

Abstract
Background
Post-traumatic stress disorder (PTSD) is characterized by acute and chronic changes in the stress response, which include alterations in glucocorticoid secretion, and critically involve the limbic system, in particular the amygdala. Important symptoms of PTSD manifest as a classical conditioning to fear, which recurs each time trauma-related cues remind the subject of the original insult. Traumatic memories based on fear conditioning can be disrupted if interfering events or pharmacological intervention are applied following their retrieval.
Methods and Results
Using an animal model, here we show that a traumatic memory is persistently disrupted if immediately after its retrieval glucocorticoid receptors are inactivated in the amygdala. The disruption of the memory is long-lasting and memory retention does not re-emerge following strong reminders of the conditioned fear.
Conclusions
We propose that a combinatorial approach of psychological and pharmacological intervention targeting the glucocorticoid system following memory retrieval may represent a novel direction for the treatment of PTSD.
Keywords: fear memory, reconsolidation, glucocorticoid receptor, amygdala, PTSD, animal model
Introduction
Because the behavioral, neuroendocrine, and autonomic nervous system manifestations of posttraumatic stress disorder (PTSD) bear strong resemblance to what is observed in fear conditioned rodents, it has been hypothesized that PTSD involves molecular, cellular and anatomical mechanisms similar to those implicated in fear conditioning (Rasmusson and Charney 1997; Shalev et al 1992; Elzinga and Bremner 2002; Rau et al 2005). Although it is not possible to precisely reproduce PTSD in an animal model, fear conditioning in rodents can be used to reproduce and elucidate some aspect of PTSD, including the processing of fearful stimuli and the encoding and processing of emotional memory (Miller and McEwen 2006; Siegmund and Wotjak 2006). Fear conditioning is readily induced in laboratory animals by pairing neutral stimuli, such as contextual cues (conditioned stimulus, CS), with a fear-inducing stimulus, such as a mild foot shock (unconditioned stimulus, US). Thereafter, presentation of the context in which the conditioning was accomplished, evokes a stereotypic response in the animal that includes fear responses, glucocorticoid response, and activation of the autonomic nervous system.
Similarly, patients with PTSD suffer from intrusive memories of the original traumatic event, which are often precipitated by contextual cues that have become associated with the event in much the same way that the neutral stimuli are associated with the US in fear conditioning. Similar to fear conditioned animals, patients with PTSD manifest abnormalities of the glucocorticoid system (Yehuda 2001), increased startle response (Grillon et al 1996) and enhanced autonomic nervous system response (Yehuda 2001; Orr et al 2002).
The anatomical pathway necessary for fear conditioning has been well characterized and a brain region that plays a major role in it is the amygdala. Recently, brain imaging studies have suggested that enhanced activity of the amygdala is involved in PTSD (Shin et al 1997; Liberzon et al 1999; Shin et al 2005; Protopopescu et al 2005). Therefore, investigators have increasingly focused their attention on what is known about fear conditioning in order to develop therapeutic interventions for PTSD. Current treatments for PTSD include medications that may work, at least in part, to decrease amygdala activity (Asnis et al 2004) and exposure-based treatments such as prolonged exposure (PE) (Foa 2006) and eye movement desensitization and reprocessing (EMDR) (Rothbaum et al 2005). The exposure-based treatments operate under the premise that repeated reactivation of traumatic memories in a controlled environment allow them to be reprocessed so that they are no longer capable of causing psychological symptoms (Deacon and Abramowitz 2004).
Studies recently carried out in both animals and humans have shown that the recall of a memory can transiently revert the stable state of the memory into a labile one and, therefore, established memories can become sensitive to disruption. Following recall (reactivation) of the memory, the process that ensures that the fragile memory returns to a stable state is known as reconsolidation (Sara 2000; Nader 2003). Many recent as well as earlier studies showed that a variety of pharmacological agents can disrupt fear memory reconsolidation if applied immediately after memory reactivation, suggesting that it might be possible to identify pharmacotherapies to be used in tandem with exposure-based therapies to weaken pathogenic memories that are responsible for PTSD (Przybyslawski et al 1999; Debiec and LeDoux 2004; Bustos et al 2006. Tronson et al 2006).
In previous studies, we used the fear conditioning-based task inhibitory avoidance (IA) as a model of a traumatic memory. In this paradigm, the animal (rat) learns to avoid a context (CS) that has previously been paired with a foot shock (US). We found that, like other fear-based memories (Nader et al 2000; Debiec et al 2002), IA can be disrupted if protein synthesis or specific molecules, such as the transcription factor CCAAT enhancer binding protein β (C/EBPβ), known to mediate the post-acquisition stabilization process (consolidation), are inhibited in the amygdala following memory reactivation (Tronel et al 2005).
The consolidation of traumatic memories is known to critically involve the glucocorticoid system (McGaugh and Roozendaal 2002; Roozendaal 2002; Sandi 1998). Indeed, post-training systemic infusions of glucocorticoids enhance inhibitory avoidance retention, and this enhancement is due to selective activation of GR (Rev in McGaugh 2004). Conversely, an intra-cerebroventricular (i.c.v.) infusion of the glucocorticoid receptor antagonist RU 38486, administered before or after training in a water maze spatial task, does not affect acquisition, but impairs retention (Roozendaal and MgGaugh 1997; Oitzl et al 1998). Moreover, pre-training BLA-injections of RU38486 disrupt contextual fear conditioning consolidation in rats (Donley et al 2005) and post-training infusion of the antagonist impairs memory for an avoidance task in chicks (Sandi and Rose 1994).
Based on our data and current knowledge of the consolidation and reconsolidation processes, we hypothesized that similar modulatory responses activated following both acquisition and memory reactivation play a critical role in mediating memory stabilization (Alberini 2005).
However, thus far few studies have investigated the role of modulatory pathways during the reconsolidation of an established memory. Some work has demonstrated that the noradrenergic system plays a critical role after memory reactivation in rats (Przybyslawski et al 1999; Debiec and LeDoux 2004), whereas other studies have provided evidence for the involvement of the dopaminergic system in chicks (Sherry et al 2005), angiotensin II in the crab Chasmagnathus (Frenkel et al 2005) and the PKA-dependent pathway in rats (Tronson et al 2006). We hypothesized that, like during fear memory consolidation, the glucocorticoid system plays a critical role during the reconsolidation of reactivated traumatic memories. Here we tested this hypothesis by examining the effect of directed injection of the glucocorticoid receptor (GR) antagonist RU 38486 in the basolateral amygdala (BLA) following the reactivation of an IA memory in rat.
Materials and Methods
Animals
Long Evans adult male rats (Harlan, Indianapolis, IN) weighing 200–250 g at the beginning of procedures were used in all experiments. Rats were individually housed and maintained on a 12 h on/12 h off light/dark cycle and underwent behavioral procedures during the light cycle. All rats were allowed free access to food and water. All protocols complied with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Mount Sinai School of Medicine Animal Care Committees.
Surgical procedure
Rats were anesthetized with ketamine (60 mg/Kg; i.p) and xylazine (7.5 mg/Kg; i.p) and implanted with cannulae (22 gauge; Plastics One, Roanoke, VA) positioned 1.5 mm above the amygdala using the following coordinates according to Paxinos and Watson (1998): AP : -2.8 mm, ML : ±5.3 mm, DV :-6.25 mm from the skull surface. Rats recovered for 7 days after surgery before undergoing any experimental procedures.
Pharmacological treatment
The specific GR antagonist RU 38486 (mifepristone, Sigma, St Louis, MO) (Teutsch et al 1981; Allen et al 1988, Brogden et al 1993) was dissolved in 100% ethanol and subsequently diluted in saline to reach the appropriate concentration. Two concentrations were used: 0.6 and 2.4 ng/μl. The final concentrations were diluted in 2% ethanol. This concentration range has been previously used in injection experiments that stereotactically targeted specific brain regions, including the amygdala, and found to include a dose-response curve of effectiveness in memory retention and extinction responses (Roozendaal et al 1999, Donley et al 2005; Yang et al 2006). The compound was infused bilaterally through the infusion cannulae at 0.4 μl/min using a pump, and a total volume of 0.5 μl was infused into each side over 1.25 minutes. Controls were injected with the same volume of the vehicle solution (2% ethanol). The injection cannulae were left in place for at least 1 min to allow the solution to completely outflow from the cannula tip.
Behavioral Procedures
Inhibitory avoidance training
The IA training procedure has been previously described in Tronel et al (2005). The IA apparatus (Med Associates, St. Albans, Vermont) consisted of a rectangular-shaped Perspex box, divided into a safe compartment and a shock compartment. The safe compartment was white and illuminated; the shock compartment was black and dark. Foot shocks were delivered to the grid floor of the dark chamber via a constant current scrambler circuit. The apparatus was located in a sound-attenuated, non-illuminated room. During training sessions, each rat was placed in the safe compartment with its head facing away from the door. After ten seconds, the door separating the compartments was automatically opened allowing the rat access to the shock chamber. Latency to enter the shock chamber was taken as a measure of fear acquisition. All acquisition latencies were below 24.3 seconds. The door closed one second after the rat entered the shock chamber, and a brief foot shock (0.9 mA for 2 s) was administered. The rat was then removed from the apparatus and returned to its home cage. IA memory was tested as detailed below. Training and testing were always carried out between 10 am and 4 pm.
Memory reactivation
Forty-eight hours after training, the fear response of the animals was tested (Test 1). This test consisted of placing the rat back in the safe compartment and measuring the latency to enter the shock chamber. The rats that entered the dark compartment were removed and returned to their home cages immediately after entering. For rats that did not enter the dark side, the test was terminated at 540 s. Foot shock was not administered during the retention test. This test reactivated the fear memory.
Memory retention tests
Forty-eight hours after Test 1, animals were re-tested for fear memory retention (Test 2). In Experiment 3, memory was re-tested two additional times as detailed below (Test 3 and Test 4). All tests (Test 1–4) were carried out as described for Test 1 in memory reactivation.
Experiment 1
Rats received cannula implants that bilaterally targeted the basolateral nucleus of the amygdala (BLA). After recovery from surgery, rats were trained in IA and their fear memory was recalled (reactivated) by testing 2 days later (Test 1). Immediately after Test 1, the animals received a bilateral injection of either 0.3 ng per side in 0.5 μl of GR antagonist RU 38486 (n=8), or the same volume of vehicle solution (n=6). All animals were again tested for IA memory retention 48 h after reactivation (Test 2). An experimenter blind to the treatment of the groups scored the latencies.
Experiment 2
Rats received cannula implants that bilaterally targeted the BLA. After recovery from surgery, rats were trained in IA and their fear memory was recalled (reactivated) by testing 2 days later (Test 1). Immediately after Test 1, the animals received a bilateral injection of either 1.2 ng per side in 0.5 μl of GR antagonist RU 38486 (n=11) or the same volume of vehicle solution (n=9). All animals were again tested for IA memory retention 48 h after reactivation (Test 2). To determine whether the effect of treatment was contingent upon reactivation, rats (n=8) were bilaterally injected with RU 38486 in the absence of reactivation (Test1). An experimenter blind to the treatment of the groups scored the latencies.
Experiment 3
Experiment 2 was repeated. Thus, rats that receive cannula implants targeting the amygdalae, were trained with IA and tested 48 hours after training (Test 1). Immediately after Test 1, the animals received a bilateral injection of either 1.2 ng per side in 0.5 μl of GR antagonist RU 38486 (n=8) or the same volume of vehicle solution (n=6). All animals were again tested for IA memory retention 48 h after reactivation (Test 2). To determine whether the effects were stable over time, one week later the animals were tested again (Test 3). To determine whether memory could re-emerge, immediately after Test 3, rats were exposed to a reminder shock of 0.9 mA in a different box. Rats were tested a final time 24 h later (Test 4). Latency to re-enter the dark compartment was always taken as the measure of memory retention. An experimenter blind to the treatment of the groups scored the latencies.
Experiment 4
Experiment 2 was repeated. Thus, rats that received cannula implants targeting the BLA, were trained in IA and tested 48 hours after training (Test 1). Immediately after Test 1, the animals received a bilateral injection of either 1.2 ng per side in 0.5 μl of GR antagonist RU 38486 (n=8) or the same volume of vehicle solution (n=7). All animals were again tested for IA memory retention 48 h after reactivation (Test 2). Subsequently, to exclude that the intra-amygdala administration of RU 38486 causes a non-specific, general inactivation or damage of the amygdala, rats underwent another IA training trial. Thus, 3 hours after Test 2, rats that received a bilateral injection of 1.2 ng per side of RU 38486 (n=8) were re-trained in IA. Forty-eight hours later, the rats were tested (Test 3).
Statistical analyses
Statistical analyses of the behavioral data were performed using an analysis of variance (ANOVA) followed by a Bonferroni-protected post-hoc t test.
Histology
At the end of the behavioral experiments, rats were anesthetized and brains were removed, frozen, sectioned and inspected for cannula placement. Brains were sectioned (40 μm) and mounted. Sections were inspected to determine the location of the injection sites. Animals with cannula placements that targeted sites outside the BLA area were excluded from the study.
Results
Figure 1 shows representative areas targeted by the stereotactic injections performed in this study. Animals with injection sites that deviated from these areas were omitted from the study.
Figure 1. Photomicrograph of a representative cannula placement and schematic representations of injection sites into the BLA at the indicated rostrocaudal planes.
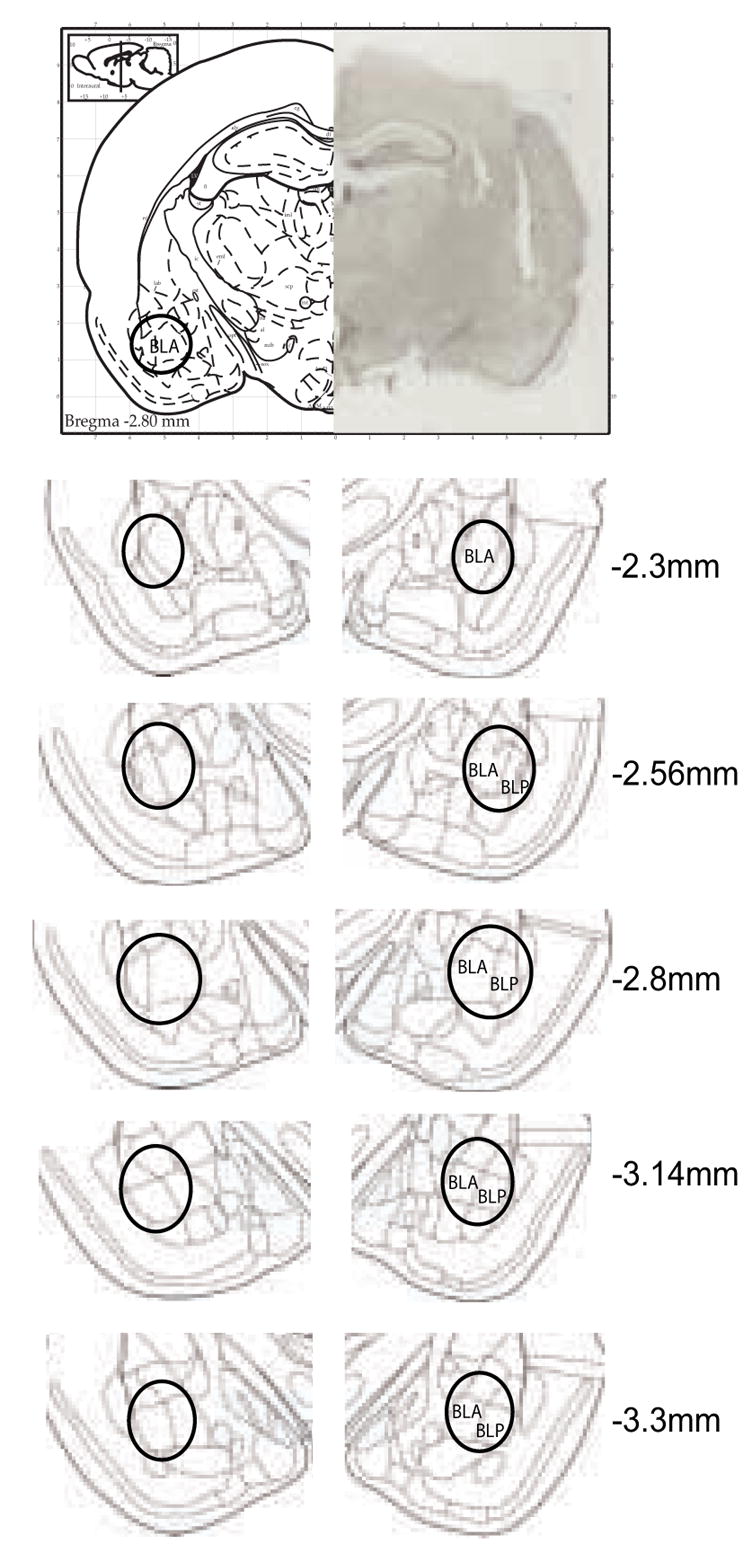
The numbers represent the coordinates from bregma (in millimeters) according to Paxinos and Watson (1998). Injections sites were contained within the black circle. BLA: basolateral amygdaloid nucleus, anterior part; BLP: basolateral amygdaloid nucleus, posterior part.
Experiment 1
As shown in Figure 2, injection of 0.3 ng of RU 38486 following memory reactivation had no effect on memory retention at a subsequent test. In fact, as revealed by the mean avoidance latencies, at Test 2, both vehicle and RU 38486 -injected groups had similar fear memory retention (RU 38486: 386.19 ± 36.96 s; Veh: 333.5 ± 33.6 s).
Figure 2. Injection of 0.3 ng of RU 38486 in the BLA after recall has no effect on IA memory retention.
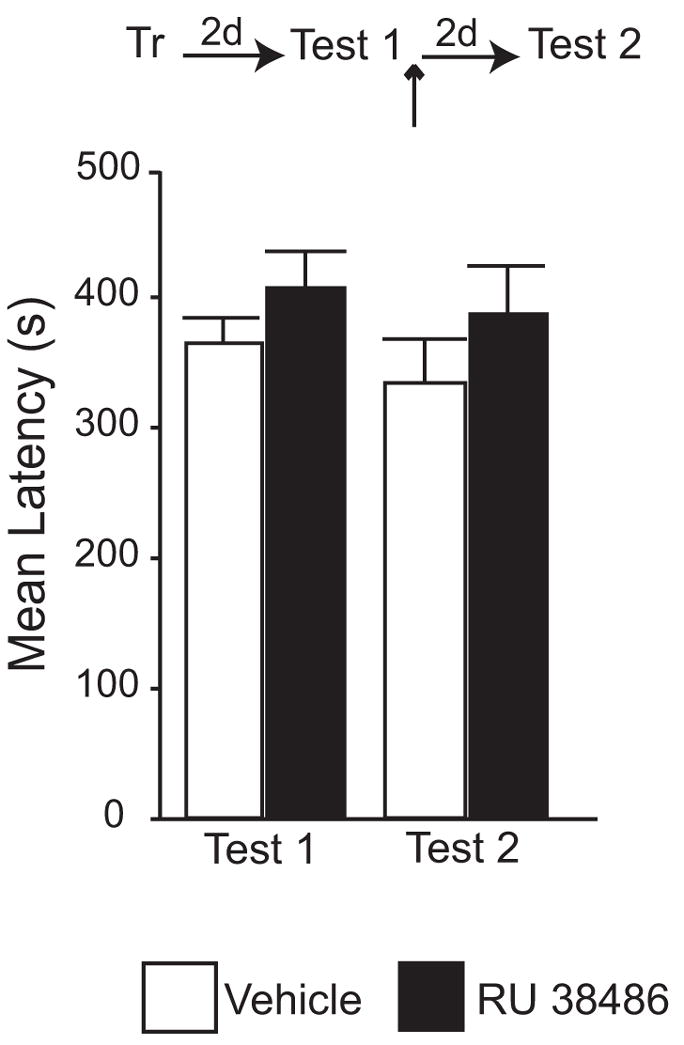
Latency to enter the shock chamber was taken as a measure of memory retention. Rats were trained in IA (Tr) and tested 2 days later (Test 1). This test recalled the memory. Immediately after Test 1, 0.3 ng of RU 38486 or the same volume of vehicle solution was bilaterally injected (↑) into the BLA. IA memory was tested again 2 days later (Test 2). No difference was observed between groups at Test 2.
Experiment 2
When bilateral BLA injections of 1.2 ng of RU 38486 were administered following IA memory reactivation (Test 1), a significant memory impairment was found at Test 2 (Figure 3). All animals, as expected, had similar latencies at Test 1 (Veh: 328.75 ± 61.65 s; RU 38486: 308.2 ± 46.81 s). A two-way ANOVA that compared the vehicle-injected and the RU 38486-injected groups at Test 1 and Test 2 showed a significant effect of treatment (F1,36 = 5.44, P < 0.05) and a significant effect of test (F1,36 = 4.53, P < 0.05). A Bonferroni-protected post-hoc test revealed that, at Test 2, the retention levels of the rats that received RU 38486 injections were significantly lower (94.46 ± 34.62 s) than those of Test1 (P < 0.01) and those of the vehicle-injected group at Test 2 (318.9 ± 68.34 s; P< 0.01) (Figure 3).
Figure 3. Injection of 1.2 ng of RU 38486 in the BLA after recall disrupts IA memory.
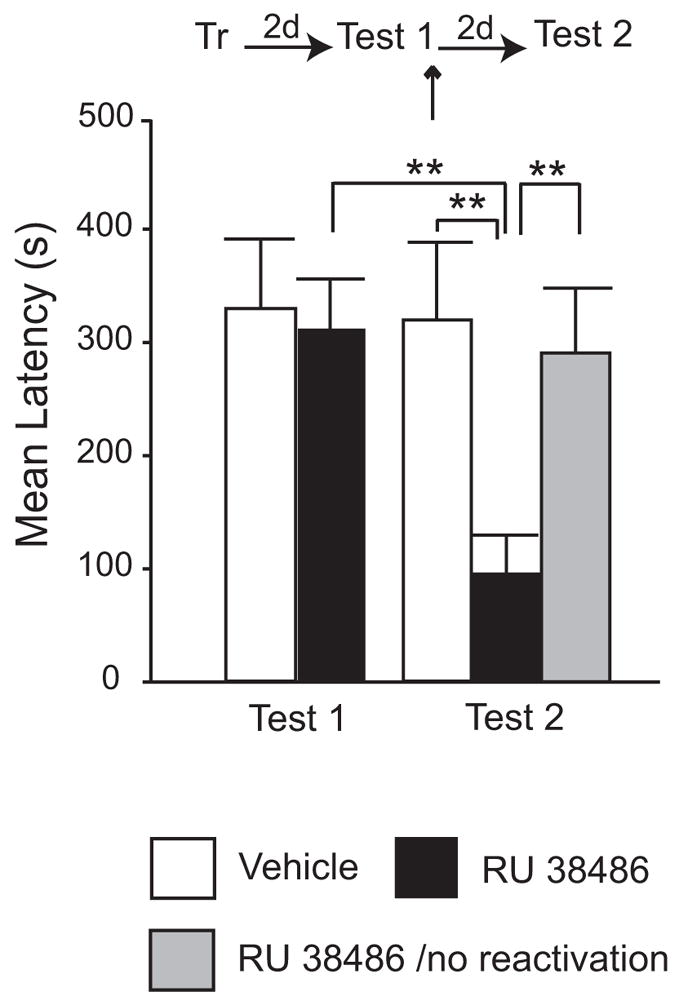
Latency to enter the shock chamber was taken as a measure of memory retention. Rats were trained in IA (Tr) and memory was recalled by testing 2 days later (Test 1). Immediately after Test 1, 1.2 ng of RU 38486 or the same volume of vehicle was bilaterally injected (↑) into the BLA. IA memory was tested again 2 days later (Test 2). RU 38486 injections impaired memory retention at Test 2 compared to vehicle or Test 1 latencies (**, P < 0.01). Injection of RU 38486 in the absence of memory reactivation had no effect.
To determine whether the memory disruption caused by RU 38486 was contingent upon memory reactivation as opposed to RU 38486 having primary memory blocking effects, rats were injected with the antagonist 48h after training in the absence of memory reactivation (Test 1). A one-way ANOVA revealed a significant effect of the treatment. No difference was found in the avoidance latencies of these rats (290.38 ± 54.04 s) compared to controls that received vehicle injections after Test 1 (318.9 ± 68.34 s) (Figure 3), but both of these groups were significantly different than the group injected with RU 38486 after Test 1 (94.46 ± 34.62 s; P < 0.05). Together, these results indicate that blocking GRs in the BLA after IA memory reactivation disrupts memory retention at a subsequent test.
Experiment 3
Some studies have reported that the loss of memory induced by agents or events administered after memory reactivation by CS presentation may be due to a transient blockade of memory retrieval. Indeed, several reports have shown that the memory loss observed after amnesic treatments applied following memory reactivation can either revert spontaneously (Lewis 1976; Judge and Quartermain 1982; Lattal and Abel 2004; Prado-Alcala et al 2006) or be restored by the administration of a reminder (Miller and Kraus 1977; Vianna et al 2001, Anokhin et al 2002; Salinska et al 2004). Therefore, we tested whether the RU 38486-induced memory disruption was i) persistent over time and ii) resistant to the effect of reminders. First, to establish whether the amnesia observed was long lasting, after replicating the previous results found in Experiment 2 at 2 days after reactivation (Test 2), we re-tested memory retention one week later (Test 3).
Furthermore, to determine whether the memory could be recovered, immediately after Test 3, animals were exposed to a reminder shock (0.9 mA in a different box). Rats were tested 24 h later (Test 4). A similar approach was used by Duvarci and Nader (2004) who showed that anisomycin administration into the later amygdala after recall persistently disrupts auditory fear reconsolidation. As shown in Figure 4, a two-way ANOVA with repeated measures followed by a Bonferroni-protected post-hoc test on latencies at all four tests demonstrated a significant effect of the treatment (F1, 36 = 14.07, P < 0.01), test (F3, 36 = 7.38, P< 0.001) and test x treatment interaction (F3, 36 = 4.71, P< 0.01). Post-hoc analysis revealed that fear memory retention of the RU 38486-injected group was significantly lower at Test 2 (169.02 ± 57.74 s), Test 3 (170.57 ± 63.97 s) and Test 4 (228.74 ± 73.6 s) compared to Test 1 (RU 38486: 478.32 ± 41.38 s; P < 0.001; Veh: 503.66 ± 27.84 s) and to the vehicle-injected group at the same testing times (Test 2: 464.11 ± 55.42 s; Test 3: 445.78 ± 70.24 s; Test 4: 540 ± 0 s, P < 0.01 for each comparison). Moreover, the retention of all rats from the vehicle-injected group that received the reminder shock exceeded the cut-off time used in our testing (540 s) at Test 4, demonstrating that, as expected, the reminder strengthened the memory in animals that did not receive RU 38486. Together, these results indicate that a dose-dependent inhibition of GRs by administration of the antagonist RU 38486 directly into the BLA following the reactivation of a traumatic memory blocks reconsolidation and persistently disrupts the memory, which is not recovered following subsequent reminders.
Figure 4. GR-antagonist-dependent disruption of a reactivated IA memory is long-lasting and memory does not recover following a shock reminder.
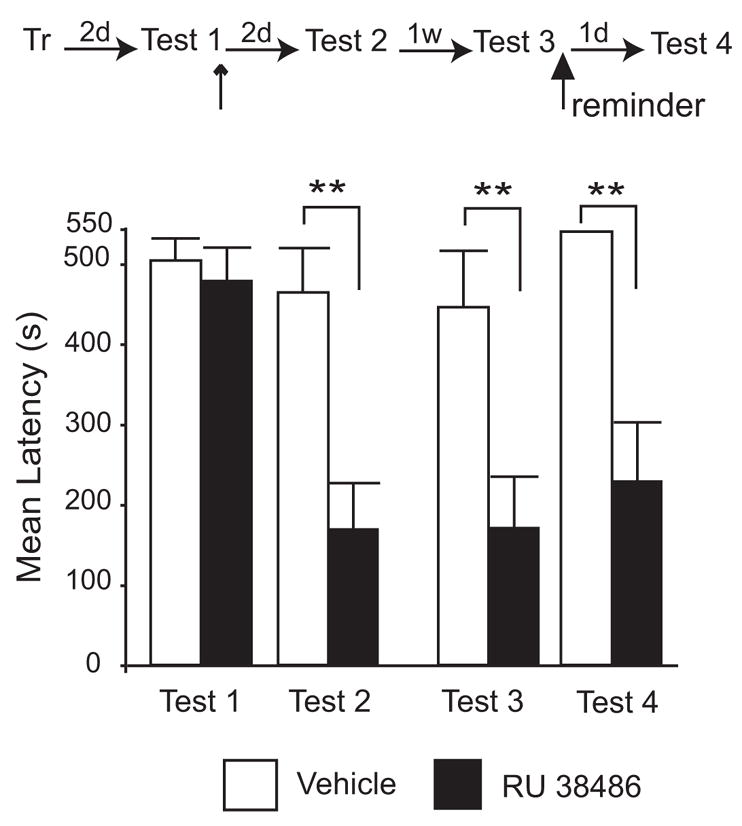
Latency to enter the shock chamber was taken as a measure of memory retention. Rats were trained in IA (Tr) and memory was recalled by testing 2 days later (Test 1). Immediately after Test 1, 1.2 ng of RU 38486 or the same volume of vehicle was bilaterally injected (↑) into their BLA. IA memory was tested 2 days (Test 2) and 1 weeks later (Test 3). Immediately after Test 3, the rats were exposed to a reminder shock. Memory was tested again 1 day later (Test 4). RU 38486-injected group showed a significantly lower retention at Test 2, Test 3 and Test 4 compared to the vehicle-injected group (**; P < 0.01).
Experiment 4
To exclude that the amnesia caused by the bilateral injection of 1.2 ng of RU38486 is caused by permanent damage or inactivation of the injected regions, we tested whether the amnesic rats could re-learn IA. Permanent damage or inactivation of the BLA would predict that the rats will be unable to re-learn IA, because the acquision of this task requires an intact and functional amygdala (Jellestad and Bakke 1985). As depicted in Figure 5, confirming the results found in Experiment 2, bilateral injection of 1.2 ng of RU 38486 impaired memory retention when tested 48h later (Test 2). All animals had similar latencies at Test 1 (Veh: 480.87 ± 40.05 s; RU 38486: 490.99 ± 29.43 s). A two-way ANOVA that compared treatments and testing time points showed a significant effect of treatment (F1,26 = 36.95; P < 0.001), a significant effect of the testing time (F1,26 = 26.61; P < 0.001) and a significant interaction (F1,26 = 41.07; P < 0.001). A Bonferroni-protected post-hoc test revealed that, at Test 2, the retention levels of the rats injected with RU 38486 were significantly lower (136.11 ± 32.23 s) than their mean Test 1 latencies (490.99 ± 29.43 s P < 0.001) and those injected with vehicle (519.23 ± 14.35 s; P < 0.001). Rats were then retrained in IA and tested 48 h later. Latency score for the RU 38486-injected group were submitted to a one-way ANOVA with repeated measures, which revealed a significant effect of the testing time (F 2,23 = 75.55; P < 0.0001). Retrained rats showed a strong memory retention (540 s). No significant difference was found in the avoidance latencies of rats injected with RU 38486 at Test 1 and Test 3, however, both of these groups were significantly different than that at Test 2 (136.11 ± 32.23 s; P < 0.001). Hence, the impairment caused by amygdala injection of RU 38486 is not due to damage or non-specific inactivation of this region.
Figure 5. The amnesic animals that received bilateral injection of RU 38486 in the BLA after recall can re-learn amygdala-dependent tasks.
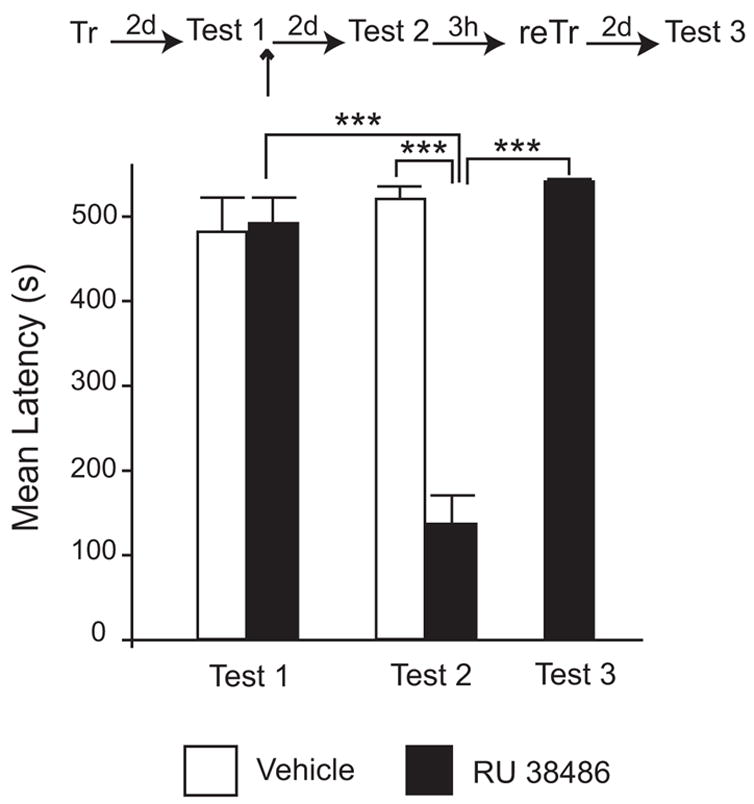
Latency to enter in the shock chamber was taken as a measure of memory retention. Rats were trained in IA (Tr) and memory was recalled by testing 2 days later (Test 1). Immediately after Test 1, 1.2ng of RU 38486 or the same volume of vehicle solution was bilaterally injected (↑) into the BLA. IA memory was tested again 2 days later (Test 2). The RU 38486-injected group was retrained three hours later with IA and tested 2 days later (Test 3). The group that received RU 38486 injections showed memory impairment at Test 2 compared to vehicle-injected or Test 1 (***, P<0.001). When exposed to another IA training session, the amnesic rats re-learned IA. At Test 3, the RU 38486-injected rats showed significant memory retention compared to the impairment found at Test 2 (***, P < 0.001).
Discussion
This study provides evidence for the critical role of the glucocorticoid system in the reconsolidation and maintenance of a fear memory following retrieval. Indeed, IA memory can be persistently disrupted following recall if GRs are inactivated in the BLA in a dose-dependent manner. Indeed, bilateral administration of 1.2 but not 0.3 ng of RU 38486 into the BLA causes amnesia. Interestingly, comparable doses showed similar effects following IA or contextual fear conditioning training, or in conditioned fear extinction (Roozendaal 2000; Donley et al 2005; Yang et al 2006). The resulting amnesia is long-lasting and robust, as it is still observed one week after treatment and remains insensitive to reminders that normally re-instate extinguished fear memories or strongly increase the retention of an established fear. We propose that GR antagonists administered following the reactivation of a traumatic memory could therefore potentially represent a novel treatment for PTSD. Thus, therapy that temporally coordinates memory reactivation and pharmacological intervention may open new therapeutic directions for the treatment of pathogenic memories.
Interestingly, a similar approach based on a combined pharmacological and exposure therapy has been used in clinical studies aiming at enhancing the extinction of fear in phobic patients (Ressler et al 2004, Hoffman et al 2006). It is important to note that both extinction and reconsolidation of a fear memory are evoked by exposure to the same unreinforced conditioned stimulus. In light of this, it could be argued that, in our experiments the GR antagonist treatment strongly and rapidly enhanced extinction rather than disrupting memory reconsolidation. However, it is becoming increasingly clear that reconsolidation and extinction exist as very distinct processes (Pedreira and Maldonado 2003): while the former mediates the re-stabilization of the memory, the latter produces a new second memory that is stored without destroying the old one but rather suppresses its expression (Bouton 2004). In agreement with this view, characteristic features of extinction are that (i) the original memory can re-emerge if the subject is again presented with the US alone (reinstatement) and (2) the extinguished memory recovers spontaneously (Bouton 2004). Because, in fact, neither reinstatement of the memory following the presentation of a strong shock reminder nor spontaneous recovery was observed in our experiments, we propose that the effect produced by RU 38486 disrupts a process of memory reconsolidation, that is, it affects the stabilization of the memory itself.
Moreover, this conclusion is also supported by two sets of previously reported observations. First, RU 38486 administered in the amygdala either before or after training disrupts memory consolidation. Since extinction reflects a new learning (Quirk et al 2006), it would be predicted that RU38486 would block rather than enhance extinction. Second, a recent study by Yang et al (2006) demonstrated that indeed intra-amygdala administration of RU 38486 impairs extinction of conditioned fear in rats.
Nevertheless, we cannot exclude that the memory impairment observed in our experiments might be due to a retrieval failure rather than an interference with the memory reconsolidation process (Sara 2000; Duvarci and Nader 2004). Our hypothesis favors the latter, as IA memory impairment was evident at testing times that were remote from the time of treatment, at which the direct effect of RU 38486 was no longer a factor. Further preclinical studies should help clarify this issue.
The finding that post-retrieval amnesia induced by the RU 38486 treatment persists even after further reminders of the trauma is important for clinical applications. Even when exposed to a strong reminder (a shock of the same intensity), no recovery of IA memory retention was observed in our amnesic animals, whereas the vehicle-injected rats strongly increased their avoidance, which reached the maximum retention score. In contrast, previous studies have shown that a variety of reminders are effective in reinstating memories following amnesic treatments that targeted consolidation, reconsolidation or extinction of a conditioned response. (Lewis 1976; Miller and Kraus 1977; Mactutus and Riccio 1978; Mactutus et al 1980; Vianna et al 2001; Anokhin et al 2002; Millin et al 2001). However, it is possible that in the consolidation and reconsolidation experiments, the amnesic treatment may not have been sufficiently effective, and therefore may have caused a temporal shift rather than a disruption of the memory. For example, experiments using protein synthesis inhibitors argue in favor of the need for sufficient blockade of protein synthesis because inadequate blockade following fear memory reactivation may result only in temporary amnesia (Milekic et al 2006).
Before applying the strategy suggested by our results to patients with PTSD, it should be emphasized that we administered RU 38486 directly into the rat amygdala, a route of administration not appropriate in humans. RU 38486 has been reported to be safe and effective when given to patients with psychotic depression and bipolar disorder (Flores et al 2006, DeBattista and Belanoff 2006), but it is unclear whether acute doses of systemic RU 38486 would successfully impede reconsolidation of traumatic memories in PTSD patients undergoing exposure therapy. An important issue that needs to be understood is, in fact, the degree of permeability of RU 38486 through the blood-brain barrier. Because it has been shown that RU 38486 poorly penetrates the blood-brain barrier (Heikinheimo and Kekkonen 1993; Donley et al 2005), it seems that very high doses would need to be administered systemically, which would greatly increase the probability of side effects. Thus, it seems that there is an impellent need for the development and testing of specific GR antagonists with high blood-bran barrier permeability.
A combination of further clinical and preclinical studies should help to answer these questions.
Acknowledgments
The authors thank Jack Gorman (McLean Hospital and Harvard Medical School) for helpful discussions and comments on the manuscript, Stephen Taubenfeld for editing the manuscript, Catia Proenca, Gabriella Pollonini for technical help and Reginald Miller and the CCMS facility of Mount Sinai. This work was supported by the National Institute of Mental Health (R01 MH65635) and Hirschl Foundation to CMA. The authors have no conflict of interest with the results shown in this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alberini CM. Mechanisms of memory stabilization: are consolidation and reconsolidation similar or distinct processes? Trend Neurosci. 2005;28:51–56. doi: 10.1016/j.tins.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Allen BD, Sutanto W, Jones MT. A correlative study of RU38486 biopotency and competition with [3H]dexamethasone for receptors in the rat central nervous system. J Steroid Biochem. 1988;30:411–415. doi: 10.1016/0022-4731(88)90133-1. [DOI] [PubMed] [Google Scholar]
- Anokhin KV, Tiunova AA, Rose SP. Reminder effects - reconsolidation or retrieval deficit? Pharmacological dissection with protein synthesis inhibitors following reminder for a passive-avoidance task in young chicks. Eur J Neurosci. 2002;15:1759–1765. doi: 10.1046/j.1460-9568.2002.02023.x. [DOI] [PubMed] [Google Scholar]
- Asnis GM, Kohn SR, Henderson M, Brown NL. SSRIs versus non-SSRIs in post-traumatic stress disorder: an update with recommendations. Drugs. 2004;64:383–404. doi: 10.2165/00003495-200464040-00004. [DOI] [PubMed] [Google Scholar]
- Bouton ME. Context and behavioral processes in extinction. Learn Mem. 2004;11:485–494. doi: 10.1101/lm.78804. [DOI] [PubMed] [Google Scholar]
- Brogden RN, Goa KL, Faulds D. Mifepristone. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs. 1993;45:384–409. doi: 10.2165/00003495-199345030-00007. Erratum in: Drugs 1993, 46:268. [DOI] [PubMed] [Google Scholar]
- Bustos SG, Maldonado H, Molina VA. Midazolam disrupts fear memory reconsolidation. Neuroscience. 2006;139:831–842. doi: 10.1016/j.neuroscience.2005.12.064. [DOI] [PubMed] [Google Scholar]
- Deacon BJ, Abramowitz JS. Cognitive and behavioral treatments for anxiety disorders: a review of meta-analytic findings. J Clin Psychol. 2004;60:429–441. doi: 10.1002/jclp.10255. [DOI] [PubMed] [Google Scholar]
- DeBattista C, Belanoff J. The use of mifepristone in the treatment of neuropsychiatric disorders. Trends Endocrinol Metab. 2006;17:117–121. doi: 10.1016/j.tem.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Debiec J, LeDoux JE, Nader K. Cellular and systems reconsolidation in the hippocampus. Neuron. 2002;36:527–538. doi: 10.1016/s0896-6273(02)01001-2. [DOI] [PubMed] [Google Scholar]
- Debiec J, Ledoux JE. Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience. 2004;129:267–272. doi: 10.1016/j.neuroscience.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Donley MP, Schulkin J, Rosen JB. Glucocorticoid receptor antagonism in the basolateral amygdala and ventral hippocampus interferes with long-term memory of contextual fear. Behav Brain Res. 2005;164:197–205. doi: 10.1016/j.bbr.2005.06.020. [DOI] [PubMed] [Google Scholar]
- Duvarci S, Nader K. Characterization of fear memory reconsolidation. J Neurosci. 2004;24:9269–9275. doi: 10.1523/JNEUROSCI.2971-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzinga BM, Bremner JD. Are the neural substrates of memory the final common pathway in posttraumatic stress disorder (PTSD)? J Affect Disord. 2002;70:1–17. doi: 10.1016/s0165-0327(01)00351-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores BH, Kenna H, Keller J, Solvason HB, Schatzberg AF. Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychofarmacology. 2006;31:628–636. doi: 10.1038/sj.npp.1300884. [DOI] [PubMed] [Google Scholar]
- Foa EB. Psychosocial therapy for posttraumatic stress disorder. J Clin Psychiatry. 2006;67 (Suppl 2):40–45. [PubMed] [Google Scholar]
- Frenkel L, Maldonado H, Delorenzi A. Memory strengthening by a real-life episode during reconsolidation: an outcome of water deprivation via brain angiotensin II. Eur J Neurosci. 2005;22:1757–1766. doi: 10.1111/j.1460-9568.2005.04373.x. [DOI] [PubMed] [Google Scholar]
- Grillon C, Southwick SM, Charney DS. The psychobiological basis of posttraumatic stress disorder. Mol Psychiatry. 1996;1:278–297. [PubMed] [Google Scholar]
- Heikinheimo O, Kekkonen R. Dose-response relationships of RU 486. Ann Med. 1993;25:71–76. doi: 10.3109/07853899309147861. [DOI] [PubMed] [Google Scholar]
- Hofmann SG, Meuret AE, Smits JA, Simon NM, Pollack MH, Eisenmenger K, Shiekh M, Otto MW. Augmentation of exposure therapy with D-cycloserine for social anxiety disorder. Arch Gen Psychiatry. 2006;63:298–304. doi: 10.1001/archpsyc.63.3.298. [DOI] [PubMed] [Google Scholar]
- Jellestad FK, Bakke HK. Passive avoidance after ibotenic acid and radio frequency lesions in the rat amygdala. Physiol Behav. 1985;34:299–305. doi: 10.1016/0031-9384(85)90119-2. [DOI] [PubMed] [Google Scholar]
- Judge ME, Quartermain D. Characteristics of retrograde amnesia following reactivation of memory in mice. Physiol Behav. 1982;28:585–590. doi: 10.1016/0031-9384(82)90034-8. [DOI] [PubMed] [Google Scholar]
- Lattal KM, Abel T. Behavioral impairments caused by injections of the protein synthesis inhibitor anisomycin after contextual retrieval reverse with time. Proc Natl Acad Sci U S A. 2004;101:4667–4672. doi: 10.1073/pnas.0306546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DJ. A cognitive approach to experimental amnesia. Am J Psychol. 1976;89:51–80. [PubMed] [Google Scholar]
- Liberzon I, Taylor SF, Amdur R, Jung TD, Chamberlain KR, Minoshima S, Koeppe RA, Fig LM. Brain activation in PTSD in response to trauma-related stimuli. Biol Psychiatry. 1999;45:817–826. doi: 10.1016/s0006-3223(98)00246-7. [DOI] [PubMed] [Google Scholar]
- Mactutus CF, Ferek JM, Riccio DC. Amnesia induced by hyperthermia: an unusually profound, yet reversible, memory loss. Behav Neural Biol. 1980;3:260–277. doi: 10.1016/s0163-1047(80)91150-4. [DOI] [PubMed] [Google Scholar]
- Mactutus CF, Riccio DC. Hypotermia-inducedretrogrde amnesia. Role of body temperature in memory retrieval. Physiol Psychol. 1978;6:18–22. [Google Scholar]
- McGaugh JL, Roozendaal B. Role of adrenal stress hormones in forming lasting memories in the brain. Curr Opin Neurobiol. 2002;12:205–210. doi: 10.1016/s0959-4388(02)00306-9. [DOI] [PubMed] [Google Scholar]
- Milekic MH, Brown SD, Castellini C, Alberini CM. Persistent disruption of an established morphine conditioned place preference. J Neurosci. 2006;26:3010–3020. doi: 10.1523/JNEUROSCI.4818-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RR, Kraus JN. Somatic and autonomic indexes of recovery from electroconvulsive shock-induced amnesia in rats. J Comp Physiol Psychol. 1977;91:434–442. doi: 10.1037/h0077326. [DOI] [PubMed] [Google Scholar]
- Miller MM, McEwen BS. Establishing an Agenda for Translational Research on PTSD. Ann N Y Acad Sci. 2006;1071:294–312. doi: 10.1196/annals.1364.023. [DOI] [PubMed] [Google Scholar]
- Millin PM, Moody EW, Riccio DC. Interpretations of retrograde amnesia: old problems redux. Nat Rev Neurosci. 2001;2:68–70. doi: 10.1038/35049075. [DOI] [PubMed] [Google Scholar]
- Nader K. Memory traces unbound. Trends Neurosci. 2003;26:65–72. doi: 10.1016/S0166-2236(02)00042-5. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Orr SP, Metzger LJ, Pitman RK. Psychophysiology of post-traumatic stress disorder. Psychiatr Clin North Am. 2002;25:271–293. doi: 10.1016/s0193-953x(01)00007-7. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, Fluttert M, Sutanto W, de Kloet ER. Continuous blockade of brain glucocorticoid receptors facilitates spatial learning and memory in rats. Eur J Neurosci. 1998;10:3759–3766. doi: 10.1046/j.1460-9568.1998.00381.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotactic coordinates. New York: Academic; 1998. [Google Scholar]
- Pedreira ME, Maldonado H. Protein synthesis subserves reconsolidation or extinction depending on reminder duration. Neuron. 2003;38:863–869. doi: 10.1016/s0896-6273(03)00352-0. [DOI] [PubMed] [Google Scholar]
- Prado-Alcala RA, Diaz Del Guante MA, Garin-Aguilar ME, Diaz-Trujillo A, Quirarte GL, McGaugh JL. Amygdala or hippocampus inactivation after retrieval induces temporary memory deficit. Neurobiol Learn Mem. 2006 doi: 10.1016/j.nlm.2006.01.006. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Protopopescu X, Pan H, Tuescher O, Cloitre M, Goldstein M, Engelien W, Epstein J, Yang Y, Gorman J, LeDoux J, Silbersweig D, Stern E. Differential time courses and specificity of amygdala activity in posttraumatic stress disorder subjects and normal control subjects. Biol Psychiatry. 2005;57:464–473. doi: 10.1016/j.biopsych.2004.12.026. [DOI] [PubMed] [Google Scholar]
- Przybyslawski J, Roullet P, Sara SJ. Attenuation of emotional and nonemotional memories after their reactivation: role of beta adrenergic receptors. J Neurosci. 1999;19:6623–6628. doi: 10.1523/JNEUROSCI.19-15-06623.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Garcia R, Gonzalez-Lima F. Prefrontal Mechanisms in Extinction of Conditioned Fear. Biol Psychiatry. 2006 doi: 10.1016/j.biopsych.2006.03.010. May 17 Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Rasmusson AM, Charney DS. Animal models of relevance to PTSD. Ann N Y Acad Sci. 1997;821:332–351. doi: 10.1111/j.1749-6632.1997.tb48290.x. [DOI] [PubMed] [Google Scholar]
- Rau V, DeCola JP, Fanselow MS. Stress-induced enhancement of fear learning: an animal model of posttraumatic stress disorder. Neurosci Biobehav Rev. 2005;29:1207–1223. doi: 10.1016/j.neubiorev.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Rothbaum BO, Tannenbaum L, Anderson P, Graap K, Zimand E, Hodges L, Davis M. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61:1136–1144. doi: 10.1001/archpsyc.61.11.1136. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McGaugh JL. Glucocorticoid receptor agonist and antagonist administration into the basolateral but not central amygdala modulates memory storage. Neurobiol Learn Mem. 1997;67:176–179. doi: 10.1006/nlme.1996.3765. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Williams CL, McGaugh JL. Glucocorticoid receptor activation in the rat nucleus of the solitary tract facilitates memory consolidation: involvement of the basolateral amygdala. Eur J Neurosci. 1999;11:1317–1323. doi: 10.1046/j.1460-9568.1999.00537.x. [DOI] [PubMed] [Google Scholar]
- Roozendaal B. 1999 Curt P. Richter award. Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology. 2000;25:213–38. doi: 10.1016/s0306-4530(99)00058-x. [DOI] [PubMed] [Google Scholar]
- Roozendaal B. Stress and memory: opposing effects of glucocorticoids on memory consolidation and memory retrieval. Neurobiol Learn Mem. 2002;78:578–595. doi: 10.1006/nlme.2002.4080. [DOI] [PubMed] [Google Scholar]
- Rothbaum BO, Astin MC, Marsteller F. Prolonged Exposure versus Eye Movement Desensitization and Reprocessing (EMDR) for PTSD rape victims. J Trauma Stress. 2005;18:607–616. doi: 10.1002/jts.20069. [DOI] [PubMed] [Google Scholar]
- Salinska E, Bourne RC, Rose SP. Reminder effects: the molecular cascade following a reminder in young chicks does not recapitulate that following training on a passive avoidance task. Eur J Neurosci. 2004;19:3042–3047. doi: 10.1111/j.0953-816X.2004.03407.x. [DOI] [PubMed] [Google Scholar]
- Sandi C, Rose SP. Corticosteroid receptor antagonists are amnestic for passive avoidance learning in day-old chicks. Eur J Neurosci. 1994;6:1292–1297. doi: 10.1111/j.1460-9568.1994.tb00319.x. [DOI] [PubMed] [Google Scholar]
- Sandi C. The role and mechanisms of action of glucocorticoid involvement in memory storage. Neural Plast. 1998;6:41–52. doi: 10.1155/NP.1998.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara SJ. Retrieval and reconsolidation: toward a neurobiology of remembering. Learn Mem. 2000;7:73–84. doi: 10.1101/lm.7.2.73. [DOI] [PubMed] [Google Scholar]
- Shalev AY, Ragel-Fuchs Y, Pitman RK. Conditioned fear and psychological trauma. Biol Psychiatry. 1992;31:863–865. doi: 10.1016/0006-3223(92)90113-e. [DOI] [PubMed] [Google Scholar]
- Sherry JM, Hale MW, Crowe SF. The effects of the dopamine D1 receptor antagonist SCH23390 on memory reconsolidation following reminder-activated retrieval in day-old chicks. Neurobiol Learn Mem. 2005;83:104–112. doi: 10.1016/j.nlm.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Shin LM, Kosslyn SM, McNally RJ, Alpert NM, Thompson WL, Rauch SL, Macklin ML, Pitman RK. Visual imagery and perception in posttraumatic stress disorder. A positron emission tomographic investigation. Arch Gen Psychiatry. 1997;54:233–241. doi: 10.1001/archpsyc.1997.01830150057010. [DOI] [PubMed] [Google Scholar]
- Shin LM, Wright CI, Cannistraro PA, Wedig MM, McMullin K, Martis B, Macklin ML, Lasko NB, Cavanagh SR, Krangel TS, et al. A functional magnetic resonance imaging study of amygdala and medial prefrontal cortex responses to overtly presented fearful faces in posttraumatic stress disorder. Arch Gen Psychiatry. 2005;62:273–281. doi: 10.1001/archpsyc.62.3.273. [DOI] [PubMed] [Google Scholar]
- Siegmund A, Wotjak CT. Toward an animal model of posttraumatic stress disorder. Ann N Y Acad Sci. 2006;1071:324–34. doi: 10.1196/annals.1364.025. [DOI] [PubMed] [Google Scholar]
- Taubenfeld SM, Milekic MH, Monti B, Alberini CM. The consolidation of new but not reactivated memory requires hippocampal C/EBPbeta. Nat Neurosci. 2001;4:813–818. doi: 10.1038/90520. [DOI] [PubMed] [Google Scholar]
- Teutsch G, Costerousse G, Deraedt R, Benzoni J, Fortin M, Philibert D. 17 alpha-alkynyl-11 beta, 17-dihydroxyandrostane derivatives: a new class of potent glucocorticoids. Steroids. 1981;38:651–665. doi: 10.1016/0039-128x(81)90084-2. [DOI] [PubMed] [Google Scholar]
- Tronel S, Milekic MH, Alberini CM. Linking new information to a reactivated memory requires consolidation and not reconsolidation mechanisms. PLoS Biol. 2005;3:e293. doi: 10.1371/journal.pbio.0030293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson NC, Wiseman SL, Olausson P, Taylor JR. Bidirectional behavioral plasticity of memory reconsolidation depends on amygdalar protein kinase A. Nat Neurosci. 2006;9:167–169. doi: 10.1038/nn1628. [DOI] [PubMed] [Google Scholar]
- Vianna MR, Izquierdo LA, Barros DM, de Souza MM, Rodrigues C, Sant'Anna MK, Medina JH, Izquierdo I. Pharmacological differences between memory consolidation of habituation to an open field and inhibitory avoidance learning. Braz J Med Biol Res. 2001;34:233–240. doi: 10.1590/s0100-879x2001000200011. [DOI] [PubMed] [Google Scholar]
- Yang YL, Chao PK, Lu KT. Systemic and intra-amygdala administration of glucocorticoid agonist and antagonist modulate extinction of conditioned fear. Neuropsychopharmacology. 2006;31:912–924. doi: 10.1038/sj.npp.1300899. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Biology of posttraumatic stress disorder. J Clin Psychiatry. 2001;62(Suppl 17):41–46. [PubMed] [Google Scholar]