); this conformational change travels across the membrane to the extracellular domain, which switches from the bent (inactive) to the extended (active) conformation (10) (Fig. 1B). The ligand-binding site also undergoes allosteric modifications, resulting in 10,000-fold increased affinity for physiological ligands, such as intercellular adhesion molecule-1 (ICAM-1) (11).
Our design for the fusion of cutinase with αL aimed to maintain proper integrin structure, function, and conformational regulation. Using structural models [based on the x-ray structure of αVβ3 (12, 13)] of LFA-1 in its inactive and active states, we determined that the N terminus of αL was incompatible with these criteria, because it localized in the vicinity of interacting molecular surfaces critical for integrin function. We searched for suitable loops connecting different regions of the ectodomain; among these, the loops connecting strands 3 and 4 on the “bottom” face of blades 2, 3, and 4 (W2, W3, and W4) of the αL β-propeller seemed ideal candidates (Fig. 1C). This surface is predicted to face away from the β2 subunit in both bent and extended conformations, as determined by structure-based sequence alignment and homology modeling (Fig. 1B). Thus, cutinase was inserted into each of these loops of the mouse αL chain, along with two short linker peptides to account for the 28.2-Å separation between the N and C termini (14) (Fig. 1B).
Synthesis and in Vitro Function of LFA-1/Cutinase.
To characterize the fusion proteins, 293T cells were cotransfected with β2-IRES-GFP and W2, W3, W4, empty vector or WT αL. Forty-eight hours after transfection, the expression of αL was quantified by flow cytometry (Fig. 2A). All three fusion constructs showed surface levels comparable to WT, suggesting that the folding and the heterodimerization of mutant αL chains were not significantly affected.
Fig. 2.

Expression and function of mouse LFA-1/cutinase. (A) 293T cells were transfected with β2-IRES-GFP and W2, W3, W4, empty vector (NT) or WT αL. Forty-eight hours later, the cells were harvested, and the expression of LFA-1 was quantified by flow cytometry. The mean fluorescence intensity (MFI) of LFA-1 after gating on GFP+ (i.e., β2+) cells is shown. n = 3. (B) Forty-eight hours after transfection, 293T cells were incubated with fluorescent multivalent ICAM-1/hFc. Data are shown as the ratio of ICAM-1-binding cells in activating (2 mM Mn2+) vs. basal (1 mM Ca2+ and 1 mM Mg2+) conditions. n = 3 transfections. (C) Representative flow cytometry plots from an ICAM-1-binding experiment. (D) Immunoprecipitation of mouse αL from surface-biotinylated (Upper) or pNPP-biotin-treated (Lower) K562 cells, either nontransfected (NT) or transfected with β2 plus W3, W4, or WT αL. The expected positions in the gel of β2, αL, and αL + cutinase (αL′) are indicated on the right (arrows). Molecular mass markers are indicated on the left (in kilodaltons); the ratio of β2 over αL signal is indicated at the bottom of each lane. (E and F) Parental K562 (Ctrl) or K562 transfected with β2 plus W3, W4, or WT αL were deposited on immobilized ICAM-1 in basal (E) or activating (F) conditions. Data are shown as the increase in specific (on immobilized ICAM-1) vs. aspecific (on immobilized BSA) adhesion. Data are representative of two experiments, each in triplicate.
Next, we sought to determine whether the inserted domain affected the conformational regulation of LFA-1/cutinase. To this end, we performed binding assays using soluble multivalent ICAM-1 ligand. We incubated transient 293T transfectants in basal (1 mM Ca2+ and 1 mM Mg2+) or activating (2 mM Mn2+) conditions (15) and quantified the increase in binding to ICAM-1 by flow cytometry (Fig. 2 B and C). Based on this functional assay, fusion proteins W2 and W4 were indistinguishable from WT, suggesting that the equilibrium between low- and high-affinity conformation of these mutants was unaffected by the presence of cutinase. In contrast, the Mn2+-dependent activation of W3 was perturbed, although the difference from WT was not statistically significant.
Given that in vivo LFA-1/ICAM-1 interactions mediate cellular adhesion, which is not an interaction with a soluble ligand, we further tested our constructs through adhesion assays using an ICAM-1-coated substrate (16). To this end, we generated K562 stable transfectants, selected for high expression of β2 and either W3, W4, or WT αL. The clone of K562 cells expressing W2 that we initially selected could not be propagated and was not included in this set of experiments.
Surface expression of LFA-1/cutinase was assessed by immunoprecipitating lysates from surface-biotinylated nontransfected K562 cells, W3, W4, or WT αL transfectants with an anti-αL mAb. Western blots with streptavidin–HRP revealed bands corresponding to the Mr of integrin β2 in WT, W3, and W4 transfectants but not in nontransfected controls (Fig. 2D). A second band, at the approximate Mr of mouse αLwas shifted in W3 and W4 transfectants by an amount corresponding to the added cutinase domain (Fig. 2D). W4 αL coprecipitated with the β2 chain better than W3 and with the same efficiency as the WT αL chain (Fig. 2D). When pNPP-biotin was used as a surface-biotinylation reagent, only bands corresponding to the cutinase-containing (W3 and W4), but not WT αL, were visible, confirming the specificity of pNPP probes for cutinase (Fig. 2D).
Next, we measured the ability of K562 transfectants to resist detachment from plastic-bound ICAM-1/hFc after centrifugation. No significant adhesion was measured in basal condition (Fig. 2E), but incubation with Mn2+ stimulated and strengthened the attachment of K562 to ICAM-1/hFc (Fig. 2F). W3 and W4 fusion proteins mediated adhesion to an extent comparable with WT αL. As predicted from its small size, binding of a pNPP derivative to the active site of cutinase did not alter these adhesion properties (data not shown). Based on these data, we chose the W4 construct for further characterization.
Conjugation of pNPP to Fluorochromes.
The -SH terminus in pNPP-SH permits coupling reactions with maleimide-based reagents, commonly used for protein conjugation. The double bond of maleimide undergoes alkylation by reacting with -SH groups to form stable thioether bonds (17). After the strategy shown in supporting information (SI) Fig. 5A, we synthesized pNPP-Alexa488, -biotin, and -Alexa633. Next, we incubated K562 cells stably expressing mouse β2 and W4 or WT αL with 1 μM pNPP-Alexa for 30 min. Although comparable levels of LFA-1 were detected by mAb staining, only cells expressing LFA-1/cutinase bound pNPP-Alexa488 (Fig. 3A). Similar results were obtained with pNPP-Alexa633 (Fig. 3B). No aspecific staining of WT-expressing cells by pNPP was observed. These observations were confirmed at a population level by flow cytometry analysis, in which K562 cells expressing W3 or W4, but not WT αL, bound pNPP-Alexa488 in direct correlation with LFA-1/cutinase expression levels (SI Fig. 6).
Fig. 3.

Specific binding of pNPP conjugates to LFA-1/cutinase. (A) K562 cells stably transfected with mouse β2 and W4 or WT αL were incubated with a phycoerythrin-conjugated anti-LFA-1 mAb and 1 μM pNPP-Alexa488 for 30 min at room temperature. (B) As in A, but the cells were incubated with 1 μM pNPP-Alexa633 for 30 min at room temperature and counterstained with FITC-conjugated anti-LFA-1 mAb. (C and D) K562 transfectants were incubated with FITC-conjugated anti-LFA-1 and 200 nM pNPP-QD655 (C) or EG-QD655 for 90 min (D) at room temperature. (Scale bars, 10 μm.) Data are representative of two or more independent experiments per group.
We followed a similar approach to couple QDs to pNPP (SI Fig. 5B). Because maleimide-QDs are not commercially available, we functionalized amino-QDs using the cross-linker succinimidyl-4-(N-maleimidomethyl) cyclohexana-1-carboxylate. Next, we incubated the maleimide-QDs with a 1:10 mixture of pNPP-SH and ethylene glycol (EG)-SH (HS-C11-EG3-OH, henceforth referred to as EG-SH, a reagent with the same structure as pNPP-SH but no pNPP active group) to obtain stable pNPP-QD conjugates. EG-SH was added to reduce the number of pNPP moieties on the surface of pNPP-QDs (decreasing their valency) and to passivate the QD surface, reducing aspecific interactions (18). After 1 h of incubation with 200 nM pNPP-QD655, only K562 cells expressing W4 but not WT αL displayed a distinct membrane staining, indicating that pNPP-QD655 bound specifically to the cutinase domain (Fig. 3C). Importantly, QDs coated with EG alone (EG-QD655) stained the W4 K562 transfectants only marginally above background (Fig. 3D).
Dynamic Imaging of LFA-1/Cutinase on Live Cells.
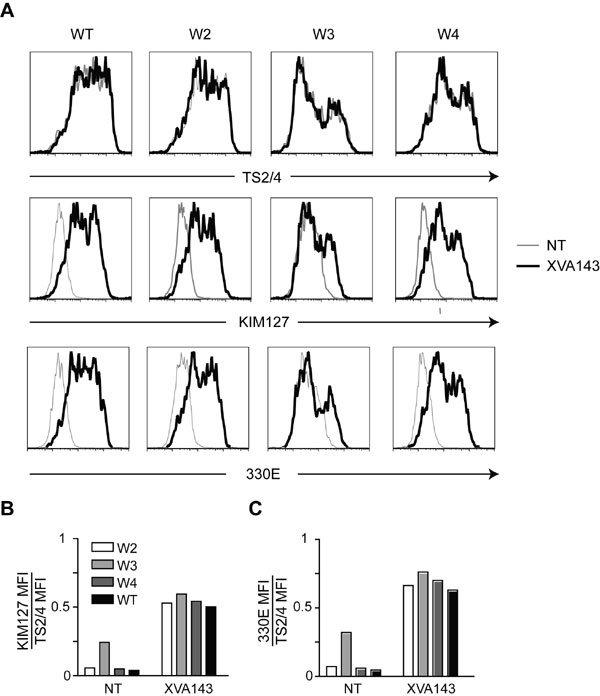
To validate our labeling strategy for use in dynamic imaging of live cells, we visualized the migration of LFA-1/cutinase expressing cells in an in vitro system that takes advantage of the motility of BAF cells, a murine B cell line (19). In contrast to K562 cells, BAF cells retain expression of the endogenous copy of (mouse) LFA-1; this compelled us to generate and use human LFA-1/cutinase constructs. To verify that the insertion of cutinase in the W4 loop of human LFA-1 was as innocuous as in the mouse construct, we used mAbs that bind differentially to the bent and extended conformation of human LFA-1 (10). These activation-sensitive antibodies recognize epitopes buried in the intersubunit contact surface, when LFA-1 is inactive (bent), but exposed and available in the extended conformation. Consistent with the mouse data, human W2 and W4 constructs exposed the activation epitopes tested as efficiently as WT LFA-1 (SI Fig. 7), after incubation with an allosteric antagonist (XVA143), known to induce binding to activation-dependent mAbs (20). The human W3 construct displayed an altered profile of binding to the activation-sensitive mAbs, confirming that the introduction of cutinase in this loop perturbs LFA-1 regulation.
To image the dynamic redistribution of LFA-1 during BAF locomotion, we coated glass slides with human ICAM-1 (for adhesion) and SDF-1 (for chemokinesis) and imaged pNPP-Alexa488-labeled BAF cells expressing either human WT LFA-1 or W4 (Fig. 4A; SI Movie 1). LFA-1 dynamics were observed by following the signal from pNPP-Alexa488 using live cell fluorescence imaging. W4-expressing BAF cells spread, polarized, and migrated on ICAM-1 (SI Movie 2). Neither the cutinase insert nor the fluorescent tag interfered with LFA-1 function, because spreading and motility of BAF cells expressing WT LFA-1 were comparable (SI Movie 1). Consistent with the observation that mouse LFA-1 does not bind to human ICAM-1 (21), the parental BAF cell line did not attach to or migrate on human ICAM-1 substrate (data not shown); therefore, transgenic human LFA-1 and LFA-1/cutinase were entirely responsible for the migratory behavior of BAF transfectants. To obtain a more quantitative picture of LFA-1 distribution during cell locomotion, we compared the signal density on migrating and static cells. Although an accumulation of LFA-1-derived signal was clearly noticeable in the uropod of translocating BAF cells (Fig. 4B), the distribution of the integrin on static cells was homogenous and nonpolarized (Fig. 4C). These results indicate that pNPP labeling can be used to obtain quantitative information about protein distribution.
Fig. 4.

In vitro imaging of LFA-1/cutinase-mediated migration. (A) Human W4-expressing BAF cells were incubated with 1 μM pNPP-Alexa488 at room temperature for 20 min and then washed. The cells were allowed to settle on a glass slide kept at 37°C and coated with human ICAM-1 and SDF-1. A time-lapse series at 45-s intervals is shown (left to right, top to bottom). (Scale bar, 10 μm.) (B and C) Pseudocoloring and quantitative analysis of pNPP-Alexa488 signal on the membrane of translocating (B) or static (C) W4-expressing BAF cells. The intensity of pNPP-Alexa488 is diplayed in the histogram plot to the right of each image. The numbering on the microphotograph corresponds to the numbering on the x axis of the histogram. The red rectangle was used to calculate the background pixel intensity (*). The rainbow bar in the top right corner of the micrographs indicates the color code from low (blue) to high (red) signal intensity. Data are representative of two experiments.
Discussion
The data presented above establish the cutinase/pNPP system as an attractive approach to covalently attach molecular labels to proteins of interest. Other strategies that combine genetic tags with exogenous labels have been described (22, 23). For example, fluorogenic biarsenical (FlAsH) compounds bind noncovalently to a 6-aa tag that can be inserted in helical sequences (24). Unfortunately, FlAsH reagents have some affinity to isolated thiols and therefore display significant background staining (25). A fluorescein derivative complexed with Zn2+ ions has also been used to label surface proteins containing hexahistidine tags (26) but requires elaborate chemical synthesis and lacks the flexibility of the pNPP-SH reagent. Yet another technique exploits the catalytic properties of human O6-alkylguanine transferase (hAGT), which binds covalently to benzylguanine (BG) derivatives (23, 27). Background issues also frustrate this approach, because AGT is normally found in mammalian cells. Mutants of hAGT have been developed that react selectively with BG probes (28), but the presence of the endogenous enzyme remains problematic and negatively influences the signal-to-noise ratio. Finally, as part of its metabolic cycle, hAGT is degraded after self-alkylation, resulting in signal attenuation over time (29).
Compared with existing methods, we believe the cutinase/pNPP system offers the following advantages: (i) Background staining in mammalian cells is negligible, most likely because no protein with significant homology to cutinase is found in the mammalian proteome; (ii) the cutinase termini are opposing the active site, thus allowing for the insertion of the enzyme in any available termini or loop of target proteins while retaining pNPP binding activity; (iii) the pNPP substrate is easily synthesized, relatively stable in water (30), and its alkyl-thio derivative (pNPP-SH) can be conjugated to commercially available maleimide-based reagents, using standard equipment found in most biology labs; (iv) the chemistry used for coupling pNPP to small organic molecules can easily be adapted to QDs. Notably, QDs have large multiphoton cross-sectional efficiency, with a multiphoton fluorescence 100–1,000 times more intense than that of organic fluorochromes (31), and they exhibit virtually no photobleaching. This makes pNPP-QD an ideal probe for high-resolution imaging with low phototoxicity in deep tissues of living organisms. Moreover, the described method is easily transferable to other nanoparticles, for example metal colloidal particles that may be used in other imaging techniques.
In conclusion, we propose the cutinase/pNPP technology as a flexible strategy to covalently label specific proteins in living cells. We predict that, accompanied by progress in QD and organic fluorochrome chemistry, this approach will provide a useful noninvasive tool for studying protein dynamics in vitro and in vivo.
Materials and Methods
αL-Cutinase Fusion Constructs.
The coding sequence of cutinase from Leu-15 to Ala-214 was amplified with primers that contained sequences coding for the peptidic linkers: GSGGGGSG for the sense primer (5′-CGGGATCCGGCGGAGGCGGCTCCGGCCTTGGTAGAACAACTCGC-3′); and GSGGGGTG for the antisense primer (5′-AATACCGGTCCCGCCTCCCCCGGAGCCAGCAGAACCACGGACAG-3′). N- and C-terminal portions of mouse integrin αL were amplified from C57BL/6 splenocytes cDNA. For the N-terminal fragments, we used a common sense primer containing an XbaI site: 5′-GCTCTAGACGCAGATGAGTTTCCG-3′. The antisense primers contained a BamHI site: W2 5′-TGGGATCCACTCTGGGGGAAGAGG-3′; W3 5′-TGGGATCCCAGGTCCAGAAAGCC-3′; W4 5′-TGGGATCCAGCCTCT G G G GCTTG-3′. For C-terminal fragments, we used a common antisense primer, starting 2 bp downstream from the stop codon and containing an XmaI restriction site: C-terminal antisense 5′-CCCCCCGGGCCTTAGTCCTTGTCAC-3′. The sense primers contained an AgeI site: W2 5′-ATCACCGGTCTGGAGGGACCTATG-3′; W3 5′-ATCACCGGTCGTGAAGACCTGCAGG-3′; and W4 5′-ATCACCGGTGGAGGACGTTGGAACC-3′. The insertion sites for the cutinase domain are: between Ser-109 and Leu-110 for W2, between Leu-359 and Gln-360 for W3, and between Ala-416 and Gly-417 for W4. WT αL was amplified by using the common sense and antisense primers. The fragments were cloned into pBluescript with a customized multicloning site and verified by sequencing. We found a conservative Val→Ala at residue 593, in the thigh domain of the W2 construct; a Met→Val at residue 813, in the calf-1 domain of the W4 construct; and a full triplet insertion, coding for Val at position 719, in a loop of the thigh domain of the W3 construct. The mutations were likely introduced during the amplification step, but considering their location and their conservative nature, we did not predict significant structural or functional consequences.
To generate the N terminus of human αL/cutinase, we used: sense primer 5′-NNNTCTAGAGTGCTGGAAGGATGAAGG-3′; W2 antisense 5′-NNNGGATCCATTCTGGCGGAAGAG-3′; W3 antisense 5′-NNNGGATCCCAGGTCTGCCTTCAGG-3′; W4 antisense 5′-NNNGGATCCGCCCTGTGGCTCTTGG-3′. For the C terminus: antisense primer 5′-NNNNCCCGGGAGTGCAGGGGAGTGTGC-3′; W2 sense 5′-NNNNACCGGTCTGCAGGGTCCCATGC-3′; W3 sense 5′-NNNNACCGGTCAGGATGACACATTTATTGG-3′; W4 sense 5′-NNNNACCGGTGGAGGACACTGGAGCCAGGTCC-3′. The insertion sites are: between Asn-110 and Leu-111 for W2, between Leu-359 and Gln-360 for W3, and between Gly-416 and Gly-417 for W4.
Plasmids.
For expression in 293T, K562, and BAF cells, αL and αL/cutinase sequences were cloned into the XhoI site of pcDNA3.1 (+) (Invitrogen, Carlsbad, CA), and β2 sequences were cloned into pIRES2-eGFP (Clontech, Mountain View, CA) (for 293T cells) or a truncated version in which IRES-eGFP sequences had been removed (for K562 and BAF cells).
Antibodies and Reagents.
Mouse anti-human LFA-1 mAbs TS2/4 have been described (32), KIM127 was kindly provided by M. Robinson (Celltech, Slough, U.K.), and 330E was a gift from D. Staunton (ICOS, Bothel, WA). The LFA-1 inhibitor, XVA143, was a gift from M. Shimaoka (20). FITC- and phycoerythrin-conjugated anti-LFA-1 mAbs (2D7) for immunofluorescence and purified anti-LFA-1 M17/4 for immunoprecipitation were purchased from BD Biosciences (San Jose, CA).
Cell Lines.
293T cells were grown in DMEM/10% FCS with l-Gln, antibiotics, and nonessential amino acids. Transient transfections were carried out with TransIT-293 (Mirus Bio, Madison, WI). K562 cells were grown in RPMI medium 1640/10% FCS with l-Gln and antibiotics. Stable transfectants were obtained by electroporation and selection in 1 mg/ml G418. Clones were obtained by single-cell sorting, followed by expansion in selective medium. BAF, a pro-B-cell line (19), was propagated in RPMI medium 1640/10% FCS, 50 μM 2-mercaptoethanol/10% WEHI-3 conditioned medium, as a source of IL-3. BAF transfectants were generated and maintained as for K562 cells.
Binding to Soluble Multimeric ICAM-1.
293T transfectants were washed once with HBSS/10 mM EDTA and twice with Hepes-buffered saline (HBS). Cells (2 × 105) were resuspended in 50 μl of HBS containing 5 μg/ml murine ICAM-1/hFc fusion protein (R&D Systems, Minneapolis, MN) pre-cross-linked with a 16:1 wt/wt excess Cy3-labeled anti-human polyclonal antibody (Jackson ImmunoResearch, West Grove, PA) and 1 mM Ca2+Mg2+ or 2 mM Mn2+. After 30 min of incubation at room temperature, the cells were washed twice with ice-cold HBS and analyzed by flow cytometry.
Immunoprecipitation.
K562 transfectants (15 × 106) were surface-biotinylated with either 0.5 mg/ml EZlink-sulfo-NHS-biotin (Sigma, St. Louis, MO) or 1 μM pNPP-biotin and then lysed with 1% Nonidet P-40. Two milligrams of cell lysate was immunoprecipitated with 20 μg of anti-LFA-1 mAb (clone M17/4) and protein G-conjugated beads (Roche, Indianapolis, IN). The samples were run on a denaturing gel and transferred to a nitrocellulose membrane. Immunoprecipitated proteins tagged with biotin were revealed with streptavidin-HRP (Sigma). The signal ratio was calculated with ImageJ, Ver. 1.37 (National Institutes of Health, Bethesda, MD).
Binding to Immobilized ICAM-1.
Adhesion of K562 to immobilized ICAM-1 was measured as described (16). Cells were labeled with 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein, acetoxymethyl ester (BCECF; Molecular Probes, Eugene, OR), washed, and resuspended in HBSS/2.5% FCS with 1 mM Ca2+Mg2+ or 2 mM Mn2+. One hundred microliters of the cell suspension was transferred to 96-well V-bottom plates, precoated at 4°C with 10 μg/ml murine ICAM-1/hFc (R&D Systems) or BSA in Tris-buffered saline, pH 9. The plates were incubated for 30 min at 37°C followed by centrifugation at 100 × g. Nonadherent cells accumulated at the bottom of the well and were quantified with a fluorescence plate reader.
Synthesis of pNPP Conjugates.
pNPP-(CH2)11-SH (pNPP-SH) and the inactive OH-EG3-(CH2)11-SH (EG-SH) were synthesized as described (30). For synthesis of pNPP-(CH2)11-EG2-biotin (pNPP-biotin) and pNPP-(CH2)11-Alexa488 (pNPP-Alexa488), a thiol-reactive reagent, either biotin-(OCH2CH2)2-maleimide (Pierce, Rockford, IL) or Alexa488-C5-maleimide (Molecular Probes), was reacted with a 5× molar excess of pNPP-(CH2)11-SH in ethanol at room temperature, overnight and under nitrogen. The final compound was purified by using HPLC with a combination of acetonitrile and H2O (0.1% trifluoroacetic acid) as mobile phase.
To couple QDs to pNPP-SH, 0.8 nmol of amino(PEG)-QD655 (Invitrogen) were activated with 1 mM sulfo–succinimidyl-4-(N-maleimidomethyl) cyclohexana-1-carboxylate (SMCC) for 1 h at room temperature; free SMCC was washed away by filtration on a 50,000 Mr cutoff membrane. To control the number of pNPP groups and to passivate the QD surface (18), we added to maleimide-QD655 10:1 EG-SH:pNPP-SH at a final thiol concentration of 100 μM. After 1 h of incubation at room temperature, the reaction was quenched with 100 μM 2-mercaptoethanol. Unreacted pNPP-SH and EG-SH were removed by filtration as above, and pNPP-QD655 were concentrated to 2 μM.
Confocal Imaging.
K562 transfectants were stained with FITC- or phycoerythrin-labeled anti-LFA-1 mAbs, 1 μM pNPP-Alexa488, 1 μM pNPP-Alexa633, 200 nM pNPP-QD655, or 200 nM EG-QD655. Cells were incubated at room temperature for 30 min (Alexa) or 90 min (QDs), washed once, fixed with formaldehyde, and analyzed on a confocal microscope (Radiance 2000; Bio-Rad, Hercules, CA).
In Vitro Live Cell Imaging.
BAF stable transfectants were labeled with 1 μM pNPP-Alexa488 for 20 min at room temperature. Cells were then allowed to settle on the surface of a 37°C ΔT Bioptechs chamber, coated with human ICAM-1/SDF-1. Live cell imaging was conducted on an Axiovert S200 epifluorescence microscope (Zeiss MicroImaging, Thornwood, NY). Pseudocoloring was performed with a custom script in Photoshop (Adobe, San Jose, CA), and quantification of pNPP signal was performed with ImageJ.
Statistical Analysis.
Data are shown as mean ± SEM. Statistical significance was assessed by two-tailed unpaired Student's t test when comparing two groups or by one-way ANOVA followed by Student–Newman–Keuls (SNK) posttest, for more than two groups. Differences were considered significant when P < 0.05.
Supplementary Material
Acknowledgments
We thank Antonio Peixoto for help with biochemistry, Can Xie (CBR Institute, Boston, MA) for donating the LFA-1 models, and Harry Leung for confocal imaging advice. R.B. was supported in part by the Armenise–Harvard foundation. K.R.L. was supported by National Institutes of Health Fellowship AI063854. C.V.C. was supported by a grant from the Arthritis Foundation.
Abbreviations
- ICAM
intercellular adhesion molecule
- LFA
lymphocyte function-associated antigen
- pNPP
p-nitrophenyl phosphonate
- QD
quantum dot
- EG
ethylene glycol.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0705201104/DC1.
References
- 1.Giepmans BN, Adams SR, Ellisman MH, Tsien RY. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 2.Ravetch JV, Clynes RA. Annu Rev Immunol. 1998;16:421–432. doi: 10.1146/annurev.immunol.16.1.421. [DOI] [PubMed] [Google Scholar]
- 3.Neri D, Carnemolla B, Nissim A, Leprini A, Querze G, Balza E, Pini A, Tarli L, Halin C, Neri P, et al. Nat Biotechnol. 1997;15:1271–1275. doi: 10.1038/nbt1197-1271. [DOI] [PubMed] [Google Scholar]
- 4.Rader C, Ritter G, Nathan S, Elia M, Gout I, Jungbluth AA, Cohen LS, Welt S, Old LJ, Barbas CF., III J Biol Chem. 2000;275:13668–13676. doi: 10.1074/jbc.275.18.13668. [DOI] [PubMed] [Google Scholar]
- 5.Tsien RY. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 6.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 7.Shaner NC, Steinbach PA, Tsien RY. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 8.Longhi S, Nicolas A, Creveld L, Egmond M, Verrips CT, de Vlieg J, Martinez C, Cambillau C. Proteins. 1996;26:442–458. doi: 10.1002/(SICI)1097-0134(199612)26:4<442::AID-PROT5>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 9.Kim M, Carman CV, Springer TA. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 10.Luo BH, Carman CV, Springer TA. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimaoka M, Xiao T, Liu JH, Yang Y, Dong Y, Jun CD, McCormack A, Zhang R, Joachimiak A, Takagi J, et al. Cell. 2003;112:99–111. doi: 10.1016/s0092-8674(02)01257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, Joachimiak A, Goodman SL, Arnaout MA. Science. 2001;294:339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 14.Hughes TE, Zhang H, Logothetis DE, Berlot CH. J Biol Chem. 2001;276:4227–4235. doi: 10.1074/jbc.M007608200. [DOI] [PubMed] [Google Scholar]
- 15.Dransfield I, Cabañas C, Craig A, Hogg N. J Cell Biol. 1992;116:219–226. doi: 10.1083/jcb.116.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weetall M, Hugo R, Friedman C, Maida S, West S, Wattanasin S, Bouhel R, Weitz-Schmidt G, Lake P. Anal Biochem. 2001;293:277–287. doi: 10.1006/abio.2001.5140. [DOI] [PubMed] [Google Scholar]
- 17.Hermanson GT. Bioconjugate Techniques. San Diego: Academic; 1996. p. 148. [Google Scholar]
- 18.Zheng M, Li Z, Huang X. Langmuir. 2004;20:4226–4235. doi: 10.1021/la035981i. [DOI] [PubMed] [Google Scholar]
- 19.Palacios R, Steinmetz M. Cell. 1985;41:727–734. doi: 10.1016/s0092-8674(85)80053-2. [DOI] [PubMed] [Google Scholar]
- 20.Shimaoka M, Salas A, Yang W, Weitz-Schmidt G, Springer TA. Immunity. 2003;19:391–402. doi: 10.1016/s1074-7613(03)00238-3. [DOI] [PubMed] [Google Scholar]
- 21.Johnston SC, Dustin ML, Hibbs ML, Springer TA. J Immunol. 1990;145:1181–1187. [PubMed] [Google Scholar]
- 22.Chen I, Ting AY. Curr Opin Biotechnol. 2005;16:35–40. doi: 10.1016/j.copbio.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Gronemeyer T, Godin G, Johnsson K. Curr Opin Biotechnol. 2005;16:453–458. doi: 10.1016/j.copbio.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 24.Griffin BA, Adams SR, Jones J, Tsien RY. Methods Enzymol. 2000;327:565–578. doi: 10.1016/s0076-6879(00)27302-3. [DOI] [PubMed] [Google Scholar]
- 25.Stroffekova K, Proenza C, Beam KG. Pflügers Arch. 2001;442:859–866. doi: 10.1007/s004240100619. [DOI] [PubMed] [Google Scholar]
- 26.Hauser CT, Tsien RY. Proc Natl Acad Sci USA. 2007;104:3693–3697. doi: 10.1073/pnas.0611713104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. Nat Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 28.Juillerat A, Gronemeyer T, Keppler A, Gendreizig S, Pick H, Vogel H, Johnsson K. Chem Biol. 2003;10:313–317. doi: 10.1016/s1074-5521(03)00068-1. [DOI] [PubMed] [Google Scholar]
- 29.Keppler A, Kindermann M, Gendreizig S, Pick H, Vogel H, Johnsson K. Methods. 2004;32:437–444. doi: 10.1016/j.ymeth.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Hodneland CD, Lee YS, Min DH, Mrksich M. Proc Natl Acad Sci USA. 2002;99:5048–5052. doi: 10.1073/pnas.072685299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larson DR, Zipfel WR, Williams RM, Clark SW, Bruchez MP, Wise FW, Webb WW. Science. 2003;300:1434–1436. doi: 10.1126/science.1083780. [DOI] [PubMed] [Google Scholar]
- 32.Sanchez-Madrid F, Krensky AM, Ware CF, Robbins E, Strominger JL, Burakoff SJ, Springer TA. Proc Natl Acad Sci USA. 1982;79:7489–7493. doi: 10.1073/pnas.79.23.7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}