

Abstract
The goals of this study were to assess the extent to which bulk genomic DNA sequences contribute to their own packaging in nucleosomes and to reveal the relationship between nucleosome packaging and positioning. Using a competitive nucleosome reconstitution assay, we found that at least 95% of bulk DNA sequences have an affinity for histone octamer in nucleosomes that is similar to that of randomly synthesized DNA; they contribute little to their own packaging at the level of individual nucleosomes. An equation was developed that relates the measured free energy to the fractional occupancy of specific nucleosome positions. Evidently, the bulk of eukaryotic genomic DNA is also not evolved or constrained for significant sequence-directed nucleosome positioning at the level of individual nucleosomes. Implications for gene regulation in vivo are discussed.
Keywords: chromatin, gene regulation, transcriptional activation
DNA in nucleosomes is tightly bent in comparison to its persistence length, a length scale of DNA stiffness (1). Substantial mechanical work must be done against the bending stiffness to package DNA in nucleosomes; this mechanical work is done at the expense of the favorable free energy of histone–DNA interactions and subtracts from the net thermodynamic stability of the particles. The free energies involved are surprisingly large. Taking the persistence length of DNA to be 50 nm and assuming that DNA in a nucleosome is uniformly bent in a circular trajectory at a radius of curvature of 4.4 nm leads to an estimated free energy cost (1) for bending DNA in a nucleosome of ≈75 kcal·mol−1. Indeed, because of previous estimates such as this, it was once considered remarkable that nucleosome exist at all. Analogous problems exist for DNA twist.
The discovery that certain DNA sequences are naturally bent (2) suggested that eukaryotic genomic DNA sequences might be evolved or constrained to facilitate their packaging in nucleosomes (3, 4). Moreover, one might anticipate the existence of a relationship between the free energy of nucleosomal packaging and the phenomenon of nucleosome positioning (such an equation is developed in the present study), suggesting the possibility that nucleosome positioning signals may be encoded in genomic DNA. This idea is interesting in part because of the relationship between nucleosome positioning and gene regulation (5).
Results from several studies support the hypothesis that genomes are evolved to facilitate their own packaging. (i) Analyses of DNA fragments present in isolated nucleosome core particles, chromatosomes, and dinucleosomes reveal nonrandom periodic distributions of certain dinucleotides and longer sequences, with particular relative phases (see ref. 6 and references therein). The results suggest that such sequences facilitate the bending of DNA that is necessary for nucleosome formation and that the bending may be produced by an anisotropic flexibility of the A+T-rich and G+C-rich regions. (ii) Shrader and Crothers (7, 8) designed sequences de novo that obeyed these rules. The designed sequences were found to bind histones (and form nucleosomes) with particularly high affinity. Moreover, they confer “rotational,” but not “translational,” positioning, consistent with the proposed explanation of their high affinity. (iii) Sets of sequences that have been found by chance to be organized in preferentially positioned nucleosomes in vivo have been analyzed (9–12) and are found to have nonrandom periodic distributions of certain dinucleotides, related to (although different in detail from) those discovered in the sequences of isolated nucleosomal DNA. (iv) Finally, a recent study using Fourier transformation to analyze data emerging from the Saccharomyces cerevisiae and Caenorhabditis elegans genome sequencing projects reveals the periodicities detected in those other studies of selected sequences and shows other dinucleotides to also be nonrandomly periodic at similar wavelengths (13).
While these results collectively suggest that genomic DNA sequences do indeed participate in their own nucleosome packaging and in nucleosome positioning, they leave open the question of what fraction of the genomic DNA may have this capability and how large the energetic contribution is.
The goal of the present study was to assess the extent to which bulk genomic DNA sequences contribute to their own packaging in nucleosomes and to nucleosome positioning. We measured the relative free energies for DNA binding to histone octamer for genomic DNA fragments and for randomly synthesized DNA, using a competitive nucleosome reconstitution assay, and we developed an equation that relates the free energy measured in nucleosome reconstitution assays to the fractional occupancy of specific nucleosome positions in vitro. The implications of our results for gene regulation in vivo are discussed.
MATERIALS AND METHODS
Random DNA.
The original pool of synthetic random DNA was the generous gift of D. Bartel and J. Szostak (14) and has been extensively characterized by them. We received 2 μg of a 294-bp double-stranded DNA fragment constructed as follows: T7 promotor–L22–N72–StyI–N76–BanI–N72–R20, where the sequence of L22 is 5′-ggaacactatccgactggcacc-3′ and the sequence of R20 is 3′-ggaaccagtaatcctagggc-5′. N72 and N76 represent segments of synthetic random DNA of 72 or 76 bp; these are joined together by two 6-bp defined restriction sequences, StyI (5′-ccaagg-3′) and BanI (5′-ggcacc-3′). We used the primers L30 (5′-gggagctcggaacactatccgactggcacc-3′, which deletes the T7 promoter) and R20 (above) to amplify molecules.
We subjected 1.6 μg of the random sequence DNA to eight cycles of PCR in 50 mM Tris·HCl (pH 9.0 at 25°C), 20 mM (NH4)2SO4, 1.5 mM MgCl2, 0.2 mM each dNTP, 0.5 mM each primer, and 50 units/ml of Tfl DNA polymerase (Epicentre Technologies, Madison, WI). PCRs were pooled and purified as described (15, 16). Our final yield of DNA was 132 μg, corresponding to ≈82 copies each of 5 × 1012 different molecules.
Molecules were cloned into the HincII site of the plasmid pGEM 3Z (Promega). When desired, individual molecules were amplified from plasmid DNA either using the PCR conditions described above, followed by labeling with T4 polynucleotide kinase, or by including [α-32P]dATP in the PCRs. Labeled fragments were purified on 5% acrylamide gels.
Genomic DNA and Histones.
Bulk chicken erythrocyte chromatin having a range of lengths from ≈200 bp to 10 kbp (weight average, ≈1 kbp) was prepared as described (17), and the DNA was extracted and digested with the restriction enzyme RsaI to a weight average length of ≈400 bp. Nucleosome core particle DNA (18) and histone octamer (19) were prepared from chicken erythrocyte nuclei as described.
Analytical Reconstitutions.
20 μg of PCR-amplified random DNA (including trace amounts of radiolabeled material) plus 1.15 μg of histone octamer were mixed in 250 μl of TE (10 mM Tris/1 mM EDTA, pH 7.5) plus 2.0 M NaCl [all buffers contain 0.5 mM phenylmethylsulfonyl fluoride (PMSF) and 2 mM benzamidine]. The mixture was dialyzed for 2 hr into TE plus 2.0 M NaCl, followed by 2 hr each of dialysis into TE plus 1.5, 1.0, and 0.5 M NaCl, successively, and finally dialysis overnight into 0.5× TE. Samples were loaded onto linear 5–30% sucrose gradients (in 0.5× TE) and spun for 24 hr at 41,000 rpm at 4°C in a Beckman SW-41 rotor. Gradients were fractionated (by pumping from the bottom) into ≈0.5-ml fractions and Cerenkov-counted.
Competitive Reconstitutions.
Histone octamer (2 μg) was mixed with 30 μg of RsaI-digested chicken erythrocyte DNA or with 20 μg of chicken erythrocyte core particle DNA plus the desired radiolabeled tracer DNA in 50 μl of TE plus 2 M NaCl (all buffers contain 0.5 mM PMSF and 1 mM benzamidine). This mixture was loaded into microdialysis buttons, which were in turn placed into a dialysis bag containing the same buffer. Samples were dialyzed at 4°C into the starting buffer for >2 hr, then into two changes of 0.5 × TE for a minimum of 12 hr each. Aliquots of the reconstituted material were run on 5% acrylamide gels containing 1/3× TBE (30 mM Tris-borate/0.67 mM EDTA). Gels were dried and quantified by PhosphorImager (Molecular Dynamics). Keq values were calculated as ratios of background subtracted counts in complex to counts in free DNA. ΔG values were calculated from each equilibrium constant, and ΔΔG values were calculated from differences between pairs of ΔG values as indicated. A lower weight concentration of core particle DNA than RsaI digest was used as competitor; this choice was intended as a compromise between equal molar and weight concentrations, which cannot both be achieved simultaneously since the average lengths are different. It appears that the RsaI-digested DNA is sufficiently long that weight concentration may have been the more relevant variable controlling the association with histone octamer. In any case, the results obtained with the two different competitors at their different concentrations do not differ significantly.
Computational Analysis.
Computations on the genome of the yeast S. cerevisiae were carried out using the data set previously described (13), which deletes telomeric regions. Sequences were analyzed for each occurrence of the motif “(A or T)3NN(G or C)3NN,” where N is any base; the number of such motifs in each 146-bp window was counted, and the counts were histogrammed.
RESULTS
Competitive Reconstitution with Natural and Random DNA.
We use a modification of the competitive reconstitution assay developed by Shrader and Crothers (7, 8) to measure the relative free energies of different DNA molecules for binding to histone octamer in nucleosomes. In this approach, radiolabeled tracer DNA competes with a large molar excess of unlabeled competitor DNA for limiting amounts of histone octamer. Stepwise dialysis from 2.0 M NaCl allows the most stable population of nucleosomes to form in an equilibrium process (7, 8), eventually “freezing in” this equilibrium at low [NaCl]. This reconstitution procedure is well established and is known to yield native-like nucleosomes. The products of reconstitution are separated by native gel electrophoresis and quantified by PhosphorImager. The ratio of radiolabeled tracer DNA incorporated in nucleosomes to free tracer DNA defines an equilibrium constant and a corresponding free energy for histone binding of the tracer DNA that are valid for that specific competitive environment. Difference free energies are measured by subtraction of the free energies for differing radiolabeled tracer DNA molecules measured in parallel experiments having identical competitor DNA and competitive environments.
For the present study, the competitor is fragments of chicken erythrocyte genomic DNA prepared in either of two different ways. (i) Chicken erythrocyte nuclei were lightly digested with micrococcal nuclease to yield very long but soluble chromatin fibers; DNA was extracted and digested with a “four-base specificity” restriction enzyme, RsaI, to yield a diverse set of fragments that give an unbiased representation of the entire genome. (ii) Chicken erythrocyte nuclei were extensively digested with micrococcal nuclease to yield nucleosome core particles, and their DNA was isolated. The core particle DNA potentially gives an unbiased representation of the genome but also might not (e.g., it is feasible that there is a physical selection for especially stable nucleosomes, during the preparative digestion down to core particles); importantly, our results revealed little difference between these two DNA samples.
The radiolabeled tracer DNAs used were: (i) the same core particle DNA used as one of the competitors; (ii) a pool of 5 × 1012 different chemically random sequences; and (iii) two individual cloned isolates of the chemically random DNA.
Each synthetic random DNA molecule is 282 bp long and includes 220 bp of random sequence DNA arranged in three blocks joined together with two 6-bp nonrandom sequences (see Materials and Methods). Two stretches of nonrandom DNA flank each end of the constructs for use in PCR amplification. An assumption inherent in this study is that these defined sequence elements should not contribute significantly to the analysis. This assumption is consistent with available information, and we show below that this is the case experimentally. These elements contain no particular unusual sequences; they are simply particular instances of arbitrary sequence. They do not contain signals previously identified as related to nucleosome packaging (see above), nor do they contain the signals that we identified in an extensive SELEX (20) analysis of DNA determinants of nucleosome stability that we have completed (unpublished work). If by chance the defined sequence elements disfavored nucleosome formation, then that is no problem because the synthetic random sequence segments are of sufficient length that the two defined sequence ends and either one of the two restriction sequence joints can be entirely excluded from the nucleosome.
We used sucrose gradient ultracentrifugation to characterize the products of reconstitution reactions in which only the synthetic random DNA is present and the histone octamer is supplied in 0.1 mol per mol of DNA. The results of such an experiment are shown in Fig. 1. The sucrose gradients reveal that ≈10% of the DNA is formed into nucleosomes, as expected, and the mobility of the nucleosomes in the gradients is consistent with their being mononucleosomes, not dinucleosomes, which sediment at or near the bottom (21). Moreover, the amount of histone octamer supplied in the reaction is sufficient to turn 10% of the DNA into only mononucleosomes, not into dinucleosomes. In other studies (data not shown; unpublished work), we found that the reconstituted nucleosomes protect ≈146 bp against digestion by micrococcal nuclease, further arguing against the possible formation of closely packed dimers. We conclude that the reconstitution procedure yields mononucleosomes, as expected.
Figure 1.

Sucrose gradient analysis. Typical sucrose gradient profile of random sequence DNA selected with 0.1 mol of histone octamer per mol of DNA. The mobilities of mononucleosomes and of free DNA are indicated. The long length of the random DNA (282 bp) reduces the separation between the mononucleosomes and naked DNA compared with our earlier work with shorter reconstituted nucleosomes (15, 16). Sedimentation was from right to left.
To measure the free energies and difference free energies of reconstitution, we prepare reconstitution reactions containing competitor DNA in ≈10-fold mass excess over histone octamer, which in turn is present in large excess over the radiolabeled tracer DNA; importantly, the competitive environment is kept identical between samples within each experiment and between experiments.
The results of such a competitive reconstitution reaction analyzed by native gel electrophoresis and PhosphorImager are shown in Fig. 2. When the radiolabeled tracer is the same DNA as the competitor (lane 5), ≈10% of the DNA is incorporated into nucleosomes, as expected from the histone:DNA stoichiometry. Similar results are obtained when RsaI-digested genomic DNA is used as competitor (lane 1). This demonstrates that there is no significant difference in free energy for these two very different preparations of genomic DNA. (We do not carry out the converse experiment using RsaI-digested DNA as tracer, because its large dispersion of lengths prevents clean resolution of nucleosomal and naked DNA.) The gel itself further reveals that the pool of synthetic random DNA molecules (lane 2) and the two individually cloned isolates of random DNA (lanes 3 and 4) all behave similarly to the two genomic DNA samples. Evidently, there is little difference in the free energy of reconstitution between any of these DNA samples.
Figure 2.

Native gel analysis of competitive reconstitutions. Competitor DNAs are: lanes 1–4, RsaI-digested genomic fragments; lane 5, core particle DNA. Tracer DNAs are: lanes 1 and 5, core particle DNA; lane 2, pool of synthetic random DNA; lanes 3 and 4, individual cloned molecules from the synthetic random pool. S, kinased 100-bp ladder. Lanes 6 and 7, two particular cloned DNA molecules having relatively high (lane 6) or lower (lane 7) affinity for histone octamer in nucleosome reconstitution. N and D, nucleosomes and naked DNA, respectively.
These qualitative results are confirmed by the quantitative data in Table 1. The pool of synthetic random DNA and the two individual clones differ in free energy from core particle DNA by 0.1 ± 0.1 kcal·mol−1. These differences are small in comparison to a benchmark of physical significance, the characteristic energies of thermal fluctuations (RT, ≈0.6 kcal·mol−1, where R is the gas constant and T is the absolute temperature). Using core particle DNA again as the tracer but with core particle competitor DNA (instead of the RsaI digest) leads to a ΔΔG of −0.2 ± 0.2 kcal·mol−1. This difference between the two different genomic DNAs as competitors is again small in comparison to the energies of thermal fluctuations, and moreover it may be entirely attributed to the use of a 1.5-fold lower DNA weight concentration (see Materials and Methods) for the core particle competitor than for the RsaI digest competitor; the effect of the changed weight concentration is given by RT ln (20/30) ≈ −0.2 kcal·mol−1.
Table 1.
Quantitative free energy measurements
| Tracer | ΔΔG, kcal·mol−1 (n) |
|---|---|
| Synthetic random pool | −0.05 ± 0.08 (7) |
| Random clone 1 | 0.05 ± 0.08 (6) |
| Random clone 2 | 0.11 ± 0.13 (6) |
| Core particle DNA | 0 |
ΔΔG ≡ ΔGsample − ΔGcore particle DNA. ΔG (= −RT ln Keq) obtained using the indicated radiolabeled tracer with RsaI-digested genomic DNA as competitor. Values given as mean ± SD; n, number of independent measurements. Using core particle DNA as both the tracer and the competitor yields ΔΔG of −0.22 ± 0.16 kcal·mol−1 (n = 8). This difference for the two different genomic DNAs as competitors is small in comparison to RT and can be entirely attributed to the use of different DNA weight concentrations.
The small free energy differences found here represent positive results of little difference, not negative results of a failure to detect differences. Earlier studies from many laboratories establish that this methodology readily detects significant free energy differences when these in fact exist. Lanes 6 and 7 (Fig. 2) show one such example for two different individual cloned tracer DNAs, one having a high affinity for histone octamer (lane 6) and one having a more modest affinity (lane 7). The differing affinities lead to differing ratios of counts in nucleosomes to counts in naked DNA, plainly visible in the raw data (lanes 6 and 7) as well as in the integrated intensities measured by PhosphorImager (data not shown).
The broad range of mobilities for the nucleosomes reconstituted with both the pool (Fig. 2, lane 2) and individual clones (lanes 3 and 4) of the synthetic random DNA is noteworthy. Mobility differences arise from variable locations of the histone octamer within the overall DNA length (22, 23), implying that on these DNA samples, many positions are represented even on individual clones (lanes 3 and 4). As will be seen below, this behavior is a corollary of the fact that both the majority of the entire random pool and representative individual clones have low free energies for nucleosome formation that moreover do not differ much for nucleosome formation in one position versus another. (Note that the high-affinity DNA molecule in lane 6 shows a very different behavior; it gives rise to a strongly biased nucleosome position with a corresponding sharp mobility on the native gel.) The core particle DNA, while somewhat disperse in length, is mostly much shorter than the 282-bp synthetic random DNA and therefore provides a more restricted range of mobilities. (The length differences do not contribute significantly to the measured free energies; similar results are obtained using the core particle DNA, which is shorter than 282 bp, and the RsaI-digested DNA, which is longer.)
Superimposed on the broad range of mobilities for the nucleosomes reconstituted with the synthetic random DNA pool (Fig. 2, lane 2) or with either of the two individually cloned isolates (lanes 3 and 4) is a discernable band at a distinct mobility (arrowhead). The existence of this band could suggest that a fraction of the nucleosomes reconstituted on each of these DNA samples may be preferentially adopting a particular position on the DNA. But for the pool of unrelated random sequences, this could only happen if some element that all molecules have in common such as the fixed sequence elements or proximity to a DNA end were contributing detectably to the reconstitution. If this effect were large, it would invalidate a key assumption underlying this study. To quantify this effect, we measured the fraction of counts in reconstituted nucleosomes due to this species having the distinct mobility. We obtained results of 15%, 23%, and 33%, for lanes 2–4, respectively. Thus the equilibrium constants for incorporating the tracer into nucleosomes is increased by 1.15-, 1.23-, and 1.33-fold, respectively by the ability to form this species. The corresponding free energies are 0.08, 0.11, and 0.16 kcal·mol−1, far lower than the benchmark of thermal energies and smaller than the likely experimental error of the differential measurements reported in Table 1. Thus effects attributable to fixed sequence elements can be seen in this assay but are quantitatively small and do not significantly affect the conclusions of this study.
Since the best (i.e., highest affinity) ≈10% of the DNA is reconstituted into nucleosomes in this procedure, these results imply that there is little difference in the free energy of reconstitution between the best 10% of the synthetic random sequences and the best 10% of the natural genomic sequences. Importantly, however, the results for the individually cloned isolates of the synthetic random DNA show that the results from the best 10% of the synthetic random sequences are representative; there is <1% chance that both individual isolates are each in the top 10th percentile of affinity in the entire pool, yet their free energy of reconstitution is no different. We conclude that there is little free energy difference between typical random synthetic DNA and the best 10% of the natural genomic DNA. Moreover, half of the best 10% of the natural genomic DNA is worse than its measured average (by definition). Hence, we conclude that at least 95% of genomic DNA has a free energy of reconstitution (i.e., affinity for histone octamer) that differs little from that of typical synthetic random DNA. The bulk of the eukaryotic genome is evidently not constrained or evolved to aid substantially in its own packing at the level of individual nucleosomes.
Relation of Histone Binding Affinity to Nucleosome Positioning.
We consider next the relation between the free energy of nucleosome formation and the preferential occupancy of specific DNA sites by the histone octamer, also known as nucleosome positioning. We distinguish translational and rotational positioning (5). Translational positioning refers to the extent to which a histone octamer selects a particular contiguous stretch of 147 bp of DNA in preference to other stretches of the same length that are translated forwards or backwards along the DNA. Rotational positioning is a degenerate form of translational positioning in which a set of discreet translational positions, differing by integral multiples of the DNA helical repeat, are all occupied in preference to the set of other possible locations. DNA sequences that are intrinsically bent or are anisotropically bendable may lead to rotationally positioned nucleosomes.
We consider a model in which a histone octamer is constrained to be bound to a DNA molecule of length L. We take 147 bp as the amount of DNA in a nucleosome core particle, assuming for simplicity that locations of the octamer that extend beyond the DNA end (leaving DNA binding sites on the octamer unsatisfied) are negligibly populated. In vitro, for nucleosome reconstitution experiments, L is well defined, and there are L − 146 different possible locations for the octamer. In vivo, there is not a unique definition of L; one appropriate definition is the nucleosome repeat length (see ref. 24). For a further simplification, we take all possible locations of the histone octamer other than the specifically positioned one(s) to be equivalent in their free energy.
Translational Nucleosome Positioning.
Translational positioning may be understood quantitatively in terms of the free energy change for transfer of histone octamer from unrestricted exploration of any of the L − 147 available (and equivalent) nonspecific positions to fixed occupancy at a single specific location. We take the nonspecifically bound state as the reference, and assign the free energy ≡ 0. Let ΔGnet be the free energy change for transfer to the specific position. Two terms contribute: ΔGnet = ΔGintr + ΔGstat, where ΔGintr is the free energy change for transmuting a nonspecific site into a specific one, and ΔGstat is a statistical contribution to the free energy change, reflecting the fact that there may be many more possible nonspecific locations than the single specific one.
ΔGintr is the same as would obtain if the process were to take place in standard state conditions; hence ΔGintr ≡ ΔΔGHO, which is defined and also measured experimentally as the difference free energy for reconstitution of histone octamer into nucleosomes, for two DNA molecules having identical lengths but where one contains a single favored (“specific”) position and the other contains no such sites.
Transfer of histone octamer from free choice among any of L − 147 nonspecific positions to fixed occupancy at a specific position reduces the entropy of the system by the amount ΔSstat = R ln (1/(L − 147)), thereby contributing ΔGstat = −TΔSstat = −RT ln (1/(L − 147)) to the net free energy change.
With these definitions, the probability of occupancy of the specific position by the histone octamer (P) is given by:
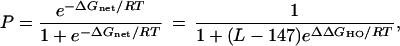 |
1 |
where R is the gas constant and T is the absolute temperature.
This equation has several important ramifications. Positioning is inherently statistical, not “precise”: it is quantified by a probability of occupancy of the specific site that is always greater than zero and less than one. P depends fundamentally on two quantities: the intrinsic free energy preference of histone octamer for the specific site compared with alternative sites, ΔΔGHO, and the number of alternative sites, L − 147. Given a particular DNA length, P is determined by ΔΔGHO.
Given a measured value for ΔΔGHO and the DNA length L, Eq. 1 allows one to calculate P. If nucleosomes prove to be freely mobile as proposed (e.g., refs.25–27), P will also equal the fraction of time that the specific position is occupied in any particular nucleosome.
Eq. 1 implies that existing “positioning sequences” have quite limited positioning power. For example, a 250-bp fragment in which occupancy at one specific position is favored by 2 kcal·mol−1 [comparable in energy to the pentamer “TG” sequence element (7, 8)] yields only ≈20% occupancy of the specific position. This limited occupancy of the specific position is nevertheless easily detected experimentally, because it stands out against a weak background; the remaining ≈80% occupancy is distributed over 103 alternative positionings, each having <1% occupancy.
Eq. 1 also provides a new way to measure ΔΔGHO. Direct experimental measurement of P, perhaps through quantitative nuclease protection studies for sites both internal and external to the positioned nucleosome, together with the known DNA length L, allows ΔΔGHO to be calculated.
Rotational Positioning.
Eq. 1 is readily generalized to account for cases in which multiple specific translational positions are preferentially populated relative to the bulk “nonspecific” positions. Rotational positioning is an example of such a case, for which there happen to be constraints on the mutual spacings of the preferred positioning sites.
For a DNA molecule of length L, let nR be the number of positions occupied by rotationally positioned histone octamers and let ΔΔGR be the intrinsic free energy for binding of histone octamer in any one of the set of preferentially occupied positions in a rotationally positioned system. (For simplicity, we let the same ΔΔGR apply to all of them.) Then, rotational positioning, too, is seen to be intrinsically statistical, quantified by a probability that any of the specific rotational positions is occupied, given by:
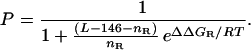 |
2 |
ΔΔGR can be measured as described above, provided that one uses for this purpose a variant of the DNA molecule that is sufficiently short (e.g., ≈150 bp), such that only one rotationally-phased position is accessible. Alternatively, a longer molecule may be used provided that the measured apparent ΔΔGR is corrected for the statistical factor RT ln nR before use in Eq. 2.
Protein-Directed Nucleosome Positioning.
It is possible that, in vivo, certain site-specific DNA binding proteins or other effects will act to influence the positioning of nucleosomes (10, 24, 28, 29). Such influences may be quantified by a coupling free energy, and Eqs. 1 and 2 may be adapted to quantify the net (but still statistical) positioning that results (J.W., unpublished work).
DISCUSSION
Nucleosome Packaging and Nucleosome Positioning of Genomic DNA.
The most important conclusion of this work is that the bulk of the eukaryotic genome (≥95%) is not evolved or constrained to aid substantially in its own packaging at the level of individual nucleosomes. Apparent differences which are detected (Table 1) are small in comparison to thermal energies, a benchmark of physical significance. The relation derived here between the free energy of nucleosome reconstitution and the probability of nucleosome positioning implies that the bulk of the eukaryotic genome is similarly not evolved or constrained to substantially bias or specify nucleosome positioning at the level of individual nucleosomes. The similarity of the results obtained with two very different representations of the genomic DNA provides further evidence that each sample reliably reflects the properties of the whole genome.
The results of this study apply to systems at equilibrium. Nucleosome core particles that result from reconstitution in vitro are kinetically trapped; in typical solution conditions they are stable against dissociation and against exchange with labeled competitor DNA for indefinitely long periods. These are not properties of an equilibrium system. Importantly, however, Shrader and Crothers (7, 8) have demonstrated experimentally that nucleosome reconstitution in vitro is an equilibrium process. The procedure through which nucleosomes are reconstituted in vitro establishes a true equilibrium distribution and then subsequently (and reversibly) freezes this in, creating stable particles that nevertheless reflect an equilibrium distribution.
It is now clear that nucleosomes are mobile even in physiological ionic conditions (25–27), and our competitive reconstitution procedure sweeps slowly through physiological ionic strength before freezing in the resulting particles at subphysiological ionic strengths. Therefore we presume that the nucleosomes resulting from our competitive reconstitutions have equilibrated at physiological ionic strengths. This means that, as regards the DNA sequence determinants of nucleosome stability and positioning, the results of our study in vitro should reflect the same histone–DNA interactions as those that obtain in vivo.
We emphasize that, in vivo, many additional factors beyond genomic DNA sequence may act to bias the positions of nucleosomes (10, 24, 28, 29). The present results serve to define and quantify just those contributions to nucleosome positioning arising from sequence dependences to histone–DNA interactions. However, our overall conclusion—that the free energies of positioning are finite and therefore that nucleosome positioning is statistical, not precise—applies also to the total effects of all of the forces that act to govern nucleosome positioning in vivo.
Relation to Previous Studies.
The present results demonstrate that the bulk of the eukaryotic genome (≥95%) is not evolved or constrained to aid substantially in its own nucleosome packaging or nucleosome positioning at the level of individual nucleosomes. Yet, at the same time, previous work by us and others demonstrates that sequence signals involved in nucleosome packaging and in nucleosome positioning are readily detectable in natural DNA. How may both of these sets of results be true at once? We imagine two limiting possibilities. (i) Perhaps signals that favor nucleosome packaging and positioning are sparsely but rather uniformly distributed along the entire genome. They can be detected by sensitive signal averaging procedures, but are quantitatively small, less than approximately ±0.2 kcal·mol−1. They may nevertheless have biological significance. Acting individually, they yield only a small bias in nucleosome positioning. Or, they may act collectively on the positioning of an entire array of nucleosomes, perhaps through constraints on the mutual packing of nucleosomes within higher order chromatin structures (24); the collective effects may be substantial. (ii) Perhaps signals involved in nucleosome packaging and positioning are concentrated into a small subset (<5%) of the genome. These are detected when enough sequence information is analyzed, while the majority of genomic sequences simply contribute incoherent “noise” to the analyses.
The second limiting possibility (above) is certainly correct to some degree; one of the best characterized “nucleosome-positioning” DNA sequences, that discovered by Simpson and Stafford (30), has a significantly higher affinity for histone octamer in nucleosome reconstitution than typical “bulk” genomic DNA (7, 8) and is itself a natural sequence.
Both of these ideas may be true at once. An example is provided in Fig. 3, which plots the probability distribution for the number of occurrences within a nucleosome-sized window of a particular sequence motif that is correlated with favorable nucleosome packaging (7, 8), averaged over the genome of the yeast S. cerevisiae. There is a single peak together with a long forward tail. The main peak reflects the predominant situations, in which signals that favor nucleosome packaging are sparsely but rather uniformly distributed along the entire genome, as in idea (i). The long forward tail reflects a minority of situations in which nucleosome packaging signals are relatively concentrated in a small subset of the genome, as in the second limiting possibility presented above. (Whether these particular genomic regions actually favor nucleosome formation remains to be tested; this example provides a demonstration in principle of how real genomic sequences plausibly conform at once to both of the ideas presented above.)
Figure 3.

Probability distribution for the number of occurrences within a nucleosome-sized window of the motif “(A or T)3NN(G or C)3NN,” where N is any base, averaged over the yeast genome. The plot is represented on a linear probability scale for ease of interpretation; a logarithmic plot (data not shown) better represents detail in the forward tail and again shows there to be a single peak at 2 on the abscissa. Only nonzero values are plotted; the last nonzero datapoint is at 25 on the abscissa. Many other motifs related to nucleosome packaging or positioning were also analyzed and yielded comparable results.
It will be important to assess experimentally where the best nucleosome-packaging sequences are in genomes, and how their locations relate to other elements of gene and genome organization.
Statistical Nature of Nucleosome Positioning.
A second important conclusion is that nucleosome positioning is inherently statistical, not precise. Briefly put, the free energies and free energy differences are finite, so probabilities are necessarily greater than zero and less than one. In this light, previous reports of precise positioning at single or multiple nearby sites must be considered as really reflecting preferential positioning at those sites. It is important to recognize this statistical property of positioning because it has substantial ramifications for proposed mechanisms of gene regulation. If positioning is not precise, then essential DNA regulatory sequences will sometimes be buried when they need to be accessible or will sometimes be accessible when they need to be repressed (buried). Mechanisms proposed for gene regulation must be robust to inevitable statistical fluctuations.
It may be helpful in this context to consider the case of prokaryotic repressor proteins as gene regulatory factors. Repressor proteins do indeed bind to and act at precise sites, but it has not proven helpful to use this language nor to consider the problem in this way. Rather, it is recognized that repressor proteins obey the laws of thermodynamics; they have a finite free energy difference for binding to the specific site versus binding to a large set of alternative nonspecific sites, and one equates their activities in gene regulation to the equilibrium fractional occupancy (i.e., the probability of occupancy) of the specific site (31).
Finally, we emphasize that DNA sequence-encoded biases in (statistical) positioning do exist and may have important biological consequences. In the context of our site exposure model for the mechanism of regulatory protein binding to nucleosomal target sites (15, 32), biases in average positioning of mobile nucleosomes substantially affect the average concentration of the regulatable (accessible) state of nucleosomes. Biases in positioning may also contribute to the stability of higher order chromatin folding (24), potentially at multiple hierarchical levels in the structure.
Acknowledgments
We acknowledge with gratitude the kind gift of the random synthetic DNA by Drs. David Bartel and Jack Szostak, and we thank Kevin Polach for helpful discussions. Research in our laboratory is supported by a grant from the National Institutes of Health.
References
- 1.Hagerman P J. Annu Rev Biochem. 1990;59:755–781. doi: 10.1146/annurev.bi.59.070190.003543. [DOI] [PubMed] [Google Scholar]
- 2.Marini J C, Levene S D, Crothers D M, Englund P T. Proc Natl Acad Sci USA. 1982;79:7664–7668. doi: 10.1073/pnas.79.24.7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Widom J. BioEssays. 1985;2:11–14. [Google Scholar]
- 4.Travers A A, Klug A. Philos Trans R Soc London B. 1987;317:537–561. doi: 10.1098/rstb.1987.0080. [DOI] [PubMed] [Google Scholar]
- 5.Simpson R T. Prog Nucleic Acid Res Mol Biol. 1991;40:143–183. doi: 10.1016/s0079-6603(08)60841-7. [DOI] [PubMed] [Google Scholar]
- 6.Travers A A, Muyldermans S V. J Mol Biol. 1996;257:486–491. doi: 10.1006/jmbi.1996.0178. [DOI] [PubMed] [Google Scholar]
- 7.Shrader T E, Crothers D M. Proc Natl Acad Sci USA. 1989;86:7418–7422. doi: 10.1073/pnas.86.19.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shrader T E, Crothers D M. J Mol Biol. 1990;216:69–84. doi: 10.1016/S0022-2836(05)80061-0. [DOI] [PubMed] [Google Scholar]
- 9.Bina M. J Mol Biol. 1994;235:198–208. doi: 10.1016/s0022-2836(05)80026-9. [DOI] [PubMed] [Google Scholar]
- 10.Staffelbach H, Koller T, Burks C. J Biomol Struct Dyn. 1994;12:301–325. doi: 10.1080/07391102.1994.10508742. [DOI] [PubMed] [Google Scholar]
- 11.Ioshikes I, Bolshoy A, Derenshteyn K, Barodovsky M, Trifonov E N. J Mol Biol. 1966;262:129–139. doi: 10.1006/jmbi.1996.0503. [DOI] [PubMed] [Google Scholar]
- 12.Bolshoy A. Nat Struct Biol. 1995;2:446–448. doi: 10.1038/nsb0695-446. [DOI] [PubMed] [Google Scholar]
- 13.Widom J. J Mol Biol. 1996;259:579–588. doi: 10.1006/jmbi.1996.0341. [DOI] [PubMed] [Google Scholar]
- 14.Bartel D P, Szostak J W. Science. 1993;261:1411–1418. doi: 10.1126/science.7690155. [DOI] [PubMed] [Google Scholar]
- 15.Polach K J, Widom J. J Mol Biol. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 16.Protacio R U, Widom J. J Mol Biol. 1996;256:458–472. doi: 10.1006/jmbi.1996.0101. [DOI] [PubMed] [Google Scholar]
- 17.Widom J. J Mol Biol. 1986;190:411–424. doi: 10.1016/0022-2836(86)90012-4. [DOI] [PubMed] [Google Scholar]
- 18.Simpson R T. Biochemistry. 1978;17:5524–5531. doi: 10.1021/bi00618a030. [DOI] [PubMed] [Google Scholar]
- 19.Feng H-P, Scherl D S, Widom J. Biochemistry. 1993;32:7824–7831. doi: 10.1021/bi00081a030. [DOI] [PubMed] [Google Scholar]
- 20.Irvine D, Tuerk C, Gold L. J Mol Biol. 1991;222:739–761. doi: 10.1016/0022-2836(91)90509-5. [DOI] [PubMed] [Google Scholar]
- 21.Yao J, Lowary P T, Widom J. Proc Natl Acad Sci USA. 1990;87:7603–7607. doi: 10.1073/pnas.87.19.7603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pennings S, Meersseman G, Bradbury M E. J Mol Biol. 1991;220:101–110. doi: 10.1016/0022-2836(91)90384-i. [DOI] [PubMed] [Google Scholar]
- 23.Dong F, Hansen J C, van Holde K E. Proc Natl Acad Sci USA. 1990;87:5724–5728. doi: 10.1073/pnas.87.15.5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Widom J. Proc Natl Acad Sci USA. 1992;89:1095–1099. doi: 10.1073/pnas.89.3.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meersseman G, Pennings S, Bradbury E M. EMBO J. 1992;11:2951–2959. doi: 10.1002/j.1460-2075.1992.tb05365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varga-Weisz P D, Blank T A, Becker P B. EMBO J. 1995;14:2209–2216. doi: 10.1002/j.1460-2075.1995.tb07215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ura K, Hayes J J, Wolffe A P. EMBO J. 1995;14:3752–3765. doi: 10.1002/j.1460-2075.1995.tb00045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kornberg R D, Stryer L. Nucleic Acids Res. 1988;16:6677–6690. doi: 10.1093/nar/16.14.6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao J, Lowary P T, Widom J. Proc Natl Acad Sci USA. 1993;90:9364–9368. doi: 10.1073/pnas.90.20.9364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simpson R T, Stafford D W. Proc Natl Acad Sci USA. 1983;80:51–55. doi: 10.1073/pnas.80.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.von Hippel P H. Science. 1994;263:769–770. doi: 10.1126/science.8303292. [DOI] [PubMed] [Google Scholar]
- 32.Polach K J, Widom J. J Mol Biol. 1996;258:800–812. doi: 10.1006/jmbi.1996.0288. [DOI] [PubMed] [Google Scholar]