

Abstract
The development of thiol-ene/thiol-epoxy hybrid networks offers the advantage of tailorable polymerization kinetics while producing a highly crosslinked, high Tg polymer that has significantly reduced shrinkage stress. Stoichiometric mixtures of pentaerythritol tetra(3-mercaptopropionate) (PETMP)/triallyl-1,3,5-triazine-2,4,6-trione (TATATO) (thiol-ene, mixture 1) and PETMP/bisphenol a diglycidyl ether (BADGE) (thiol-epoxy, mixture 2) were prepared and hybrid mixtures of 75/25, 50/50, 25/75, and 10/90 w/w of mixtures 1 and 2 were polymerized using a combination of both radical and anionic initiation. The light exposure timing and the relative initiation conditions of the two types were used to control the order and relative rates of the radical and anionic polymerizations. The 50/50 w/w thiol-ene/thiol-epoxy hybrid material exhibited a final stress of only 0.2 MPa, which is 90 % lower than the stress developed in a control dimethacrylate resin. Kinetic analysis indicates composition affects network development in thiol-ene/thiol-epoxy hybrid networks and produces materials with robust mechanical properties.
Keywords: dental restorative material, thiol-ene, polymerization stress
1. Introduction
The development of materials with tailorable properties as well as low shrinkage and stress has been a thrust of the photopolymer industry for decades, with particular importance in dental materials. Towards this goal, interest has turned to the evaluation of alternative polymerization schemes, such as hybrid or interpenetrating networks (IPNs). More specifically, hybrid polymerizations, systems where one or both of the monomers used in the polymerization contain functional groups which are polymerizable by multiple curing methods[1], and IPNs, polymer networks that are cured simultaneously using separate polymerization mechanisms and physically interlock [2–4], offer synergistic material properties in the resultant material.
Previous investigations have focused on dual curing modes in acrylate/vinyl ether[5, 6], vinyl ether/epoxy[7, 8], or acrylate/epoxy systems[9–11], providing tailorable mechanical properties and improved polymerization kinetics. Investigations have shown the importance of monomer composition in controlling the final conversion and the polymer glass transition temperature[4], as well as the importance of cure sequence in acrylate/epoxy hybrid systems on the final resin conversion and morphology[12, 13]. In addition, hybrid systems have demonstrated reduced oxygen inhibition and sensitivity to impurities, allowing for improved hardness and reduced swelling[10].
However, many applications, including the in situ formation of dental composites, require fast cure times and low reaction temperatures, which is difficult to achieve using several reaction combinations. In addition, the chain growth polymerizations of the acrylate or vinyl ether networks lead to significant polymerization shrinkage and stress, despite the advantages of the epoxy ring opening polymerization. Hence, investigation of alternative hybrid systems, which produce lower polymerization shrinkage and stress, is of great interest.
This investigation presents hybrid networks of dual step-growth networks, represented by a thiol-ene/thiol-epoxy hybrid system. The inclusion of a thiol-ene network offers numerous advantages such as a lack or oxygen inhibition[14], delayed gel points and improved control of the polymerization[15–20]. In addition, thiol-enes exhibit reduced polymerization shrinkage and stress[21] when compared with methacrylate systems commonly used in dental restorative materials[21, 22]. However, the bulk of the commercially available non-acrylated vinyl monomers and thiol monomers typically have a flexible core, making the development of high glass transition temperature, highly crosslinked materials more difficult[22, 23]. Furthermore, epoxy resins are valued for their chemical resistance, mechanical strength, excellent adhesion and low shrinkage; however, they produce brittle materials when cured without a comonomer[4]. Thus, the development of a thiol-ene/thiol-epoxy hybrid material has the possibility of low shrinkage and low stress polymeric resin with desirable mechanical properties such as a high glass transition temperature and strength. In particular, this work addresses the impact of thiol-ene and thiol-epoxy monomer ratio and cure conditions on the material properties and stress development, and subsequently compares those results to a control dimethacrylate resin typically used in dental restorative materials.
2. Experimental Section
2.1. Materials
The monomers used in this investigation, Figure 1, were pentaerythritol tetra(3-mercaptopropionate) (PETMP), triallyl-1,3,5-triazine-2,4,6-trione (TATATO), and bisphenol a diglycidyl ether (BADGE) (all obtained from Aldrich, Milwaukee, WI). 2,2-dimethoxy-2-phenylacetophenone (DMPA) (Ciba-Geigy, Hawthorn, NY) was used as the UV photoinitiator and 2,4,6-tris(dimethylaminomethyl)phenol was the anionic polymerization catalyst. All monomers and photoinitiators were used without additional purification. Samples were studied in varying weight ratios of thiol-ene to thiol-epoxy networks. More specifically, samples of stoichiometric mixtures of PETMP/TATATO (thiol-ene, mixture 1) and PETMP/BADGE (thiol-epoxy, mixture 2) were prepared and tested, and hybrid mixtures of 75/25 wt %, 50/50 wt%, 25/75 wt%, and 10/90 wt% of mixtures 1 and 2 were prepared and analyzed. All systems were prepared with 0.1 wt% DMPA and 5 wt% amine, except for the study of the effect of the amine catalyst for which the addition of 10 wt% amine in the 50/50 wt% mixture was also studied.
Figure 1.
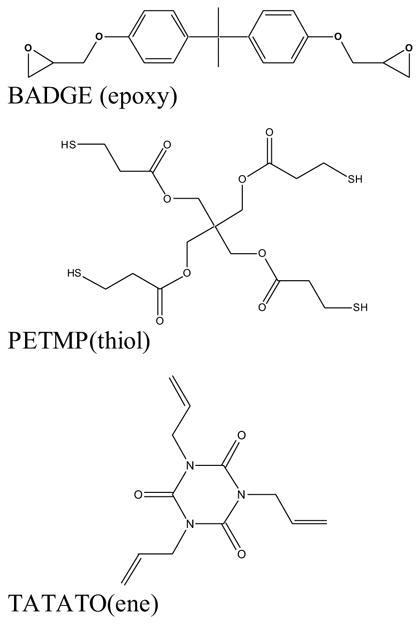
Monomers used in this study; bisphenol a diglycidyl ether (BADGE, epoxy), pentaerythritol tetra(3-mercaptopropionate) (PETMP, thiol), and triallyl-1,3,5-triazine-2,4,6-trione (TATATO, ene).
2.2. Mid-IR Analysis
Fourier Transform Infrared Spectroscopy (FTIR) studies were conducted to monitor thiol, ene, and epoxy conversion using a Nicolet Magna 750 FTIR spectrometer (Nicolet Instrument Corp., Madison, WI). A 365 nm light source (EFOS Ultracure 100ss Plus, Mississauga, Ontario, Canada) with a 320–500 nm bandpass filter was used for initiation of radical polymerization, using an irradiation intensity of 15 mW/cm2 at the surface of the sample, which was monitored by a radiometer (9811, Cole-Parmer Instruments Co., Vernon Hills, IL). Samples were placed between salt crystals and cured inside a horizontal transmission apparatus[24]. A mid-IR spectrum (4000–600 cm−1) of the sample was collected before and after irradiation, and series spectra were collected during photocuring at approximately 2 scans per second. Samples were irradiated for 300 seconds, and monitored for eighty minutes to ensure complete reactions of both networks. For the sequential anionic-radical study, samples underwent 60 minutes of dark cure,during which the anionic reaction was proceeding, which was followed by 300 s of 15 mW/cm2 UV light irradiation. Polymerization kinetics were observed using the allyl absorption peak at 1643 cm−1, the epoxy absorption peak at 916 cm−1, and the thiol absorption peak at 2572 cm−1, The degree of conversion was calculated using the area of the monitored absorption peak.
2.3. Stress Measurement
Simultaneous conversion and polymerization shrinkage stress measurements were conducted using a tensometer (American Dental Association Health Foundation), which monitors stress development using cantilever beam deflection theory. A detailed description of the tensometer and the measurement technique is found elsewhere[25, 26]. Simultaneous conversion measurements are facilitated using remote near-IR transmitted through the polymer sample via fiber optic cables. Samples measuring 6 mm in diameter and 1.5 mm in thickness were prepared using the initiator concentrations and monomer ratios as described above. Samples were irradiated using 15 mW/cm2 UV light (NOVAcure, Mississauga, Ontario, Canada) for 60 seconds, and the stress profile monitored for 80 minutes in all of the experiments.
2.4. Polymerization Shrinkage Measurements
Post-gel volumetric shrinkage was monitored using a linometer (ACTA, Amsterdam, Netherlands), which measures axial displacement using a contact-free transducer system. 6–8 mm in diameter and 1.5 mm in thickness were prepared and irradiated using UV light as described in the mid-IR section.
2.5. Dynamic Mechanical Analysis
Thermal analyses were conducted using a dynamic mechanical analyzer (TA instruments, DMA Q800). Monomer specimens of uniform size (4 mm wide, 1.0 mm thick, and 15 mm long) were prepared using glass slide molds and cured under the same conditions as used in the IR analysis. DMA studies were conducted over a temperature range of –20 to 120 °C, with a ramping rate of 5 °C/min using extension mode (sinusoidal stress of 1 Hz frequency). Tan δ (the ratio of loss to storage modulus) was monitored as a function of temperature. The glass transition temperature (Tg) was taken to be the maximum of the loss tangent-temperature curve. All reported peak width values were taken at peak half height.
2.6. Flexural Strength and Elastic Modulus
Samples were prepared using steel molds measuring 2 mm x 2 mm x 25 mm at the conditions presented in the IR analysis section. Polymer flexural strength and modulus were calculated using a 3-point flexural test, carried out with a hydraulic universal test system (858 Mini Bioix, MTS Systems Corporation, Eden Prairie, MN, USA) using a span width of 10 mm and a crosshead speed of 1mm/min.
3. Results and Discussion
The utilization of hybrid polymerizations offers the advantage of tailoring material properties by varying polymerization kinetics or monomer ratios. To facilitate the discussion of the experimental results, the pertinent reaction mechanisms for the thiol-ene and thiol-epoxy polymerizations are discussed first.
Traditional thiol-ene polymers cure using a step growth radical polymerization, in which a thiol adds to a carbon-carbon double bond[16]. More specifically, the accepted initiation, propagation and termination steps are illustrated below[27, 28].
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
Initiation in radical thiol-ene polymerizations is achieved through the generation of radical centers, which is accomplished by one of several different mechanisms. Here, 2,2-dimethoyxy-2-phenylacetophenone (DMPA), which is a Norrish type I cleavage radical initiator, is used as a photoinitiator(PI). Upon absorption of a photon of light, the photoinitiator cleaves, producing two radicals (step 1).
The radical abstracts a hydrogen from a thiol monomer (step 2) producing a thiyl radical which either propagates or terminates. The thiyl radical most frequently propagates through a carbon-carbon double bond, which then abstracts a hydrogen from a thiol, recreating a thiyl radical (steps 3 and 4). This sequential propagation and chain transfer is characteristic of the thiol-ene step growth mechanism. Traditionally, the vinyl functional group does not homopolymerize in a thiol-ene polymerization, thus the thiol and ene functional groups should be consumed at the same rate. However, for some thiol-vinyl polymerizations the vinyl monomer participates in both step growth propagation-chain transfer events as well as chain growth homopolymerization (step 5). In this case, the vinyl monomer will achieve a higher final consumption rate than that of the thiol monomer. Termination by radical-radical recombination (Steps 6–8) has been assumed to be the primary termination mechanism[20]. Recent work suggests that all possible radical-radical recombinations occur[29].
The second polymerization occurring in the hybrid polymerizations discussed in this paper is a thiol-epoxy reaction. Thiols react with epoxies in a base catalyzed addition reaction or nucleophilic displacement reaction as shown in equations 9 through 13 [30, 31]. More particularly, a thiol functional group creates a reactive intermediate with a tertiary amine, resulting in a thiol anion (step 9). The thiol anion attacks an epoxide group via an anionic addition (step 10). Additionally, the tertiary amine reacts with the epoxide group creating an epoxide anion. In the presence of a hydrogen donor, such as a thiol or hydroxyl group, the polymerization proceeds in a nucleophilic displacement (steps 11 and 12). Here, reaction 11 is expected to occur more readily than hydrogen abstraction from pendant hydroxyl groups, step 12. More specifically, the pKa of methyl 3-mercaptopropionate[32] is 10.4 as compared to isopropanol , which has a pKa of 17.1, making the thiol anion a better nucleophile. Furthermore, Streitweiser has demonstrated that the relative rate of nucleophilic displacement of a thiol anion is over 3 orders of magnitude faster than a similar alcohol anion[30]. In the absence of abstractable hydrogens, an anionic epoxy homopolymerization ensues via initiation by the tertiary amine-epoxy zwitterionic product[31]. For the hybrid reactions discussed in this paper, the predominant products are formed via equations 10 and 11.
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
Hence, as demonstrated by the above mechanisms, monomer concentrations, polymerization conditions, and initiator concentrations are variables which may be altered to tailor the polymerization mechanism and, ultimately, material properties. First, the analysis of the effects of monomer composition on polymerization kinetics was conducted, Figure 2b. A careful inspection of the epoxy conversion profiles shown in Figure 2b suggests that the extent of epoxy conversion is not affected by the initial thiol-ene polymerization, although the time required to reach the final epoxy conversion increases as the epoxy amount decreases. However, observation of the thiol conversion in Figure 2a reveals little difference in final thiol conversion despite the dual curing schemes. This suggests that as the epoxy concentration is decreased, the epoxy functional group participates in more homopolymerization events rather than copolymerizing with the thiol. For the hybrid systems studied, the ratio of epoxy homopolymerization to copolymerization, represented thus forth by Repoxy, was determined to increase from 1.1 for the purely thiol-epoxy resin to 1.7 for the 25/75 wt%thiol-epoxy/thiol-ene resin. This increase is attributed to the reduced mobility of the unreacted thiol functional groups in the hybrid systems that contain higher concentrations of vinyl monomer as a result of a higher crosslinking extent.
Figure 2.
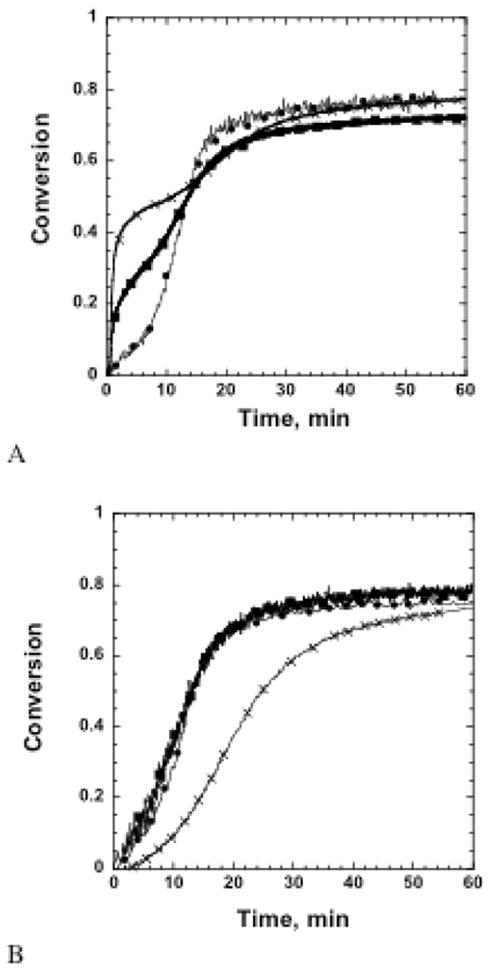
Functional group conversion as a function of time for hybrid systems initially cured for 300 s using 15 mW/cm2 320–500 nm light, 0.1 wt % DMPA, and 5 wt % amine. (A) Thiol conversion and (B) epoxy conversion for (X) 50/50 wt% thiol-epoxy/thiol-ene (■)75/25 wt% thiol-epoxy/thiol-ene; and (●)100 wt% thiol-epoxy.
Changing the sequence of polymerization such that the thiol-epoxy is polymerized before the thiol-ene significantly affects the overall conversion and network development in the hybrid resins. As observed in Figure 3, the initial rate of epoxy polymerization is faster, and the epoxy is able to reach higher final conversions. In addition, the epoxy homopolymerization to copolymerization ratio, Repoxy, stays near one, suggesting that the thiol concentration and increased monomer mobility prior to the thiol-ene reaction is conducive to nearly ideal thiol-epoxy polymerization. However, the cure of the thiol-epoxy network first decreases monomer mobility such that the final conversion of the thiol and vinyl in the hybrid systems is significantly lower than in the hybrid systems where the thiol-ene network is cured first. Specifically, the vinyl conversion only reaches 38% conversion in the 50/50 wt% thiol-epoxy/thiol-ene hybrid material, and no vinyl conversion is observed in the75/25 wt% thiol-epoxy/thiol-ene hybrid material as a result of the formation of the highly crosslinked thiol-epoxy network. These results suggest that optimal conversion of both the thiol-ene and thiol-epoxy networks are achieved in the sequential thiol-ene thiol-epoxy polymerizations.
Figure 3.
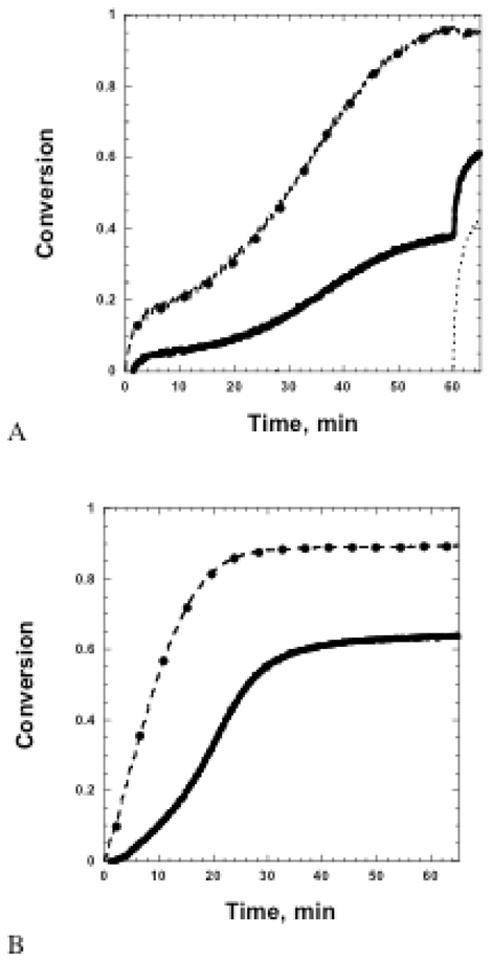
Functional group conversion as a function of time for sequential curing, where the epoxy/thiol was cured first for 50/50 wt% thiol-epoxy/thiol-ene (A) and 75/25 wt% thiol-epoxy/thiol-ene (B). The graphs depict epoxy conversion (-.-●-.-), thiol conversion(—), and ene conversion (…..).Samples underwent 60 minutes of dark cure, during which the anionic reaction was proceeding, which was followed by 300 s of 15 mW/cm2 UV light irradiation.
Thirdly, the effect of the amine catalyst on the thiol-epoxy polymerization was studied for the 50/50 wt% thiol-epoxy/thiol-ene system. As shown in Figure 4, increasing the amount of amine from 5 wt% to 10 wt% significantly increases the rate of epoxy polymerization but does not significantly change the final conversion values for any of the monomers. This increased epoxy polymerization rate does not affect the thiol-epoxy reaction.
Figure 4.
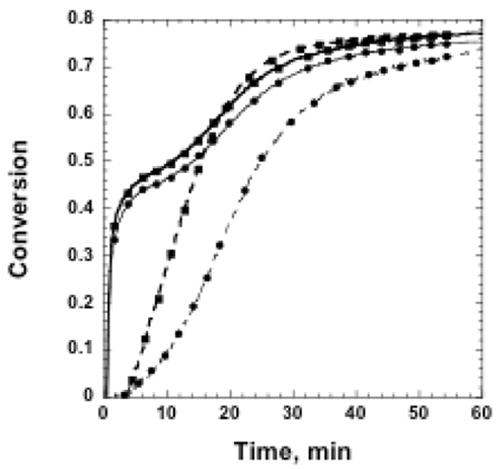
Functional group conversion as a function of time for 50/50 wt% thiol-epoxy/thiol-ene hybrid systems initially cured for 300 s with 15 mW/cm2 320–500 nm light, and 0.1 wt% DMPA with (●)5 wt% amine or (■) 10 wt% amine. The graph depicts (—)thiol conversion and (----)epoxy conversion.
In addition to polymerization kinetics, the impact of functional group ratio on the material properties was studied. The trend in glass transition temperature, shown in Table 2, is attributed to the changes in network development noted for the sequential thiol-ene thiol-epoxy reactions. More specifically, the 50/50 wt% thiol-epoxy/thiol-ene sample achieved the highest glass transition temperature as a result of the maximized final conversion of all monomers and the increased extent of epoxy homopolymerization as compared to hybrid systems with a higher epoxy concentration. While the flexural modulus was determined not to be significantly affected by monomer concentration in the hybrid resins, it should be noted that the mechanical properties of the hybrid resins are similar to that of the common dimethacrylates resin used in current dental restorations, a highly crosslinked dimethacrylates homopolymer.
Table 2.
Glass transition temperature and flexural modulus for hybrid systems initially cured for 300 s using 15 mW/cm2 320–500 nm light. 0.1 wt% DMPA, 5 wt% amine. Glass transition temperature analysis was performed using samples measuring 1 mmx2.5 mmx10 mm; flexural modulus was determined using samples measuring 2x2x25 mm.
| Resin System | Tg, °C | Flexural Modulus, GPa |
|---|---|---|
| 25/75 wt % thiol-epoxy/thiol-ene | 71(2) | 1.1(0.1) |
| 50/50 wt % thiol-epoxy/thiol-ene | 77(2) | 1.6(0.2) |
| 75/25 wt % thiol-epoxy/thiol-ene | 71(2) | 1.7(0.2) |
| 100 % thiol-ene | 55(2) | 1.7(0.2) |
| 100 % thiol-epoxy | 75(1) | 1.8(0.1) |
| Bis-GMA/TEGDMA(70/30 wt%) | 77(1) | 2.2(0.1) |
Furthermore, observation of the polymerization shrinkage and stress development in thiol-ene/thiol-epoxy hybrid systems suggests thiol-ene/thiol-epoxy hybrid materials are excellent resins for low stress polymeric material applications. The time dependent polymerization shrinkage in the hybrid materials, shown in Figure 5, exhibits shrinkage relaxation that is a result of the sequential radical-anionic cure. More specifically, as the epoxy concentration is increased, there is a larger amount of shrinkage relaxation that occurs prior to shrinkage development from the thiol-epoxy reaction due to the lower crosslinking density. As a result, as much as a 40% reduction of the shrinkage developed by the thiol-ene reaction in 50/50 wt% thiol-epoxy/thiol-ene is observed. This shrinkage relaxation translates into low stress development in the hybrid materials. As seen in Figure 6a, there is lower initial stress development (before the onset of the epoxy polymerization around 10 minutes, Figure 6b) produced by the thiol-ene polymerization in hybrid systems. Interestingly, we see a minimum in the final stress values for the 50/50 wt% thiol-epoxy/thiol-ene hybrid resin. Possible explanations for this result include decreased shrinkage as a result of higher levels of homopolymerization, or phase separation. Given the high values of conversion in the hybrid systems and no evidence of reduction in volume shrinkage in the later stages of polymerization, phase separation seems less likely. Currently, additional experiments are being conducted to elucidate this effect. Furthermore, all of the hybrid systems tested perform better than a bisGMA/TEGDMA (70/30 wt%) resin, which under identical cure conditions results in a final polymerization stress of 1.8 MPa (± 0.2 MPa) with a methacrylate conversion of 64% (± 3%) .
Figure 5.
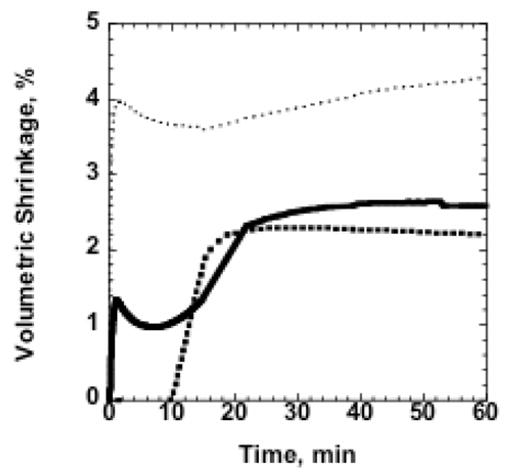
Polymerization shrinkage as a function of time for hybrid samples during stress measurements. Samples were cured using 15 mW/cm2 of 320-500 nm UV light for 60 s. Shrinkage is shown for (…..)25/75 wt% thiol-epoxy/thiol-ene; (—)50/50 wt% thiol-epoxy/thiol-ene; and(■ ■ ■) 100 wt% thiol-epoxy.
Figure 6.
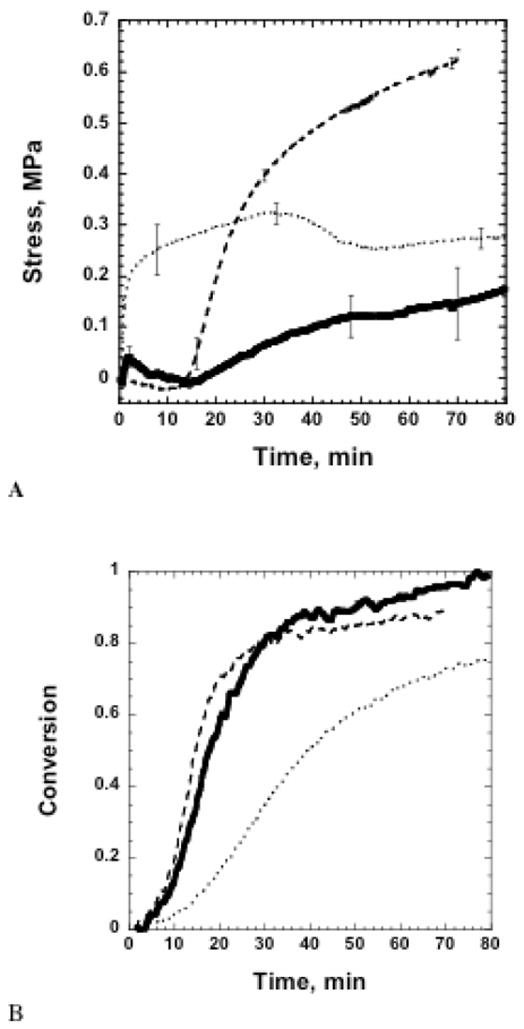
(A) Polymerization stress as a function of time for hybrid samples during stress measurements. (B) Epoxy conversion as a function of time for hybrid samples during stress measurements. Samples were cured using 15 mW/cm2 of 320–500 nm UV light for 60 s. (…..)25/75 wt % thiol-epoxy/thiol-ene; (—)50/50 wt % thiol-epoxy/thiol-ene; and(- - -)75/25 wt % thiol-epoxy/thiol-ene.
4. Conclusions
The results of this study indicate that the monomer ratio in thiol-ene/thiol-epoxy hybrid polymerizations affects resin conversion and mechanical properties. More specifically, hybrid resins of equal weight percent of thiol-ene and thiol-epoxy achieved the highest Tg and lowest polymerization stress, although polymerization rates of the epoxy were slower than hybrid samples of higher epoxy weight percent. In addition, sequential polymerization of the thiol-ene and thiol-epoxy network produced the highest final conversions of both networks, suggesting polymerization order is an important consideration in thiol-ene/thiol-epoxy hybrid polymerizations. The investigation of the mechanical properties of thiol-ene/thiol-epoxy resins suggests their strength and glass transition temperatures are similar to typical dimethacrylates dental restorative resins, while producing significantly lower polymerization shrinkage and stress. Thus, the results presented here demonstrate the utility of the thiol-ene/thiol-epoxy hybrid systems, which upon further development of photolatent anionic initiators could be used for dental restorative resins.
Table 1.
Final thiol, vinyl and epoxy conversion values and, Repoxy, the ratio of epoxy homopolymerization to thiol copolymerization. Samples cured using initial cure of 300 s of 15 mW/cm2 UV light. Standard deviation in parentheses, n=3.
| Resin System | Ene Conversion | Thiol Conversion | Epoxy Conversion | Repoxy |
|---|---|---|---|---|
| 25/75 wt % thiol-epoxy/thiol-ene | 90(2) | 82(3) | 81(4) | 1.7 |
| 50/50 wt % thiol-epoxy/thiol-ene | 93(4) | 78(1) | 86(2) | 1.6 |
| 75/25 wt % thiol-epoxy/thiol-ene | 90(8) | 70(6) | 81(1) | 1.3 |
| 90/10 wt % thiol-epoxy/thiol-ene | 80(6) | 74(2) | 81(4) | 1.1 |
| 100 % thiol-ene | 89(2) | 74(1) | NA | NA |
| 100 % thiol-epoxy | NA | 77(1) | 76(7) | 1 |
| Bis-GMA/TEGDMA(70/30 wt%) | NA | NA | NA |
Acknowledgments
Support from the US Department of Education's Graduate Assistantships in Areas of National Need (GAANN) Fellowship awarded to Jacquelyn A. Carioscia and an NIH Research Grant #DE10959 are acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Itoh H, Kameyama A, Nishikubo T. Journal of Polymer Science Part A-Polymer Chemistry. 1996;34(2):217–225. [Google Scholar]
- 2.Hua YJ, Crivello JV. Journal of Polymer Science Part A-Polymer Chemistry. 2000;38(19):3697–3709. [Google Scholar]
- 3.Rajaraman SK, Mowers WA, Crivello JV. Journal of Polymer Science Part A-Polymer Chemistry. 1999;37(21):4007–4018. [Google Scholar]
- 4.Vabrik R, Czajlik I, Tury G, et al. Journal of Applied Polymer Science. 1998;68(1):111–119. [Google Scholar]
- 5.Lin Y, Stansbury JW. Polymer. 2003;44(17):4781–4789. [Google Scholar]
- 6.Lin Y, Stansbury JW. Polymers for Advanced Technologies. 2005;16(2–3):195–199. [Google Scholar]
- 7.Chappelow CC, Pinzino CS, Power MD, et al. Journal of Applied Polymer Science. 2002;86(2):314–326. [Google Scholar]
- 8.Dean K, Cook WD, Rey L, et al. Macromolecules. 2001;34(19):6623–6630. [Google Scholar]
- 9.Eick JD, Kostoryz EL, Rozzi SM, et al. Dental Materials. 2002;18(5):413–421. doi: 10.1016/s0109-5641(01)00071-9. [DOI] [PubMed] [Google Scholar]
- 10.Cho JD, Hong JW. Journal of Applied Polymer Science. 2004;93(3):1473–1483. [Google Scholar]
- 11.Pojman JA, Elcan W, Khan AM, et al. Journal of Polymer Science Part A-Polymer Chemistry. 1997;35(2):227–230. [Google Scholar]
- 12.Dean K, Cook WD. Macromolecules. 2002;35(21):7942–7954. [Google Scholar]
- 13.Dean KM, Cook WD. Polymer International. 2004;53(9):1305–1313. [Google Scholar]
- 14.O'Brien AK, Cramer NB, Bowman CN. Journal of Polymer Science Part A-Polymer Chemistry. 2006;44(6):2007–2014. [Google Scholar]
- 15.Cramer NB, Bowman CN. Journal of Polymer Science Part A-Polymer Chemistry. 2001;39(19):3311–3319. [Google Scholar]
- 16.Jacobine AF. Thiol-Ene Photopolymers. In: Fouassier JP, Rabek JF, editors. Radiation Curing in Polymer Science and Technology. Elsevier Applied Science; London: 1993. pp. 219–265. [Google Scholar]
- 17.Cramer NB, Davies T, O'Brien AK, et al. Macromolecules. 2003;36(12):4631–4636. [Google Scholar]
- 18.Reddy SK, Cramer NB, Rydholm A, et al. Abstracts of Papers of the American Chemical Society. 2004;228:U383–U383. [Google Scholar]
- 19.Reddy SK, Cramer NB, O'Brien AK, et al. Macromolecular Symposia. 2004;206:361–374. [Google Scholar]
- 20.Morgan CR, Magnotta F, Ketley AD. Journal of Polymer Science Part A-Polymer Chemistry. 1977;15(3):627–645. [Google Scholar]
- 21.Lu H, Carioscia JA, Stansbury JW, et al. Dental Materials. 2005;21(12):1129–1136. doi: 10.1016/j.dental.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Patel MP, Braden M, Davy KWM. Biomaterials. 1987;8(1):53–56. doi: 10.1016/0142-9612(87)90030-5. [DOI] [PubMed] [Google Scholar]
- 23.Pappas SP, editor. Science and Technology. Plenum Press; New York: 1992. Radiation Curing. [Google Scholar]
- 24.Lovell LG, Berchtold KA, Elliott JE, et al. Polymers for Advanced Technologies. 2001;12(6):335–345. [Google Scholar]
- 25.Lu H, Stansbury JW, Dickens SH, et al. Journal of Materials Science-Materials in Medicine. 2004;15(10):1097–1103. doi: 10.1023/B:JMSM.0000046391.07274.e6. [DOI] [PubMed] [Google Scholar]
- 26.Lu H, Stansbury JW, Dickens SH, et al. Journal of Biomedical Materials Research Part B: Applied Biomaterials. 2004;(71B):206–221. doi: 10.1002/jbm.b.30088. [DOI] [PubMed] [Google Scholar]
- 27.Chiou BS, Kham SA. Macromolecules. 1997;30(23):7322–7328. [Google Scholar]
- 28.Kharasch MS, Nudenberg W, Mantell GJ. Journal of Organic Chemistry. 1951;16:524. [Google Scholar]
- 29.Reddy SK, Cramer NB, Bowman CN. Macromolecules. 2006;39(10):3673–3680. [Google Scholar]
- 30.Tanaka YMTF. Epoxide-Curing Reactions. In: May CT, editor. Epoxy Resins Chemistry and Technology. Marcel Dekker; New York: 1973. pp. 135–239. [Google Scholar]
- 31.Ashcroft WR. Curing Agents for Epoxy Resins. In: Ellis B, editor. Chemistry and Technology of Epoxy Resins. Blackie Academic & Professional; Bishopbriggs: 1993. pp. 37–72. [Google Scholar]
- 32.Bernasconi CF, Killion RB. Journal of the American Chemical Society. 1988;110(22):7506–7512. [Google Scholar]