

Abstract
We hypothesized that progressive decline in myocardial performance would correlate with upregulation of markers for apoptotic mechanisms following increased duration of polymicrobial sepsis in the rat. Male Sprague-Dawley rats (350-400 g) were randomized into sham, 1-, 3- and 7-day sepsis groups. Each septic rat received 200 mg/kg cecal inoculum intraperitoneally (i.p). The post-mortem analysis showed a severely inflamed peritoneum with the presence of pus in all septic animals that was directly proportional to the duration of sepsis. We observed 10, 33 and 42% mortality in the 1-, 3- and 7-day sepsis groups, respectively. Septic animals at 3 and 7 days exhibited an increased wet lung/total body weight and heart weight/total body weight. A significant increase in total cardiac troponin I (cTnI) and C Reactive Protein (CRP) and endothelin-1 (ET-1) was also observed with an increased duration of sepsis. Myocardial ET-1 concentration in the 7-day post-sepsis group was significantly elevated compared to the sham and 1-day post-sepsis groups. Sepsis also produced a significant decrease in the mean arterial pressure in the 7-day post-sepsis group and tachycardia in the 1-, 3-, and 7-day post-sepsis groups compared to the sham group. A significant prolongation of the left ventricular isovolumic relaxation rate constant, tau, and left ventricular end-diastolic pressure in the 1-, 3- and 7-day post-sepsis groups compared to the sham group was observed. In addition, a significant decrease in the rates of left ventricular relaxation (−dP/dt) and contraction (+dP/dt) in the 3- and 7-day post-sepsis groups compared to the sham and 1-day post-sepsis group was observed. Sepsis produced a significant upregulation in the expression of myocardial TRADD, cytosolic active caspase-3, the Bax/Bcl2 ratio, and the mitochondrial release of cytochrome C in the 3- and 7-day post-sepsis groups. We observed a progressive increase in the number of TUNEL positive nuclei, cytosolic caspase-3 activation and co-localization of PARP in the nuclei at 1, 3 and 7 days post-sepsis. These data suggest that the progression of sepsis from 1 day to 3-7 days produce distinct cardiodynamic characteristics with a more profound effect during later stages. The sepsis-induced decline in myocardial performance correlates with the induction of myocardial apoptosis.
Keywords: Apoptosis, caspase-3, polymicrobial sepsis, left ventricle, MAPK, rat
Introduction
Sepsis is known as a circulatory pathology arising as sequelae to a serious systemic infection. The incidence of sepsis has increased during the last 20 years, with more than 500,000-1 million patients developing sepsis each year in the United States. According to the National Vital Statistics Report (2004), sepsis (including septicemia and bacteremia) is now the 10th leading cause of death in the United States, as opposed to being the 13th leading cause in 1990 (Kochanek and Smith, 2004). The mortality from septic shock syndrome ranges from 20% to 90%, depending upon the patient's age and associated pathologies (Wenzel and Edmond, 2001). Using clinical markers for myocardial damage such as cardiac troponin I (cTnI), various research laboratories have suggested that almost 50% of septic patients exhibit myocardial dysfunction in ICUs (Charpentier et al., 2004; Fernandes et al., 1999; Maeder et al., 2006).
Sepsis is characterized as a hyperdynamic state, with normal-to-low blood pressure, normal-to-high cardiac index and low systemic vascular resistance (Parrillo et al., 1990; Holcroft et al., 2001), which are considered to be among the major complications in septic patients. Parker et al., (1984, 1990) described the phenomenon of sepsis-induced myocardial dysfunction (SIMD) in septic ICU patients, which is characterized by biventricular impairment of intrinsic myocardial contractility left and right ventricular contractile dysfunction. The clinically relevant polymicrobial septic rat model that has been characterized and standardized in our laboratory produces a prolonged hyperdynamic state of sepsis (Sam II et al., 1997). In this model, sepsis causes an increase in the ventricular filling pressure (left ventricular end diastolic pressure, LVEDP) and the time constant for left ventricular relaxation rate constant (tau) during the hyperdynamic state (that occurs at 1 day post-sepsis), suggesting an early stage of heart failure (Brahmbhatt et al., 2005). However, the cardiodynamic profile during prolonged sepsis in a rat model has not been studied. A number of mechanisms regulated by vasoactive mediators, such as endothelin-1 (ET-1), nitric oxide and mitogen-activated protein kinase pathways, have been proposed to affect myocardial performance during sepsis. Our earlier studies demonstrated that hyperdynamic sepsis produced an elevated concentration of ET-1 up to 12 h post-sepsis, which returned to basal levels at 24 h (Sharma et al., 1997). In vivo, we found that ET-1 precursor bigET-1 produced cardiac dysfunction in the septic rat heart (Brahmbhatt et al., 2005). Using isolated adult rat ventricular myocyte preparation, we demonstrated that bigET-1 produced a decompensatory contractile response via upregulation of p38-MAP phosphorylation, which correlated with an upregulated expression of cytosolic active caspase-3 (Gupta et al., 2004, 2005a). The regulation of molecular signaling and apoptosis in the myocardium during early (1-day, hyperdynamic state) and late (progressive hypodynamic state) sepsis are unknown. Therefore, in the present study, we examined whether prolonged duration of sepsis produces cardiodynamic alterations that correlate with the modulation of various markers of signaling (MAPKs, p38-MAPK, JNK) and apoptotic cascade.
Materials and Methods
Preparation of animal model
Male Sprague-Dawley rats (Harlan, IN, USA) weighing 350-400 g were used in the study. The rats were acclimatized to the laboratory conditions for at least 7 days following their arrival. On the day of the experiment, the rats were anesthetized with an i.p. injection of pentobarbital sodium (Nembutal®, Abbott; 50 mg/kg). All experiments were conducted in compliance with humane animal care standards outlined in the NIH Guide for the Care and Use of Experimental Animals and were approved by the Institute of Animal Care and Use Committee of Baylor College of Dentistry, Texas A&M Health Science Center.
Induction of sepsis
Sepsis was induced in the animals using an i.p. injection of cecal inoculum (200 mg/kg) as described previously (Brahmbhatt et al., 2005; Sharma et al., 1997; Gupta et al., 2004, 2005a,b). Briefly, cecal inoculum was prepared by suspending 200 mg of freshly removed cecal material in 5 mL of sterile 5% dextrose water (D5W). The cecal material was obtained from a healthy donor rat euthanized with Nembutal® (100 mg/kg, i.p.). Sham animals received an i.p. injection of sterile D5 W (5 mL/kg). The cecal inoculum was prepared fresh each day, and cecal material from one donor rat was used within two hours for three to five experimental animals.
Measurement of hemodynamics
The sham and septic animals were laid ventrally and catheterized through a cervical midline incision, right jugular vein and right carotid artery using a polyethylene cannula (PE-50, internal diameter 0.58 mm, external diameter 0.965 mm, Clay Adams Division, Becton Dickinson, Parsippany, NJ). The catheter inserted in the right carotid artery was further advanced to the left ventricle to obtain left ventricular (LV) pressure tracings. Once the presence of the cannula tip in the left ventricle was confirmed (as evident by the decline in diastolic blood pressure, DBP, to near zero values on the waveform tracing on the computer), the cannula was kept in position. Tail artery cannulation was performed for arterial blood pressure recording. The hemodynamic data were collected using pressure transducers (TSD 104A) connected to LV cannula and tail artery cannula. Transducers were connected to a multichannel MP100 system (BIOPAC Systems, Inc., Santa Barbara, CA) attached to a computer. The data were collected at a sampling rate of 1000 Hz. The first derivative of the LV pressure (±dP/dt) was calculated from the LV pressure tracing using the AcqKnowledge™ software. The time constant of the LV isovolumic relaxation (tau) was calculated (Brahmbhatt et al., 2005). LV pressure from the time of maximum negative dP/dt to 5 mmHg above LVEDP was fitted with the monoexponential equation p(t) = p·e−t/T where t = time obtained, e = natural logarithm and p = pressure. For the calculation of tau, the data from 5 consecutive beats for each time point were analyzed and averaged. Prior to acquisition, the transducers were calibrated at 0 mm Hg and 200 mm Hg using a standard mercury sphygmomanometer. The mean arterial pressure (MAP) was calculated by the software as [MAP= (SBP+2·DBP)/3]. The catheters were tunneled subcutaneously and exited at the back of the neck of the animals. The surgical wounds were sutured using sterile non-absorbable silk sutures (Ethicon©, Johnson & Johnson, Somerville, NJ).
Blood samples were obtained from the sham, 1-, 3- and 7-day post-septic animals. Following pentobarbital (100 mg/kg, iv) euthanasia, the animals were sacrificed and the organs (heart and lung) harvested for further biochemical analysis.
Determination of c-Reactive Protein (CRP) and cTnI concentration
The plasma concentration of CRP and cTnI were determined using an ELISA kit obtained from Life Diagnostics, Inc. In brief, the plasma samples were diluted 100-fold for CRP and four-fold for the cTnI experiments. CRP or cTnI HRP conjugate (100 μl) was also added to each well, mixed at 150 rpm and incubated at 25°C for 60 min. After the incubation, the contents of the coated wells were removed and washed five times with distilled water. The wells were dried and incubated with 100 μl of 3, 3′, 5, 5′- tetramethyl benzidine, a substrate for 20 min, and the optical density was read at 450 nm.
Determination of the concentration of ET-1 in plasma and left ventricles
The estimation of the ET-1 concentration in the plasma and LV tissue supernatant was performed using an enzyme immuno assay (EIA) kit for ET-1 obtained from Assay Designs (Ann Arbor, MI) as described earlier (Brahmbhatt et al., 2005; Sharma et al., 1997; Gupta et al., 2004; Gupta et al., 2005a,b).
Immunohistochemistry of LV tissues
Determination of apoptosis using DNA fragmentation technique
Apoptosis detection was performed using the APO-BrdU™ TUNEL Assay Kit (Invitrogen). DNA fragmentation was detected in the paraffin embedded heart tissue sections. After deparaffinization and rehydration of the tissue, its permeability was increased using Proteinase K and 10mM Tris. The endogenous peroxidases were inactivated using methanol prior to the labeling reaction. Following this procedure, the specimen was subjected to the labeling reaction using the terminal deoxynucleotidyl transferase (TdT) enzyme, BrdUTP, and reaction buffer and then incubated for 1.5 h at 37°C. After the incubation, the heart tissue sections were washed with rinse buffer and further incubated for 1 h with antibody staining solution containing Alexa Fluor 488 dye-labeled anti-BrdU antibody. The nuclei were counterstained with TO-PRO to further aid in differentiating the apoptotic cells (green) from the non-apoptotic cells using a Leica SP2 Confocal Microscope.
Determination of cytosolic active caspase-3 and PARP expression using FITC-conjugated antibody
Following the deparaffinization and rehydration procedures on the LV tissue, the specimen was incubated in 10% normal blocking serum in phosphate-buffered saline (PBS) for 20 min to suppress the non-specific binding of IgG. After repeated washing with PBS, the specimen was incubated with primary antibody for caspase-3 or PARP (1:50) along with 1.5% normal blocking serum for 60 min. After the incubation, the specimen was washed with PBS and incubated with goat antirabbit IgG-FITC (1:500) in PBS for 45 min. The specimen was then counterstained with TO-PRO for the nucleus and stored at 4°C to be visualized for confocal microscopy.
Immunoblot analysis
At the end of the experiment, the heart was harvested; LV was isolated and rapidly frozen in liquid nitrogen, homogenized in lysis buffer (containing pepstatin (2 μg/μL), aprotinin (0.1 μg/μL), leupeptin (2 μg/μL), benzamidine (16 μg/μL) and bacitracin (0.5 %) in Tris/glycine buffer), and centrifuged. The protein content in the LV tissue was determined using the Bradford method before performing gel electrophoresis. Tissue sample preparations were separated on 10% denaturing sodium dodecyl sulfate (SDS) polyacrylamide gels. A prestained marker (Kaleidoscope prestained standards, Biorad, CA) was run with each gel to determine the molecular weight of the bands. The proteins were transferred to polyvinylidene difluoride (Gelman Sciences, Rockford, IL) membranes. The membrane was blocked overnight at 4°C with 5% nonfat dry milk in tris buffered saline containing 0.2% Tween 20. The membranes were probed with the primary antibody for 1 h at room temperature. The blots were then washed and incubated with the secondary antibody for 1 h at room temperature. The specific proteins were detected by enhanced chemiluminescence (ECL detection reagent, Amersham Pharmacia Biotech). Equal protein loading was confirmed using β-actin blots. The blots were analyzed using UN-SCAN-IT™ software (Silk Scientific Inc., Orem, UT) to measure the density of the blots in pixels. The data were reported as fold change for each protein expression, normalized to β-actin in each gel.
Determination of the mitochondrial release of cytochrome C
The cytosolic and mitochondrial fractions of the LV tissue were isolated using a mitochondrial isolation kit for tissues (Pierce, Rockford, IL). Briefly, the tissue was homogenized on ice using a tissue grinder and centrifuged at 1000 ×g for 3 min at 4°C. The pellet was suspended in 800 μl of BSA in reagent A solution and vortexed for 2 min. Subsequently, mitochondrial isolation reagent B (10 μl) and reagent C (800 μl) were added and centrifuged at 700×g for 10 min at 4°C. The pellet was discarded, and the supernatant was transferred to a new tube. The supernatant was centrifuged at 3000×g for 15 min at 4°C to obtain the cytosolic fraction. The mitochondrial pellet obtained was stored at 4°C for further analyses.
The protein concentration in the mitochondrial and cytosolic fractions of the LV tissues was determined using the Bradford method. The proteins (15 μg/well) were loaded and electrophoresed. The purity of the mitochondrial and cytosolic fractions was determined using HSP-60 (rabbit polyclonal IgG, Santa Cruz, CA) and β-tubulin (goat polyclonal IgG, Santa Cruz, CA). The expression of cytochrome C and β-actin were determined in a separate gel. The blots were analyzed using UN-SCAN-IT™ software (Silk Scientific Inc., Orem, UT) to measure the density of the blots in pixels.
Chemicals and antibodies
The following primary antibodies were obtained from Santa Cruz Biotechnology, CA: mouse monoclonal p38-MAPK antibody; phosphorylated p38-MAPK: rabbit monoclonal antibody; JNK: rabbit polyclonal antibody; phosphorylated JNK: rabbit polyclonal antibody; Bax: rabbit polyclonal antibody; Total and Phosphorylated Bcl2: rabbit polyclonal antibody; TRADD: rabbit polyclonal antibody; Cyto c: rabbit polyclonal IgG antibody; Caspase-3: rabbit polyclonal IgG antibody; PARP: rabbit polyclonal IgG antibody; HSP-60: rabbit polyclonal IgG antibody; β-tubulin: rabbit polyclonal IgG antibody; β-actin: Mouse monoclonal antibody. The following secondary antibodies were obtained from Santa Cruz Biotechnology, CA: goat antirabbit IgG-HRP; goat antirabbit IgG-FITC. Sheep antimouse IgG antibody was purchased from MP Biomedicals, Inc., Germany.
Statistical Analyses
The hemodynamic and protein expression data were analyzed using two-way ANOVA repeated or one-way ANOVA (using SPSS software). After obtaining a significant F-value, a post hoc multiple range Student-Newman-Keuls test was performed. A probability value p ≤ 0.05 was considered statistically significant.
Results
Effect of sepsis on animal characteristics and biomarkers
The septic animals exhibited persistent piloerection, periocular discharge and lethargic movement, decreased food and water intake along with loose stools. In contrast, the vehicle-treated sham animals freely moved around and showed no signs of distress. The post-mortem analysis of the peritoneal cavity from the vehicle-treated septic animals revealed the presence of ascites and infarcts on the peritoneal organ. With the progression of infection, several peritoneal organs, including the liver, were found severely damaged (Fig. 1A). Sepsis produced 10, 33 and 42% mortality in the 1-, 3- and 7-day post-sepsis groups, respectively (Fig. 1B).
Figure 1.

A Chronic peritonitis as seen during postmortem examination of peritoneal cavity following polymicrobial sepsis in sham, 1-, 3- and 7-days post-septic rat. B. Percentage animals survived at 1, 3 and 7-days post-sepsis. SP-1D, 1-day sepsis; SP-3D, 3-days sepsis; SP-7D, 7-days sepsis.
Sepsis produced a significant increase in the plasma concentration of lactate, cTnI and CRP in the 1-, 3- and 7-day post sepsis groups. The plasma concentration of cTnI at 3 and 7 days post-sepsis significantly increased in the 3- and 7-day post-sepsis groups compared to the 1-day post-sepsis group. Sepsis also produced an increase in the plasma and LV concentration of ET-1 in the 3- and 7-day post-sepsis groups compared to the sham and 1-day post-sepsis groups (Table 1, N=6 in each group).
Table 1.
The effect of sepsis on hemodynamics and the concentration of various clinical and experimental biomarkers in sham, 1-day, 3-days and 7-days post-sepsis groups. Heart rate, HR; Mean arterial pressure, MAP; rat cardiac troponin I, cTnI; C-reactive protein, CRP; endothelin-1, ET-1.
| Hemodynamic charactristics and clinical parameters | ||||
|---|---|---|---|---|
| Parameters | Non-septic | Sepsis | ||
| Sham | 1-day | 3-days | 7-days | |
| HR (beats/min) | 355±13 | 457±21* | 409±13* | 449±7* |
| MAP (mmHg) | 116±5 | 104±6 | 101±4 | 91±3* |
| Clinical and experimental biomarkers | ||||
| cTnI (ng/ml) | 2.50±0.30 | 5.95±1.76* | 4.51±1.67* | 8.34±4.39* |
| CRP(ng/ml) | 0.15±0.04 | 0.61±0.06* | 0.37±0.05* | 0.47±0.15* |
| Plasma ET-1 (pg/ml) |
0.84±0.05 | 0.79±0.08 | 5.67±2.07* | 12.8±6.99* |
| Heart tissue ET-1 (pg/g) |
208±20 | 320±45 | 1037±331*# | 1251±84*# |
p<0.05 Vs. Sham
p<0.05 Vs. 1-day sepsis group, N=6 in each group.
Effect of sepsis on hemodynamics
Sepsis produced tachycardia in the 1-, 3- and 7-day post-sepsis groups compared to the sham group. The MAP was not altered in the 1- and 3-day post-sepsis groups but significantly decreased in the 7-day post-sepsis group compared to the sham group (Table 1). Sepsis produced a significant increase in the LVEDP in the 1-, 3- and 7-day post-sepsis groups compared to the sham groups (Fig. 2). The tau was significantly elevated in the 3- and 7-day post-sepsis groups compared to the sham as well as the 1-day post-sepsis group (Fig. 2). We also found depressed rates of LV contraction and relaxation at 3 days and 7 days post-sepsis compared to the sham and 1-day sepsis (Fig. 2).
Figure 2.

The effect of sepsis on cardiodynamics in sham, 1-, 3- and 7-day post-sepsis groups. LV isovolumic relaxation rate constant, tau; LV end diastolic pressure, LVEDP; first derivative of LV contraction, +dP/dt and first derivative of LV relaxation, −dP/dt. *p ≤ 0.05 as compared to the sham group, #p ≤ 0.05 as compared to the 1-day sepsis group. SP-1D, 1-day sepsis; SP-3D, 3-days sepsis; SP-7D, 7-days sepsis.
Effect of sepsis on markers of apoptosis cascades
To determine the effect of sepsis on the activation of the apoptotic cascade, we examined the expression of various proteins that play a key role in the induction of apoptosis in the myocardium. The increased duration of sepsis (from 1 to 7 days) upregulated the expression of myocardial active caspase-3 compared to the sham group (Fig. 3). Significant increases in the ratio of caspase-3 and procaspase-3 were observed in the 3- and 7-day post-sepsis groups compared to the sham and 1-day groups (Fig. 3). Sepsis produced a significant increase in myocardial TRADD and Bax expression, but down-regulated BCl2 in the 3- and 7-day post-sepsis groups compared to the 1-day post-sepsis and the sham groups (Fig. 4). A significant increase in mitochondrial-dependent apoptosis (ratio between Bax and BCl2) was observed (Fig. 4).
Figure 3.

Upper panel exhibits representative blot (from 5 experiments) for the expression of LV procaspase and active caspase-3 in sham, 1-, 3- and 7-days post sepsis groups. Alterations (fold change) in cytosolic active caspase-3/ procaspase ratio normalized to β-actin in sham, 1-, 3- and 7-days post sepsis groups. *p ≤ 0.05 as compared to the sham group, #p ≤ 0.05 as compared to the 1-day sepsis group.
Figure 4.

Upper panel exhibits representative blot (from 5 experiments) for the expression LV TNF-α receptor associated death domain (TRADD, Bax and BCl2 in sham, 1, 3- and 7-days post-sepsis as compared to sham and 1-day post-sepsis. Alterations (fold change) in Bax/ Bcl2 (lower left panel) and TRADD (lower right panel), normalized to β-actin in sham and septic groups. *p ≤ 0.05 as compared to the sham group, #p ≤ 0.05 as compared to the 1-day sepsis group.
Effect of sepsis on the mitochondrial release of cytochrome C
We observed a higher expression of β-tubulin and HSP60 in the mitochondrial and cytosolic fractions, respectively (Fig. 5A). The increased duration of sepsis produced an incremental decrease in the mitochondrial expression of cytochrome c and an increase in the cytosol (Fig. 5B). A significant increase in the ratio of cytosolic cytochrome C and mitochondrial cytochrome C was seen at 7 days post-sepsis compared to the sham group (Fig. 5C).
Figure 5.

A. Expression of HSP60, β-tubulin in the cytosolic and mitochondrial fractions in LV tissues obtained from sham, 1-, 3- and 7-days post-sepsis as compared to sham and 1-day post-sepsis. B. Representative blot (from 5 experiments) for the expression of Cytochrome C in the cytosolic and mitochondrial fractions of LV tissues. C. Ratio of the expression of cytochrome C normalized to β-actin in the cytosolic and mitochondrial fractions in sham, 1-, 3- and 7-days post-sepsis groups. *p ≤ 0.05 as compared to the sham group.
Effect of sepsis on phosphorylation of p38-MAPK and JNK
Sepsis also upregulated the phosphorylation of p38-MAPK and JNK (Fig. 6) in the 1-, 3- and 7-day post-sepsis groups compared to the sham group.
Figure 6.

Representative blot (from 5 experiments) for the expression of LV p38-MAPK, JNK, phosphorylated p38-MAPK (pp38-MAPK) and phosphorylated JNK (pJNK) in sham, 1-, 3- and 7-days post-sepsis groups. Alterations (fold change) in JNK & pJNK (lower panel), and p38-MAPK & pp38-MAPK (middle panel) normalized to β-actin in sham, 1-, 3- and 7-days post-sepsis groups. *p ≤ 0.05 as compared to the sham group, #p ≤ 0.05 as compared to the 1-day sepsis group.
Effect of sepsis on DNA fragmentation and expression of cytosolic caspase-3
A large number of cells exhibited TUNEL positive nuclei (green co-localized in blue) in the septic groups compared to the sham group (Fig. 7A). To determine whether TUNEL positive cells were cardiac myocytes, NOMARSKY DIC (Differential Interference Contrast) along with confocal microscopy were performed in the same sections. It was observed that the majority of the TUNEL positive cells were non myocytes that were seen in the vicinity of the blood vessels. It was observed thorough the analysis that 2-5% of the myocytes at the most demonstrated TUNEL positive nuclei by the seventh day post-sepsis. When subjected to FITC-conjugated PARP antibody, the LV sections exhibited co-localization of PARP and TO-PRO in the 7-day post-septic group, suggesting DNA breaks in the septic nuclei (Fig 7B). An increased immunofluorescence of caspase-3 distributed throughout the cell cytosol was observed in the septic cardiac myocytes (Fig. 7C).
Figure 7.
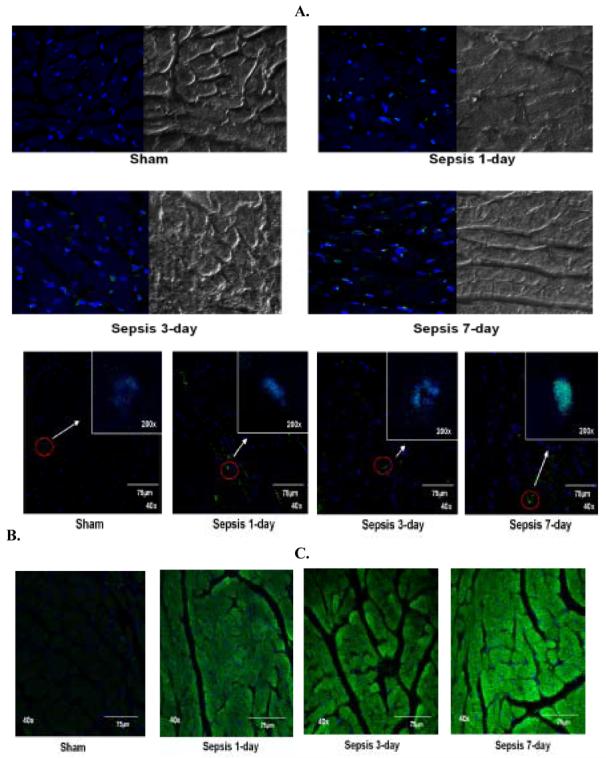
Immunohistochemistry performed in paraffinized LV tissue sections and visualized by using confocal microscopy. A. Representative photomicrographs (magnification 63x) of LV tissue sections from sham, 1-, 3- and 7-day post-sepsis stained for DNA fragmentation using APO-BrdU TUNEL kit (Invitrogen). TUNEL positive cells are stained green (Alexa Flour 488) in blue nuclei (TO-PRO) along with NOMARSKY DIC (Differential Interference Contrast) images of LV tissue sections.
B. Representative pictomicrographs (magnification 40x) of LV tissues from sham, 1-, 3- and 7-days post-sepsis stained for caspase-3 expression using FITC-conjugated caspase-3 antibody. The cytosol of the LV tissue is stained green (FITC) and the nuclei are counterstained blue with TO-PRO.
C. Representative pictomicrographs (magnifications 200x and 40x) of LV tissues from sham, 1-, 3- and 7-days post-sepsis stained for PARP expression using FITCconjugated caspase-3 antibody. The nucleus of the LV tissue exhibits green (FITC) fluorescence depicting DNA breaks in the blue nuclei which are counterstained with TO-PRO.
Discussion
Almost two decades ago, Parker et al., described the phenomenon of SIMD, characterized by left and right ventricular contractile dysfunction, in septic patients in ICU (Parker et al., 1984, 1990). It was found that the duration of hypotension during sepsis (Arlati et al., 2000) and the vasopressors administered had been found to correlate with the plasma concentration of cTnI, a regulatory protein of the thin actin filament of cardiac muscle (Bertichant et al., 1999). cTnI is a well known diagnostic marker for myocardial injury following congestive heart failure and myocardial infarction (Turner et al., 1999). These studies established the diagnostic and prognostic potential of cardiac troponin levels in the assessment of the severity of sepsis expressed by the Acute Physiology and Chronic Health Evaluation II (APACHE II) score (VerElst et al., 2000; Ammann et al., 2001). In the present study we also observed that sepsis not only increased the CRP levels but also elevated the cTnI levels, suggesting that the increased severity of sepsis is associated with myocardial dysfunction.
Clinically, sepsis has been characterized by a hyperdynamic state with normal-to-low blood pressure, a normal-to-high cardiac index, and a low systemic vascular resistance. Although cardiac output is usually maintained in volume-resuscitated septic patients, a number of investigators (Parrillo et al., 1990; Holcroft et al., 2001; Parker et al., 1984, 1990) have demonstrated impairment of cardiac function. This myocardial dysfunction is characterized by decreased ejection fraction, ventricular dilation, impaired contractile response to volume loading, and a low peak systolic pressure to end-systolic volume ratio, a load-independent measure of ventricular function (Holcroft et al., 2001; Schaer et al., 1985; Reinelt et al., 1997). Chronic peritoneal sepsis in our model produces a prolonged hyperdynamic stage of sepsis and has clinical relevance (Sam II et al., 1997). In this model, we observed that the induction of sepsis caused progression of cardiac dysfunction in vivo (Brahmbhatt et al., 2005). Sepsis produced an increase in cardiac output and vascular resistance during the hyperdynamic stage (1 day post-sepsis) of sepsis (Sam II et al., 1997). It was observed that sepsis decreased +dP/dt and −dP/dt in an isolated heart preparation (Sharma et al., 1997). During the hyperdynamic stage, sepsis increased LVEDP and tau (Brahmbhatt et al., 2005). In the present study, we observed that the progression of sepsis from 1 day to 7 days deteriorated the left ventricular performance and caused changes in LVEDP, Tau, −dP/dt and +dP/dt at 3 and 7 days post-sepsis. Although MAP was not altered at 3 days, it decreased at 7 days post-sepsis. These results suggest that sepsis produces a distinct cardiodynamic profile during hyperdynamic (1-day post-sepsis) and a progressive hypodynamic state of sepsis (during 3 to 7 days post-sepsis) in our model.
It appears that SIMD, as seen in a septic rat and human model, is distinct from other pathologies such as myocardial infarction and ischemic reperfusion injury. We have provided evidence that an early stage of heart failure was evident in a non-resuscitated model of sepsis (Brahmbhatt et al., 2005). Classical theories about heart failure, which are primarily related to loss of myocardial cells, were unable to explain the pathophysiology of sepsis-induced heart dysfunction. Apparently, sepsis-induced heart failure could be related to pacing (tachycardia)-induced heart failure; however, the characteristics of both pathologies are distinct. Whereas tachycardia-induced heart failure produces left ventricular myocardial hypertrophy, this condition was not reported in sepsis. The rapid progression of heart dysfunction during sepsis-induced tachycardia could be due to a cumulative physiological response to sympathetic stimulation, elevated levels of cytokines, ET-1, and activation of signaling molecules at cellular level. In the present study, sepsis produced an increase in plasma concentration of ET-1 at 3 and 7 days post-sepsis. Similar to our earlier studies (Sharma et al., 1997), we did not find any change in the ET-1 levels at 1 day post-sepsis. These findings suggest that sepsis produces a triphasic response on ET-1. The first occurs between 4-12 h during the first 24 h post-sepsis (hyperdynamic sepsis), and the second peak is seen at 3-7 days post-sepsis (progressive hypodynamic state of sepsis). In this study, the first peak of plasma ET-1 did not correlate with the levels of myocardial ET-1; however, the second peak not only correlated with elevated myocardial ET-1 but also with progressive myocardial dysfunction and an increase in cTnI. It appears that ET-1 may play a role in the development of SIMD during the hypodynamic state of sepsis.
Signaling and apoptosis mechanisms during SIMD
The MAPK signaling cascade has been implicated in the induction of apoptosis, hypertrophy, and contractile dysfunction (Clerk et al., 1998a, b; Tanaka et al., 2001). MAPKs are a family of serine/threonine protein kinases that consist of three members: p42/p44 extracellular receptor kinases (ERKs), c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK; p54/p46), and p38-MAPK. The p38-MAPK family has four isoforms (α, β, γ and δ) that are phosphorylated and activated by MAP kinase kinase 6 (MKK6) (Nebreda et al., 2000). Under pathological conditions, ERKs, p38-MAPK and JNK are activated by proinflammatory cytokines like interleukin-1 (IL-1) and TNF-α. The phosphorylation of MAPKs (p38-MAPK in particular induces the activation of pretranscription factors, such as activated protein-1, ATF-2, and cellular inhibitory apoptosis protein) can contribute to alterations in cellular contractility, hypertrophy, and apoptosis (Clerk et al., 1998a,b; Nebreda et al., 2000; Kan et al., 2004). Although not directly related to polymicrobial sepsis, lipopolysaccharide (LPS)-induced activation of p38-MAPK and ERK stimulates NOS II in cardiac myocytes (LaPoint et al., 1999). We have demonstrated that the blockade of ET-1 biosynthesis at the time of induction of endotoxemia downregulates the phosphorylation of myocardial p38-MAPK and NOS II (Gupta and Sharma, 2003). Song et al. (2000, 2001) demonstrated that p38-MAPK inhibits immune suppression in splenocytes obtained from the CLP mouse model of polymicrobial sepsis. Although there was no detectable JNK signal from the lymphocytes, they could specifically block the cytokine IL-10 release by the p38-MAPK inhibitor SB203580 (Song et al., 2000, 2001). In another study, the activation of p38-MAPK was demonstrated to induce a negative inotropic effect in ARVM by decreasing the response of myofilaments to Ca2+ during the onset of heart failure (Liao et al., 2002). In addition, during hypertensive end-organ damage, p38-MAPK phosphorylation was found to be related to premature mortality in a rat model of heart failure (Behr et al., 2001). Our laboratory demonstrated that bigET-1 upregulated p38-MAPK, but not ERK phosphorylation, in septic adult rat ventricular myocytes. Further, septic ARVMs failed to show a positive inotropic response to bigET-1 at 24 h, as observed at 3 h and in sham ARVMs (Gupta et al., 2005b). In the current study, sepsis produced an elevated ET-1 at 3 and 7 days post-sepsis, which correlated with increased phosphorylation of both myocardial JNK and p38-MAPK and myocardial dysfunction. These data suggest that activation of a signaling mechanism could be responsible for SIMD.
Besides changes in myocardial contractile function, p38-MAPK and JNK have also been shown to play a role in the induction of apoptosis. Under various pathologies including heart failure and sepsis, two major apoptosis cascades have been identified: the first pathway is activated through the fas-activated TNF receptor-associated death domain (TRADD) receptors and the second pathway via stress-induced stimuli (Bratton et al., 2000). Activation of a cell surface death receptor involves the tumor necrosis receptor super-family (TNF-related apoptosis-inducing ligands), TRAIL, and results in activation of caspase-8, which produces active caspase-3 in the cell cytosol via activation of the BCl2 family of proteins such as Bad and t-Bid (Gonzalez et al., 2003; Oberholzer et al., 2001). On the other hand, stress-induced apoptosis results due to mitochondrial perturbations and release of proapoptotic proteins such as Cyto C and Smac (Macfarlan et al., 2002). Once released, Cyto C binds with Apaf-1, ATP and caspase-9 to form an apoptosome that causes generation of active caspase-3 from its precursor, procaspase-3. It has been suggested that stimulation of JNK and p38-MAPK has a proapoptotic effect, whereas ERK has an anti-apoptotic effect (Gonzalez et al., 2003). Apoptosis signal-regulating kinase -1 (ASK-1), a member of the MAPK family, activates JNK and p38-MAPK phosphorylation and through mitochondria-dependent caspase-3 activation executes apoptosis (Gonzalez et al., 2003). This process involves upregulation of anti-apoptotic BCl2 phosphorylation along with the release of Cyto c (Matsuzawa and Ichijo, 2001). In the presence of p38-MAPK activation, proapoptotic ATF-2 phosphorylates and then translocates nuclear factor kB (NFkB) from the cytosol to the nucleus. NFkB-mediated transcription is also co-activated by Poly (ADP-ribose) polymerase (PARP). Caspase-induced cleavage of PARP (indicating DNA breaks) is implicated in human heart failure studies (Molnar et al., 2006). Phosphorylation at Thr-69 and Thr-71 of ATF-2 is mediated through both p38-MAPK as well as JNK (Raingeaud et al., 2006; Xia et al., 1995). Activation of p38-MAPK-mediated upregulation of BCl2, BCl2 family-related protein Bax, and activation of caspase-3 have been reported to be among the main pathways for apoptosis and hypertrophy in cardiac myocytes obtained from spontaneously hypertensive rats (Cheng et al., 1995; Chien et al., 1999; Hein et al., 2003).
In the present study, we demonstrated that the increased duration of sepsis (from 1 to 7 days) upregulated the expression of myocardial active caspase-3 and the ratio of caspase-3 and procaspase-3 at 3 and 7 days post-sepsis. Sepsis also produced upregulated myocardial TRADD and Bax, but down-regulated BCl2 at 3 and 7 days post-sepsis. A significant increase in mitochondrial-dependent apoptosis (ratio between Bax and BCl2) was observed. We also observed the mitochondrial release of Smac, characterized by an increased expression of Smac in both cytosolic and mitochondrial fractions at 3 and 7 days post-sepsis. These data suggest that 3-day sepsis (that elevated ET-1 in plasma and heart) produced upregulation of mitochondrial-dependent (Bax/BCl2 ratio and release of Smac) and independent (TRADD and phosphorylation of p38-MAPK and JNK) mechanisms, which could be responsible for an increase in cytosolic active caspase-3 and depressed myocardial performance.
A large number of cells exhibited TUNEL positive nuclei (green co-localized in blue) in septic groups compared to the sham group. NOMARSKY DIC (Differential Interference Contrast) along with confocal microscopy revealed that the majority of the TUNEL positive cells were non myocytes and were seen in the vicinity of the blood vessels. It was observed during analysis that 2-5% myocytes at the most demonstrated TUNEL positive nuclei by 7 days post-sepsis. These data are consistent with an in vivo study, in which 8% apoptotic nuclei were reported in a global cardiac ischemia reperfusion injury model in the rabbit (Yeh et al., 2006). An increased immunofluorescence of caspase-3 distributed throughout the cell cytosol was observed in the septic LV sections. When subjected to FITC-conjugated PARP antibody, LV sections exhibited co-localization of PARP and TO-PRO in the 7-day post-septic group, suggesting DNA breaks in the septic nuclei. Since elevated cytosolic active caspsase-3 could be responsible for DNA fragmentation (review by Oberholzer et al., 2001), it appears that increased expression of PARP and TUNEL in septic nuclei occurs due to elevated myocardial active caspase-3 following prolonged septic insult to the heart. The present study suggest that elevation of caspase-3 along with release of cytochrome c could be one of the main pathways responsible for the elevated expression of myocardial PARP bound to DNA breaks. These data support our contention that prolonged sepsis produces myocardial apoptosis and thus could be a major factor for SIMD.
Conclusions
In summary, the data obtained in the present study demonstrated that an increased number of myocardial apoptotic cells correlated with progressive LV dysfunction, elevated concentration of ET-1, expression of proapoptotic proteins (Bax and Cyto C), active caspase-3, TRADD, and mortality at 3 and 7 days post-sepsis. These data suggest that the progression of sepsis from 1 day to 3-7 days produce distinct cardiodynamic characteristics with a more profound effect during later stages. An increase in ET-1 may be one factor that precipitates changes seen at 7-day-only such as MAP decrease, increased ratio of cytosolic cytochrome C to mitochondrial Cytochrome C and PARP DNA breaks (Table 2). We concluded that sepsis-induced decline in myocardial performance correlates with the induction of myocardial apoptosis.
Table 2.
Summary of sepsis-induced alterations on the hemodynamic and biochemical parameters during early (1-day post-sepsis) and later (3- to 7-days post sepsis) stages. + denotes significant change compared to the sham group; ++ denotes significant change compared to the 1-day post-sepsis group.
| Parameters | 1-day post sepsis | 3-7 days post sepsis |
|---|---|---|
|
Hemodynamic | ||
| HR | + | + |
| MAP | No Change | No change to decrease |
| +dP/dt | No change | No change |
| −dP/dt | No change | Decrease |
| LVEDP | + | + |
| Tau | + | ++ |
|
Biochemical markers | ||
| cTnI | + | + |
| CRP | + | + |
| ET-1 | No change | ++ |
|
Apoptosis proteins | ||
| Active Caspase-3 | + | ++ |
| Bax/BCl2 | + | ++ |
| TRADD | + | ++ |
| Release of Cyto C | No change | Increase |
|
Signaling proteins | ||
| Phosphorylation of | ||
| p38-MAPK | No change | ++ |
| JNK | + | ++ |
Acknowledgements
The present study was supported by the funds obtained from the National Institutes of Health (NHLBI, HL 66016), AHA (160444Z) and [in part] from a grant from the Texas A&M Health Science Center Research Development and Enhancement Program. The authors also acknowledge the technical assistance provided by Dr. Sachin Brahmbhatt in obtaining hemodynamic data of the study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ammann P, Fehr T, Minder EI, Gunter C, Bertel O. Elevation of troponin I in sepsis and septic shock. Intensive Care Medicine. 2001;27:965–969. doi: 10.1007/s001340100920. [DOI] [PubMed] [Google Scholar]
- Arlati S, Brenna S, Prencipe L, Marocchi A, Casella GP, Lanzani M, Gandini C. Myocardial necrosis in ICU patients with acute non-cardiac disease: a prospective study. Intensive Care Medicine. 2000;26:31–37. doi: 10.1007/s001340050008. [DOI] [PubMed] [Google Scholar]
- Behr TM, Nerurkar SS, Nelson AH, Coatney RW, Woods TN, Sulpizio A, Chandra S, Brooks DP, Kumar S, Lee JC, Ohlstein EH, Angermann CE, Adams JL, Sisko J, Sackner-Bernstein JD, Willette RN. Hypertensive end-organ damage and premature mortality are p38 mitogen-activated protein kinase-dependent in a rat model of cardiac hypertrophy and dysfunction. Circulation. 2001;104:1292–1298. doi: 10.1161/hc3601.094275. [DOI] [PubMed] [Google Scholar]
- Bertinchant JP, Larue C, Pernel I, Ledermann B, Fabbro-Peray P, Beck L, Calzolari C, Trinquier S, Nigond J, Pau B. Release kinetics of serum cardiac troponin I in ischemic myocardial injury. Clinical Biochemistry. 1996;29:587–594. doi: 10.1016/s0009-9120(96)00105-1. [DOI] [PubMed] [Google Scholar]
- Brahmbhatt S, Gupta A, Sharma AC. Bigendothelin-1 (1-21) fragment during Early Sepsis Modulates tau, p38-MAPK phosphorylation and Nitric Oxide Synthase Activation. Molecular and Cellular Biochemistry. 2005;271:225–237. doi: 10.1007/s11010-005-6416-3. [DOI] [PubMed] [Google Scholar]
- Bratton SB, MacFarlane M, Cain K, Cohen GM. Protein complexes activate distinct caspase cascades in death receptor and stress-induced apoptosis. Experimental Cell Research. 2000;256:27–33. doi: 10.1006/excr.2000.4835. [DOI] [PubMed] [Google Scholar]
- Charpentier J, Luyt CE, Fulla Y, Vinsonneau C, Carious A, Grabar S, Dhainaut JF, Mira JP, Chiche JD. Brain natriuretic peptide: a marker of myocardial dysfunction and prognosis during severe sepsis. Critical Care Medicine. 2004;32:660–665. doi: 10.1097/01.ccm.0000114827.93410.d8. [DOI] [PubMed] [Google Scholar]
- Cheng W, Li B, Kajstura J, Li P, Wolin MS, Sonnenblick EH, Hintze TH, Olivetti G, Anversa P. Stretch-induced programmed myocyte cell death. Journal of Clinical Investigation. 1995;96:2247–2259. doi: 10.1172/JCI118280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien K. Stress pathways in heart failure. Cell. 1999;98:555–558. doi: 10.1016/s0092-8674(00)80043-4. [DOI] [PubMed] [Google Scholar]
- Clerk A, Michael A, Sugden PH. Stimulation of the p38 mitogen-activated protein kinase pathway in neonatal rat ventricular myocytes by the G-protein coupled receptor agonists, endothelin-1 and phenylephrine: A role in cardiac myocyte hypertrophy? Journal of Cellular Biology. 1998a;142:523–535. doi: 10.1083/jcb.142.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerk A, Sugden PH. Stress-responsive mitogen activated protein kinases (c-jun N-terminal kinases and p38 mitogen activated protein kinases) in the myocardium. Circulation Research. 1998b;83:345–352. doi: 10.1161/01.res.83.4.345. [DOI] [PubMed] [Google Scholar]
- Fernandes CJ, Jr., Akamine N, Knobel E. Cardiac troponin: a new serum marker of myocardial injury in sepsis. Intensive care Medicine. 1999;25:1165–1168. doi: 10.1007/s001340051030. [DOI] [PubMed] [Google Scholar]
- Gonzalez A, Fortuno MA, Querejeta R, Ravassa S, Lopez B, Lopez N, Diez J. Cardiomyocyte apoptosis in hypertensive cardiomyopathy. Cardiovascular Research. 2003;59:549–562. doi: 10.1016/s0008-6363(03)00498-x. [DOI] [PubMed] [Google Scholar]
- Gupta A, Sharma AC. Metalloendopeptidase inhibition regulates phosphorylation of p38-mitogen-activated protein kinase and nitric oxide synthase in heart after endotoxemia. Shock. 2003;20:375–381. doi: 10.1097/01.shk.0000087202.34916.0c. [DOI] [PubMed] [Google Scholar]
- Gupta A, Brahmbhatt S, Sharma AC. Left ventricular mitogen activated protein kinase signaling following polymicrobial sepsis during streptozotocin-induced hyperglycemia. Biochimica et Biophysica Acta. 2004;1690:42–53. doi: 10.1016/j.bbadis.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Gupta A, Aberle N, Kapoor R, Ren J, Sharma AC. BigET-1 induced contractile function is mediated via p38-MAPK phosphorylation in adult rat ventricular myocytes during sepsis. Biochimica et Biophysica Acta. 2005a;1741:127–139. doi: 10.1016/j.bbadis.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Gupta A, Aberle N, Ren J, Sharma AC. Endothelin-converting enzyme–1-mediated signaling in adult rat ventricular myocyte contractile function and apoptosis during polymicrobial sepsis. Journal of Molecular and Cellular Cardiology. 2005b;38:527–37. doi: 10.1016/j.yjmcc.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Hein S, Arnon E, Kostin S, Schonburg M, Elsasser A, Polyakova V, Bauer EP, Klovekorn WP, Schaper J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–91. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- Holcroft JW. Hypertonic saline for resuscitation of the patient in shock. Advances in Surgery. 2001;35:297–318. [PubMed] [Google Scholar]
- Kan H, Xie Z, Finkel MS. p38 MAP kinase-mediated negative inotropic effect of HIV gp120 on cardiac myocytes. American Journal of Physiology. 2004;286:C1–C7. doi: 10.1152/ajpcell.00059.2003. [DOI] [PubMed] [Google Scholar]
- Kochanek KD, Smith BL. Deaths: Preliminary data for 2002. National Vital Statistics Report. 2004;52:1–48. [PubMed] [Google Scholar]
- LaPointe MC, Isenovic E. Interleukin-1beta regulation of inducible nitric oxide synthase and cyclooxygenase-2 involves the p42/44 and p38-MAPK signaling pathways in cardiac myocytes. Hypertension. 1999;33:276–282. doi: 10.1161/01.hyp.33.1.276. [DOI] [PubMed] [Google Scholar]
- Liao P, Wang SQ, Wang S, Zheng M, Zheng M, Zhang SJ, Cheng H, Wang Y, Xiao RP. p38 Mitogen-activated protein kinase mediates a negative inotropic effect in cardiac myocytes. Circulation Research. 2002;90:190–196. doi: 10.1161/hh0202.104220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlan M, Merrison W, Bratton SB, Cohen GM. Proteasome-mediated degradation of Smac during apoptosis: XIAP promotes Smac ubiquitination in vitro. Journal of Biological Chemistry. 2002;277:36611–36616. doi: 10.1074/jbc.M200317200. [DOI] [PubMed] [Google Scholar]
- Maeder M, Fehr T, Rickli H, Ammann P. Sepsis-Associated Myocardial Dysfunction, Diagnostic and Prognostic Impact of Cardiac Troponins and Natriuretic Peptides. CHEST. 2006;129:1349–1366. doi: 10.1378/chest.129.5.1349. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Ichijo H. Molecular mechanisms of the decision between life and death: regulation of apoptosis by apoptosis signal-regulating kinase 1. Journal of Biochemistry (Tokyo) 2001;130:1–8. doi: 10.1093/oxfordjournals.jbchem.a002947. [DOI] [PubMed] [Google Scholar]
- Molnár A, Tóth A, Bagi Z, Papp Z, Édes I, Vaszily M, Galajda Z, Papp GJ, Varró J, Szüts V, Lacza Z, Gerö D, Szabó C. Activation of the poly (ADP-Ribose) polymerase pathway in human heart Failure. Molecular Medicine. 2006;12:143–152. doi: 10.2119/2006-00043.Molnar. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebreda AR. p38 MAP kinases: Beyond the stress response. Trends in Biochemical Sciences. 2000;25:257–259. doi: 10.1016/s0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- Oberholzer C, Oberholzer A, Clare-Salzler M, Moldawer LL. Apoptosis in sepsis: a new target for therapeutic exploration. FASEB J. 2001;15:879–892. doi: 10.1096/fj.00-058rev. [DOI] [PubMed] [Google Scholar]
- Parker MM, McCarthy KE, Ognibene FP, et al. Right ventricular dysfunction and dilatation, similar to left ventricular changes, characterize the cardiac depression of septic shock in humans. Chest. 1990;97:126–131. doi: 10.1378/chest.97.1.126. [DOI] [PubMed] [Google Scholar]
- Parker MM, Shelhamer JH, Bacharach SL, et al. Profound but reversible myocardial depression in patients with septic shock. Annals of Internal Medicine. 1984;100:483–490. doi: 10.7326/0003-4819-100-4-483. [DOI] [PubMed] [Google Scholar]
- Parrillo JE, Parker MM, Natanson C, Suffredini AF, Danner RL, Cunnion RE, Ognibene FP. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Annals of Internal Medicine. 1990;113:227–242. doi: 10.7326/0003-4819-113-3-227. [DOI] [PubMed] [Google Scholar]
- Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3-and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Molecular and Cellular Biology. 1996;16:1247–55. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinelt H, Radermacher P, Fischer G, Geisser W, Wachter U, Wiedeck H, Georgieff M, Vogt J. Effects of a dobutamine-induced increase in splanchnic blood flow on hepatic metabolic activity in patients with septic shock. Anesthesiology. 1997;86:818–824. doi: 10.1097/00000542-199704000-00012. [DOI] [PubMed] [Google Scholar]
- Sam AD, II, Sharma AC, Law WR, Ferguson JL. Splanchnic vascular control during sepsis and endotoxemia. Frontiers in Bioscience. 1997;2:e72–e92. doi: 10.2741/a229. [DOI] [PubMed] [Google Scholar]
- Schaer GL, Fink MP, Parrillo JE. Norepinephrine alone versus norepinephrine plus low-dose dopamine: enhanced renal blood flow with combination pressor therapy. Critical Care Medicine. 1985;13:492–496. doi: 10.1097/00003246-198506000-00011. [DOI] [PubMed] [Google Scholar]
- Sharma AC, Motew SJ, Farias S, Alden KJ, Bosmann HB, Law WR, Ferguson JL. Sepsis alters myocardial and plasma concentrations of endothelin and nitric oxide in rats. Journal of Molecular and Cellular Cardiology. 1997;29:1469–1477. doi: 10.1006/jmcc.1997.0386. [DOI] [PubMed] [Google Scholar]
- Song GY, Chung CS, Chaudry IH, Ayala A. Immune suppression in polymicrobial sepsis: Differential regulation of Th1 and Th2 responses by p38-MAPK. Journal of Surgical Research. 2000;91:141–146. doi: 10.1006/jsre.2000.5928. [DOI] [PubMed] [Google Scholar]
- Song GY, Chung CS, Chaudry IH, Ayala A. MAPK p38 antagonism as a novel method of inhibiting lymphoid immune suppression in polymicrobial sepsis. American Journal of Physiology - Cell Physiology. 2001;281:C662–C669. doi: 10.1152/ajpcell.2001.281.2.C662. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Honda M, Takabatake T. Redox regulation of MAPK pathways and cardiac hypertrophy in adult rat cardiac myocyte. Journal of the American College of Cardiology. 2001;37:676–68. doi: 10.1016/s0735-1097(00)01123-2. [DOI] [PubMed] [Google Scholar]
- Turner A, Tsamitros M, Bellomo R. Myocardial cell injury in septic shock. Critical Care Medicine. 1999;27:1775–1780. doi: 10.1097/00003246-199909000-00012. [DOI] [PubMed] [Google Scholar]
- Ver Elst KM, Spapen HD, Nguyen DN, et al. Cardiac troponin I and T are biological markers of left ventricular dysfunction in septic shock. Clinical Chemistry. 2000;46:650–657. [PubMed] [Google Scholar]
- Wenzel RP, Edmond MB. Managing antibiotic resistance. New England Journal of Medicine. 2001;343:1961–1963. doi: 10.1056/NEJM200012283432610. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yeh C, Chen T, Lee C, Wu Y, Lin Y, Lin PJ. Cardiomyocytic apoptosis following global ischemia and reperfusion can be attenuated by peroxisome proliferator-activated receptor α but not γ activators. Shock. 2006;29:262–270. doi: 10.1097/01.shk.0000225863.56714.96. [DOI] [PubMed] [Google Scholar]