

Escherichia coli, a venerable workhorse for biochemical and genetic studies and for the large-scale production of recombinant proteins, is one of the most intensively studied of all organisms. The natural habitat of E. coli is the gastrointestinal tract of warm-blooded animals, and in humans, this species is the most common facultative anaerobe in the gut. Although most strains exist as harmless symbionts, there are many pathogenic E. coli strains that can cause a variety of diseases in animals and humans. In addition, from an evolutionary perspective, strains of the genus Shigella are so closely related phylogenetically that they are included in the group of organisms recognized as E. coli (1, 2). Pathogenic E. coli strains differ from those that predominate in the enteric flora of healthy individuals in that they are more likely to express virulence factors — molecules directly involved in pathogenesis but ancillary to normal metabolic functions. Expression of these virulence factors disrupts the normal host physiology and elicits disease. In addition to their role in disease processes, virulence factors presumably enable the pathogens to exploit their hosts in ways unavailable to commensal strains, and thus to spread and persist in the bacterial community.
It is a mistake to think of E. coli as a homogenous species. Most genes, even those encoding conserved metabolic functions, are polymorphic, with multiple alleles found among different isolates (1). The composition of the genome of E. coli is also highly dynamic. The fully sequenced genome of the laboratory K-12 strain, whose derivatives have served an indispensable role in the laboratories of countless scientists, shows evidence of tremendous plasticity (3). It has been estimated that the K-12 lineage has experienced more than 200 lateral transfer events since it diverged from Salmonella about 100 million years ago and that 18% of its contemporary genes were obtained horizontally from other species (4). Such fluid gain and loss of genetic material are also seen in the recent comparison of the genomic sequence of a pathogenic E. coli O157:H7 with the K-12 genome. Approximately 4.1 million base pairs of “backbone” sequences are conserved between the genomes, but these stretches are punctuated by hundreds of sequences present in one strain but not in the other. The pathogenic strain contains 1.34 million base pairs of lineage-specific DNA that includes 1,387 new genes; some of these have been implicated in virulence, but many have no known function (5).
The virulence factors that distinguish the various E. coli pathotypes were acquired from numerous sources, including plasmids, bacteriophages, and the genomes of other bacteria. Pathogenicity islands, relatively large (>10 kb) genetic elements that encode virulence factors and are found specifically in the genomes of pathogenic strains, frequently have base compositions that differ drastically from that of the content of the rest of the E. coli genome, indicating that they were acquired from another species. Here, we explore some of the known virulence factors that contribute to the heterogeneity of E. coli strains, and we review what is known regarding the origin and distribution of these factors.
Pathotypes and pathogenic clones
Pathogenic forms of E. coli associated with human and animal diseases are remarkably diverse. Certain pathogenic strains cause enteric diseases ranging in symptoms from cholera-like diarrhea to severe dysentery; other E. coli may colonize the urinary tract, resulting in cystitis or pyelonephritis, or may cause other extraintestinal infections, such as septicemia and meningitis. In discussing the diversity of pathogenic forms of this versatile species, we distinguish between an isolate’s pathotype, a classification of E. coli into groups that have a similar mode of pathogenesis and cause clinically similar forms of disease, and the pathogenic clone, bacteria of a genetic lineage within a bacterial species that share similarities because of recent descent from a common ancestral cell. There are at least eight recognized pathotypes of E. coli (Table 1) but many more distinct pathogenic clones (see Figure 1). Bacteria of the same pathogenic clone represent a monophyletic branch of an evolutionary tree and typically carry many of the same mobile genetic elements, including those that determine virulence.
Table 1.
Clinical and epidemiological features and virulence factors of various E. coli pathotypes
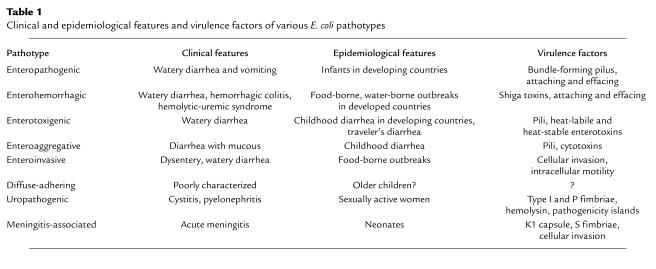
Figure 1.
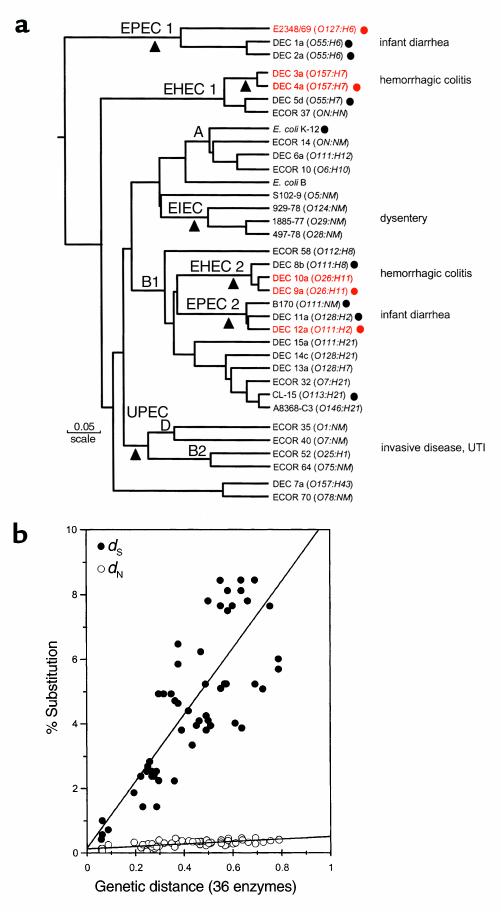
(a) The dendrogram is based on analysis of polymorphism at 36 protein loci studied by multilocus enzyme electrophoresis. Isolates mentioned repeatedly in the text are shown in red. The number of differences between strains is converted to a genetic distance assuming that each difference results from at least one amino acid–altering mutation at the DNA level. The diagram can be interpreted as a hypothetical phylogeny of strains that can be tested by gathering independent data. Main branches representing pathotypes are labeled. The A, B1, B2, and D groups are the clusters from the ECOR set. The triangles mark positions at which major acquisition of virulence factors are postulated to have occurred. (b) Nucleotide substitutions for seven housekeeping genes plotted against genetic distance. Nucleotide differences were analyzed separately for synonymous sites (dS), positions in codons where point mutations do not predict amino acid replacements, and nonsynonymous sites (dN), where point mutations result in amino acid changes. The points are averages of the comparison of pairs of strains (marked with circles) in a. UTI, urinary tract infection.
Early evidence for the clonal nature of pathogenic E. coli was seen in the repeated recovery of identical serotypes and biotypes from separate outbreaks of disease. The idea of widespread pathogenic clones gained support from the study of protein polymorphisms, first with patterns of the major outer proteins and then through the broad application of multilocus enzyme electrophoresis (1). Recent sequence comparisons have shown that a phylogenetic approach based on the clone concept, however, is complicated by recombination events, which, like mutations, contribute to the divergence of bacterial genomes in nature (reviewed in refs. 6, 7).
The diversity of pathotypes and their genetic relatedness are illustrated in the dendrogram (Figure 1). This analysis, based on multilocus enzyme electrophoresis, includes strains of the pathotypes associated with enteric disease and strains representing the major phylogenetic groups (groups A, B1, B2, and D) of the E. coli Reference (ECOR) collection, a set of natural isolates chosen to represent genetic variation in the E. coli species as a whole. The dendrogram includes pathogenic strains of the most common clones of five serogroups (O26, O111, O55, O128, and O157) associated with infectious diarrheal disease; these widespread clones are referred to as the DEC (diarrheagenic E. coli) clones. In addition, there are representatives of the common clones of enteroinvasive E. coli (8). The genetic distance between clones based on alleles detected by enzyme electrophoresis strongly correlates with the amount of sequence divergence in housekeeping genes (Figure 1b). The sequence data indicate that the deepest branches in the dendrogram reflect about 8% divergence at synonymous sites. It should be emphasized that because of past recombination, the dendrogram cannot be a true phylogeny but can only serve as a framework for investigating the evolution of the various clones.
Pathotypes of E. coli are concentrated in clonal groups, although some pathotypes are found in multiple lineages (Figure 1). In particular, there are two clusters of enteropathogenic E. coli (EPEC) that are associated with infantile diarrhea and two clusters of enterohemorrhagic E. coli (EHEC) that are associated with hemorrhagic colitis. The EPEC 1 and EHEC 1 clusters are highly divergent, whereas both EPEC 2 and EHEC 2 are more closely related to one another and fall into the B1 group of ECOR. The finding that independent lineages harbor the same virulence factors and cause clinically similar disease indicates that certain pathotypes have evolved multiple times in different clonal groups (7). EPEC and EHEC groups are phylogenetically distinct from the enteroinvasive E. coli (EIEC), bacteria that cause dysentery and are most closely related to strains of the ECOR group A. The clonal groups associated with enteric diseases are also different from those recovered in extraintestinal infections including uropathogenic E. coli (UPEC), which are found near the bottom of the dendrogram in the B2 and D groups of ECOR (9, 10).
Below, we focus on the virulence factors and pathogenic mechanisms of two major pathotypes, EPEC and EHEC, for which data exist both on the genetic basis of disease and on the phylogenetic history of the strains. These examples are put forward to demonstrate how genetic polymorphisms among E. coli strains profoundly influence disease. The reader is referred elsewhere for reviews of diarrheagenic (11) and extraintestinal E. coli pathogenesis (12).
Enteropathogenic E. coli
EPEC was the first group of strains recognized as pathogens, an insight that followed from serological studies comparing strains cultured from devastating outbreaks of neonatal diarrhea with other strains isolated from healthy infants. Although such outbreaks are now rare in developed countries, EPEC strains continue to be a leading cause of diarrhea among infants from developing countries worldwide (11). In recent years, the pathogenesis of EPEC infection has proved to be amenable to genetic dissection, and several themes have emerged.
A plasmid-encoded, type IV bundle-forming pilus critical for virulence.
In the early 1980s, investigators in the laboratory of James Kaper reported that a particular pattern of adherence to tissue culture cells by EPEC strains was associated with the presence of a large EPEC adherence factor (EAF) plasmid (11). Rather than covering tissue culture cells uniformly, EPEC strains form densely packed three-dimensional clusters on the surface of the cell, a pattern known as localized adherence. This pattern of adherence is so characteristic of and specific to EPEC strains that it can be used as the basis for diagnosis (11). The ability to perform localized adherence can be transferred to nonpathogenic laboratory E. coli strains by transformation with the EAF plasmid. Conversely, EPEC strains lose this ability and demonstrate attenuated pathogenicity when cured of this plasmid.
The principal factor responsible for the localized adherence phenotype is a surface appendage known as the bundle-forming pilus (BFP), a member of the type IV fimbria family that is encoded on the EAF plasmid (Figure 2a) (13). EPEC cells cluster because of the ability of BFP to reversibly aggregate into ropelike bundles. If any of the genes required for the formation of BFP are inactivated by mutation, the bacteria fail to form aggregates and do not display localized adherence (14). The major structural subunit of BFP is bundlin, a highly polymorphic protein encoded by bfpA of the EAF plasmid-borne bfp operon. Another protein, BfpF, which is predicted to be a cytoplasmic nucleotide-binding protein, plays a special role in aggregation. When bfpF is mutated, the bacteria continue to make pili that aggregate and allow the bacteria to do the same (15); however, the pili fail to form higher-order bundles and the bacteria remain trapped in aggregates (16). Interestingly, despite the fact that they remain capable of further steps in pathogenesis, bfpF mutants are significantly attenuated in their ability to cause diarrhea (17). Thus, it appears that not only the BFP structure, but also intact BFP function, is required for full virulence.
Figure 2.
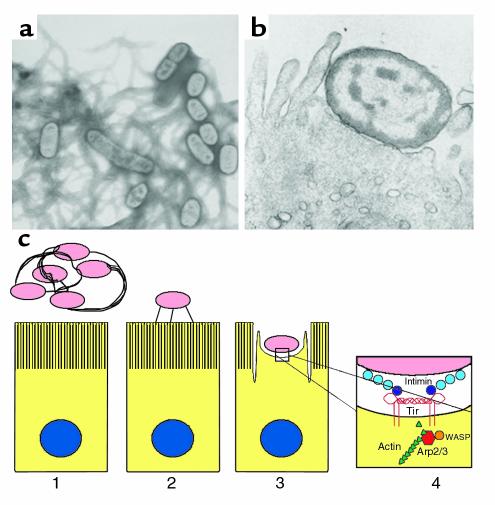
Pathogenesis of EPEC infection. (a) Electron micrograph of a culture of EPEC bacteria grown under conditions that lead to the production of type IV fimbria known as bundle-forming pili (BFP). BFP are required for bacterial aggregation and localized adherence to epithelial cells. (b) Electron micrograph of an EPEC bacterium engaged in attaching and effacing activity with a host intestinal epithelial cell. Note the loss of microvilli and the formation of a cuplike pedestal to which the bacterium is intimately attached. (c) A model of EPEC pathogenesis. A bacterial aggregate, connected by bundles of BFP fibers, is shown near an intestinal epithelial cell (panel 1). As infection proceeds, the bacteria detach from the pilus fibers, disaggregate, and become connected to the host cell through a surface appendage that contains EspA (panel 2). It is believed that Tir, EspB, and EspF travel through this appendage to the host cell. EspF is not required for attaching and effacing activity but plays a role in disruption of intestinal barrier function and host cell death. EspB and Tir are required for attaching and effacing activity (panel 3). The bacterial outer membrane protein intimin, composed of three immunoglobulin-like extracellular domains (D0–D2, light blue) and a receptor-binding lectin-like domain (D3, dark blue), binds to Tir in the host cell membrane (panel 4). Tir forms a four-helix bundle composed of two molecules each containing two antiparallel α helices connected by a hairpin loop. One intimin molecule binds to each loop of the dimer. Wiskott-Aldrich syndrome protein (WASP) is recruited to the pedestal where it activates the Arp2/3 complex to nucleate and polymerize actin.
A chromosomal pathogenicity island encoding a type III secretion system and the ability to alter the host cytoskeleton.
The histopathological hallmark of EPEC infection is the formation of intestinal lesions caused by the ability of bacterial cells to attach intimately to the host cell membrane, destroy microvilli, and induce the formation of cuplike pedestals composed of cytoskeletal proteins upon which the bacteria sit (Figure 2b). This ability, known as attaching and effacing activity, has been observed in vitro and in duodenal and rectal biopsies from infants with EPEC infection (11, 13).
A 35-kb genetic element known as the locus of enterocyte effacement (LEE) is necessary for this effect and, when cloned from EPEC strain E2348/69 #into a nonpathogenic E. coli strain, is sufficient to confer attaching and effacing activity (18). The LEE is considered to be a pathogenicity island because it contains virulence loci, it is not found in nonpathogenic E. coli strains, it is inserted into the genome of E. coli at specific sites (tRNA genes), and finally because its distinctive G+C content (38%) indicates its origin in another species. The LEE is inserted near different tRNA loci in different EPEC strains (18). The LEE from strain E2348/69 carries 41 genes, which encode a type III secretion system and various proteins secreted via this system, including an adhesin and its cognate receptor, a regulator, and several proteins of unknown function. Type III secretion systems are found in bacteria from several Gram-negative genera that have close relationships with eukaryotic hosts (19). These systems can transport bacterial proteins across the inner and outer membranes of the bacteria and the host cell plasma membrane and can deliver effector proteins to the surface or interior of host cells.
The proteins secreted via type III systems can be divided into two classes: the effector proteins, which are translocated to the host cell, and the components of the translocation apparatus, which are required to deliver the effector proteins into the host cell. The best-characterized EPEC effector protein is called Tir, for translocated intimin receptor. Tir is encoded by the LEE and is translocated via the type III system into host cells, where it is inserted in the plasma membrane (18, 20). Mutations in components of the type III secretion system or in the genes encoding two of the secreted proteins, EspA and EspB, prevent the translocation of Tir. Thus, EspA and EspB can be classified as part of the translocation apparatus. Tir has two membrane-spanning domains and is oriented so that both the amino- and the carboxy-termini protrude into the host cell cytoplasm (21). Once inserted into the host cell membrane, Tir serves as a receptor for intimin, an outer membrane protein required for virulence. Intimin is the product of the eae gene, located just downstream of tir in the LEE. Thus, EPEC have evolved an adherence mechanism in which the bacteria synthesize both the adhesin (intimin) and its receptor (Tir); the latter is inserted directly into the host cell by the LEE secretion apparatus.
Luo et al. (22) recently determined the three-dimensional structure of the extracellular domain of intimin bound to the extracellular domain of Tir. They identified a series of immunoglobulin-like domains (D0–D2) that give intimin a rigid, roughly cylindrical shape and a distal carboxy-terminal domain (D3) consisting of an incomplete C-lectin structure. In the cell membrane, Tir forms a dimer with each molecule consisting of a pair of antiparallel α helices separated by a hairpin turn. The entire structure is a four-helix bundle with the hairpin loops protruding from either side (Figure 2c). Intimin binds to Tir principally at the loops, such that each Tir dimer binds to two intimin molecules. Tir forms contacts with intimin along one side of the C-lectin domain. To achieve this configuration, both intimin and Tir appear to be oriented roughly parallel to both the bacterial and the eukaryotic cell membranes. This orientation accounts for the close contact (∼10 nm) between the bacteria and host cells in intimate adherence.
While Tir is clearly an effector protein, the roles of three other proteins, EspA, EspB, and EspD, which are encoded in an operon in the LEE and are secreted by EPEC via the type III system, are still being defined. EspA appears to be purely a component of the translocation apparatus. EspA molecules form a surface appendage that can be seen by electron microscopy bridging the bacteria and host cells (18). There is no evidence that EspA molecules penetrate the host cell cytoplasm or membranes. EspD has several putative transmembrane domains and has been observed in the host cell membrane (23). Because it is required for the translocation of EspB, EspD is also a part of the translocation apparatus. Interestingly, when espD is mutated, EspA filaments are much shorter than normal, suggesting a role for EspD in formation or stabilization of the translocation apparatus.
The function of the EspB protein is more enigmatic. While EspB is required for the translocation of Tir, indicating that it is a component of the translocation apparatus, EspB is itself translocated to the host cell. The protein has a hydrophobic stretch that could act as a transmembrane domain, and EspB molecules have been detected in the host cell membrane. Based on these observations, some investigators have suggested that EspB forms part of a pore that enables the passage of Tir into the host cell (18). However, when host cells are transfected with a vector that enables them to express EspB, their shape is radically altered and they lose stress fibers, suggesting that EspB also acts as an effector protein and affects cytoskeletal regulation (24).
What triggers the molecular events in the host cells that lead to the attaching and effacing activity? A recent study shows that the Arp2/3 complex, which nucleates and polymerizes actin, is localized within the actin-rich pedestals of attaching and effacing lesions (25). Members of the Wiskott-Aldrich syndrome protein (WASP) family, which activate the Arp2/3 complex, are also localized within the pedestals, and dominant-negative forms of WASP prevent attaching and effacing activity. Thus it has been proposed that EPEC activates WASP to stimulate the polymerization of actin (Figure 2c).
Recent work has shed light on the role in pathogenesis of another secreted protein, EspF. An espF mutant strain exhibits normal attaching and effacing activity (26) but fails to provoke a decrease in transepithelial electrical resistance — a phenotype, found in wild-type EPEC strains, that may be related to loss of intestinal barrier function and diarrhea in vivo (in this issue, ref. 27). In addition, the espF mutant fails to induce apoptosis in host cells, another feature of the EPEC–host cell interaction (28). Application of EspF to the exterior of cells has no effect, but synthesis of EspF in transfected cells results in rapid cell death. Interestingly, EspF contains proline-rich repeats that may serve as Src-homology 3 binding domains, allowing it to interact with as-yet unidentified host proteins. These domains could mediate the effects of EspF on intestinal barrier function and host cell apoptosis.
A large toxin that inhibits lymphocyte activation.
Several years ago, a factor was described that is produced by EPEC and related strains of E. coli and that inhibits lymphocyte activation. This heat-labile factor blocks lymphocyte proliferation and the production of IFN-γ, IL-2, IL-4, and IL-5. Although lymphocytes exposed to the factor are nonresponsive, there is no evidence that they undergo apoptosis or are killed. When the gene encoding this factor, lymphostatin, was cloned and mutated, the resulting strain could no longer inhibit lymphocyte function (29). A relatively short stretch of the sequence from this very large protein is homologous to the enzymatic domain of the large Clostridial cytotoxins, which covalently inactivate members of the Rho family of small mammalian GTPases. Sequences homologous to this gene are widespread but are distributed sporadically among EPEC and EHEC strains. The mechanism by which lymphostatin blocks lymphocyte activation and the role, if any, of lymphostatin in disease have not been established.
Two divergent groups of EPEC
As seen in Figure 1, two distinct phylogenetic groups have been identified that have a concentration of EPEC. Strains belonging to each of these groups display the serotypes that were first implicated in outbreaks of infantile diarrhea in the 1940s and 1950s.
The first group, EPEC 1, includes some of the originally identified adherent strains, most notably, strain E2348/69 (serotype O127:H6), the widely used model organism of human EPEC infection. This group comprises widespread clones with EPEC serotypes O55:H6, O119:H6, O125:H6, O127:H6, and O142:H6 (30). Bacteria of these clones usually carry both the LEE and the EAF plasmid, and they display typical localized adherence. EPEC 2 consists of other classical EPEC serotypes, such as O111:H2, O114:H2, O126:H2, and O128:H2. Some of these clones are common and very widespread. For example, DEC 12 (serotype O111:H2) has historically been the most common E. coli recovered from outbreaks of infantile diarrhea in the US and is the most frequently recovered O111 clone associated with diarrheal disease in Brazil (31). The EPEC 2 group also includes strain B171, an intensively studied O111 strain originally recovered from a diarrhea outbreak (17, 32).
The divergence between EPEC 1 and EPEC 2 is seen not only in their allelic differences in housekeeping genes, but also in their distinct intimin alleles and the sites at which the LEE pathogenicity island is inserted into the bacterial genome (18). In both EPEC groups, the bfp operon is carried on highly related EAF plasmids. Some of these plasmids are self-transmissible, but the single member of the group that has been sequenced in its entirety lacks genes for transmission (32). The bfpA gene, which encodes bundlin, the major structural subunit of BFP, shows considerable sequence variability. The eight known alleles can be separated into two groups (α and β). Because α and β bundlin alleles are distributed in both EPEC groups, it appears that the plasmids have recently spread horizontally (33). Comparison of the sequences of α and β bundlin also indicates an excess of nonsynonymous substitution in the 3′ end of the gene. This finding suggests the influence of positive selection for amino acid replacements and enhanced polymorphism in bundlin, which could be a source of variation in virulence among EPEC clones.
In summary, the interactions between EPEC and the host are complex (Figure 2c). A plasmid-encoded type IV BFP is essential for full virulence, but exactly how it facilitates infection is not clear. The LEE pathogenicity island encodes a type III secretion system, an outer membrane adhesin, and its cognate receptor necessary for attaching and effacing activity. An additional protein translocated to host cells induces host cell death and a loss of intestinal barrier function. A large toxin with lymphocyte inhibitory activity may aid the bacteria in forestalling an immune response. Finally, the combination of virulence factors that define EPEC has emerged at least twice in the evolutionary radiation of pathogenic E. coli. The extent to which these EPEC groups differ genetically and in virulence or epidemiological properties has not been fully explored.
Enterohemorrhagic E. coli
EHEC was first recognized as a cause of infectious diarrheal disease as a result of several outbreaks of severe bloody diarrhea (hemorrhagic colitis) in the early 1980s. Since then, EHEC strains, particularly serotype O157:H7, have been implicated worldwide in outbreaks of food- and water-borne disease in developed countries. The nomenclature for this group of organisms is confusing. EHEC belong to a larger group of pathogenic strains known as Shiga toxin–producing E. coli (STEC), which are defined by their ability to produce Shiga toxins (Stx). (For historical reasons, these same toxins are alternatively referred to as verotoxins and the organisms that produce them as VTEC.) EHEC are a subset of STEC that carry the LEE and exhibit attaching and effacing activity. EHEC strains of serotype O157:H7 have caused both the largest number of outbreaks and epidemics that have involved the greatest numbers of patients. Strains with this serotype have also caused the majority of sporadic STEC infections (34–36). Although EHEC O157:H7 strains contain large plasmids similar to those of EPEC, they lack the genes required for synthesis of BFP. Instead, EHEC plasmids carry a homologue of the lifA gene encoding lymphostatin, genes encoding a type II secretion system, catalase-peroxidase (katP), a secreted serine protease (espP), and a hemolysin operon (37, 38). It is not clear what, if any, proteins are secreted by the type II system. Indeed, the role of the EHEC plasmid in pathogenesis has not been confirmed in animal models of infection (11).
The most serious complication of EHEC infection is hemolytic-uremic syndrome (HUS). HUS is a microangiopathic hemolytic anemia characterized by disseminated capillary thrombosis and ischemic necrosis (34). The kidneys are the end organs most severely affected, but ischemic necrosis of the intestines, central nervous system (stroke), and indeed any organ may occur. Approximately 15% of those with HUS either die or are left with chronic renal failure (35). Because of the danger of HUS, EHEC strains and mutants cannot be tested in volunteers. EHEC strains of serotype O157:H7 can tolerate acidic environments and have a very low infectious dose. These strains colonize the gastrointestinal tract of cattle and may contaminate ground beef during processing. A variety of other foods including milk, juices, lettuce, and sprouts have been involved in outbreaks. The infection is acquired by ingestion of contaminated food or water or by person-to-person spread through close contact.
The recently completed genomic sequence of EHEC O157:H7 suggests that there exist many potential virulence genes, including fimbrial operons, other adhesins, toxins, secretion systems, and iron acquisition systems that have yet to be explored (5). In this review we will concentrate on two potential virulence systems that have been relatively well studied.
Production of Shiga toxins.
The Shiga toxins are the most important factor that differentiates EHEC from EPEC (11). These toxins are encoded by bacteriophages related to the classic λ phage, which lysogenize these strains. Stx1 and Stx2, the two most prevalent forms of the toxin found in EHEC strains pathogenic for humans, are encoded by closely related bacteriophages. Each toxin is composed of a single A subunit noncovalently associated with a pentamer composed of identical B subunits. The B subunits bind specifically to globotrioacyl ceramide and related glycolipids on host cells. The A subunit is taken up by endocytosis and transported to the endoplasmic reticulum. The toxin target is the 28S rRNA, which is depurinated by the toxin at a specific adenine residue, causing protein synthesis to cease and infected cells to die by apoptosis (39). Receptors for Stx are found on endothelial cells. Renal microvascular endothelial cells appear to be particularly sensitive to the toxin. It is presumed that Stx enter the systemic circulation after translocation across the intestinal epithelium (11) and damage endothelial cells, which leads to activation of coagulation cascades, formation of microthrombi, intravascular hemolysis, and ischemia.
The EHEC LEE.
The composition of the EHEC LEE from a serotype O157:H7 strain is very similar to that of a distantly related EPEC O127:H6 strain (18). The gene order of the elements from the two strains is identical and the predicted amino acid sequences of most of the proteins that compose the type III secretion systems are nearly identical. Three differences between the LEE elements of EPEC and EHEC stand out. The EHEC LEE is larger than that of EPEC due to the presence in the former of the remnants of a lysogenic bacteriophage. It appears that this phage entered the EHEC LEE subsequent to acquisition of the LEE by a progenitor of the EHEC strain. The EPEC and EHEC LEE show much greater sequence divergence in the sequences encoding intimin and the secreted proteins than in those for the secretion system. It has been suggested that this divergence could be the result of selective pressure exerted by the immune system of the host on the bacteria. The EHEC LEE has an espF gene, which could be involved in cell death and loss of intestinal barrier function. Interestingly, the predicted EHEC EspF protein has four proline-rich motifs, rather than three, as does the EPEC protein. Finally, unlike the LEE from EPEC strain E2348/69, the divergent LEE from O157:H7 EHEC does not confer attaching and effacing activity upon nonpathogenic strains of E. coli (40).
Evolution of EHEC groups.
Like EPEC, EHEC strains fall into two divergent clonal groups. EHEC 1 includes the O157:H7 clone complex and the closely related O55:H7 clone (DEC 5), an atypical EPEC clone. Bacteria of the O55:H7 clone (DEC 5) have the eae gene encoding intimin but most lack the EAF plasmid encoding BFP, and they do not typically display localized adherence (41). Bacteria of this clone invariably carry the eae gene, but otherwise they display a diverse array of virulence traits, suggesting that this pathogenic clone has a propensity to acquire new virulence factors.
In addition to its distinct virulence traits, E. coli O157:H7 is unusual in that these organisms do not ferment sorbitol rapidly or exhibit β-glucuronidase (GUD) activity, in contrast to most commensal E. coli (42). However, one sorbitol-positive (Sor+), nonmotile (H–) O157 clone that carries the eae gene and produces Stx2 has been implicated in an outbreak of HUS in Germany. Because the restriction digests of these Sor+ O157:H– strains differed from typical O157:H7 in pulsed field gel electrophoresis, Feng and coworkers (42) used multilocus enzyme electrophoresis to assess the clonal relationships among a variety of Stx-producing O157 strains. Their analysis revealed that these strains comprise a cluster of five closely related electropherotypes that differ from one another by only one or two enzyme alleles. The Sor+ O157:H– strains from Germany belong to the most divergent clone of the complex and appear to represent a new clone with similar virulence properties to those of O157:H7.
Stepwise evolution of E. coli O157:H7.
From the genotypic and phenotypic data, Feng and colleagues formulated an evolutionary model that posits a series of steps that led to the emergence of O157:H7 (42). The model is based on the assumption that during divergence, the probability of loss of function greatly exceeds that of gain of function for metabolic genes, that the gain of function usually occurs via lateral transfer of genes, and that the sequence of events invoking the fewest total steps is the most likely model.
The evolutionary steps are outlined in Figure 3a, which begins at the left with the ancestral or primitive states and progresses to the right to the contemporary or derived states. The model begins with an EPEC-like ancestor that is assumed to resemble most present-day E. coli in its ability to express β-glucuronidase (GUD+) and to ferment sorbitol (Sor+). From this EPEC-like ancestor, the immediate ancestor with the O55 somatic and the H7 flagellar antigens evolved. This ancestral cell, labeled A1, represents the most recent common ancestor of the EPEC O55:H7 clone and of EHEC O157:H7 and its relatives. A1 is assumed to have inherited its LEE (which is found near the selC gene in bacteria of this lineage) from an early EPEC-like ancestor carrying the γ variant of the eae gene. The next step, A1 to A2, was the acquisition of stx2, presumably by transduction by a toxin-converting bacteriophage, resulting in Stx2-producing O55:H7 strains. The next stage involved two changes, the acquisition of the EHEC plasmid and a switch in somatic antigen from O55 to O157. From here, the model proposes that two distinct lines evolved. In the lower path, the bacterial lineage lost motility, but it retained the Stx2 and the GUD+Sor+ primitive phenotypes, to give rise to the German O157:H– clone. Along the upper path, the lineage lost GUD activity and the ability to ferment sorbitol, and acquired the stx1 gene (presumably by phage conversion) to give rise to the phenotype of the common O157:H7 clone that has spread globally. Recent loss of stx genes and motility, in nature or during isolation and culture, would account for the variants among isolates of this clone.
Figure 3.
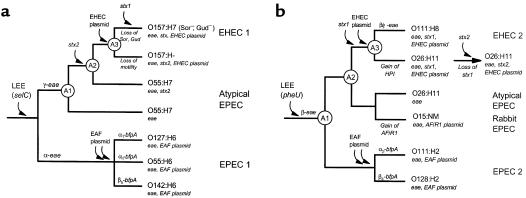
Cladograms of major evolutionary steps in the divergence of EPEC and EHEC clones. The two cladograms are based on the presence of the LEE at the selC (a) or pheU (b) loci. The diagrams are models of a branching order for the ancestry of the chromosomal backgrounds or clonal frames inferred from multilocus analysis. Branch lengths are arbitrary and not set to an evolutionary scale. Points of acquisition of principal virulence factors that define EPEC and EHEC are marked on the branches. Gains and losses of genes or phenotypes are marked below branches. The circles designate ancestral nodes referred to in the text. The EPEC (EAF) plasmid has two arrows to denote the possibility that it may have been acquired multiple times, a hypothesis to account for the α and β bundlin (bfpA) alleles occurring in both EPEC groups. HPI, high pathogenicity island.
The stepwise model of Feng et al. (42) makes specific predictions about the history of descent and the order of acquisition of virulence factors in the emergence of the EHEC pathotype. The model predicts that both O157:H7 and the German O157:H– were derived from an EPEC-like O55:H7 ancestor that carried the LEE and acquired the stx2 gene. This proposition is supported by the similarities between these strains in eae sequence (43) and by the presence of identical mutations in the gene for β-glucuronidase (42). The German O157:H– clone, however, represents an early-diverging member of the EHEC clone complex, which retained the ancestral ability to ferment sorbitol and to express GUD activity. The hypothesis of early divergence of this nonmotile clone is also supported by the observation that there are multiple mutations in fliC, presumably a result of the long-term silencing of flagellin expression.
The model also stipulates that stx2 was acquired once, before the somatic antigen transition to O157 and prior to the acquisition of the EHEC plasmid and stx1. Recent evidence, however, indicates that different O157:H7 strains harbor diverse Stx2-encoding phages (44). The relative significance of mutation and of recombination or gene conversion in explaining the diversity of toxin-converting phages remains to be elucidated.
A second group of EHEC.
An unexpected finding of the evolutionary analysis was that the O157:H7 cluster is only distantly related to a second group of Stx-producing strains (primarily serotypes O26:H11 and O111:H8), which were originally classified as EHEC along with O157:H7. Bacteria of these two EHEC groups have in common a large plasmid (pO157) that encodes a variety of putative virulence factors (37).
Much less is known about the virulence properties, epidemiology, and evolution of the EHEC 2 group, the most commonly isolated group of non-O157, Stx-producing strains. Although they often have serotypes O111:H8, O111:H–, O26:H11, or O26:H–, members of EHEC 2 include diverse O:H serotypes, and many of these strains are nonmotile or nontypable with standard antisera. Because members of this group have the same principal virulence factors as E. coli O157:H7 (i.e., Stx and the LEE) and are recovered from patients with hemorrhagic colitis and HUS, they have been classified together with O157:H7 as EHEC. However, evolutionary genetic analysis indicates that this group is sufficiently divergent from E. coli O157:H7 (7) to be considered as a second group of EHEC (30).
EHEC 2 includes several widespread clones, including, for example, a common nonmotile O111 clone that occurs in both North and South America (29). Members of this clone have eae and produce both Stx1 and enterohemolysin (31). Interestingly, the EHEC 2 group also includes some non–Stx-producing pathogens, such as RDEC-1, an O15:NM isolate from a case of rabbit diarrhea that has been used as a model organism for human EPEC infection.
A stepwise evolutionary model can be hypothesized to explain the radiation of the various clones of the EHEC 2 group (Figure 3b). The emergence of this pathogenic lineage is thought to begin with the acquisition of a LEE island, which is located at the pheU site, because this is a conserved characteristic found in both EPEC 2 and EHEC 2 strains (7). This ancestral LEE carried an ancestral β intimin gene, which is found among the diverse serotypes in these groups. From the ancestral EPEC-like strain (A1), one lineage led to the EPEC 2 group of strains characterized by the localized adherence phenotype encoded on the EAF plasmid, and the other lineage (A2) led to the EHEC 2 group of strains.
The subsequent stages in the evolution of the EHEC 2 group are not yet clear but apparently involved multiple gains and losses of Shiga-toxin genes and pathogenicity islands. In Figure 3b, we have assembled the information into a sequence of events that is highly speculative and requires further study. We posit that A2 was an ancestral O26:H11 strain that eventually acquired an stx1 phage and an EHEC plasmid to give rise to the widespread EHEC O26:H11 clone. A2 was also the recent ancestor that experienced an antigenic shift to O111 to produce the EHEC O111 clone. Data from multilocus sequencing and multilocus enzyme electrophoresis show that these two EHEC clones are closely related genetically, indicating that these events occurred recently in evolution.
Other important genetic changes have also occurred. Karch and colleagues (45) have recently shown that the O26:H11 clone carries a pathogenicity island homologous to sequences from pathogenic Yersinia, and that this so-called high pathogenicity island (HPI) is not found in the closely related O111 strains. From an evolutionary perspective, this observation suggests that the HPI was either very recently acquired in the O26 lineage or recently lost in the O111 lineage. Further divergence of the O26:H11 and O111:H8 EHEC clones also appears to have involved recombination within the intimin gene. EHEC O111 strains carry a mosaic intimin allele (β/γ-eae) with the sequence of the conserved trans-membrane domain resembling the β-eae gene and the sequences encoding the variable external domains resembling γ-eae (C.L. Tarr and T.S. Whittam, unpublished results). The nature of the recombination event and its influence on the intimin-Tir interaction has yet to be illuminated.
The dynamic nature of clonal evolution in the EHEC 2 group is perhaps best seen in a recent finding that there has been a dramatic replacement of O26 clones in Europe in the past decade (46). The clonal replacement, detected with pulsed field gel electrophoresis comparisons of O26 strains, indicates that a new subclone with stx2 and a distinct EHEC plasmid variant has spread over the past several years to high frequency (46). Presumably this new type has been recently derived from the common O26:H11 EHEC clone (Figure 3b).
Because EHEC 2 strains share the prominent virulence factors of O157:H7 and cause similar disease, and are also common in the bovine reservoir, it is possible that these organisms will emerge as important food-borne pathogens in North America.
Conclusions
E. coli serves as a prime example of the role of polymorphisms within a bacterial species in human disease. A multitude of E. coli pathotypes cause distinct diseases. Genetic variation, both acquired through the horizontal spread of virulence factors and present in certain lineages that are inherently more pathogenic, is responsible for these diverse clinical entities. Studies of two pathotypes, EPEC and EHEC, have been particularly revealing, and the molecular and cellular basis of pathogenesis for both of these pathotypes is emerging. In addition, studies of clonal relationships have illuminated the evolution of these pathogens. One of the important themes that has emerged from studies of polymorphisms within virulence factor genes is the presence of increased rates of nonsynonymous substitution (amino acid–altering mutations) in surface-exposed and secreted proteins, implying the influence of diversifying selection on polymorphism. This effect is seen in the divergence of the LEE-borne genes of EPEC and EHEC: the genes for Tir, intimin, and several of the Esp’s have levels of nonsynonymous change five to ten times greater than seen in housekeeping genes. Bundlin is also highly polymorphic and has experienced an accelerated rate of nonsynonymous substitution in the 3′ end of the gene. Presumably the increased diversity helps the individual organism to escape the immune response within a host or favors spread of a variant in a population against the effects of herd immunity. Evidence for recombination within virulence factor genes also illustrates the potential for reintroduction of mobile genetic elements containing virulence factors into established pathogens to increase diversity. E. coli may thus be viewed as a rapidly evolving species capable of generating new pathogenic variants that can foil host protective mechanisms and result in new disease syndromes.
Acknowledgments
This work was supported by Public Health Service awards AI-32074, AI-37606, and DK-49720 (to M.S. Donnenberg) and AI-43291 (to T.S. Whittam) from NIH. The authors are grateful to Rick Blank for supplying the electron micrograph shown in Figure 2a. An earlier, more extensively referenced version of this review is available from the author at http://medschool.umaryland.edu/infeMSD/som.html.
References
- 1.Whittam, T.S. 1996. Genetic variation and evolutionary processes in natural populations of Escherichia coli. In Escherichia coli and Salmonella: cellular and molecular biology. F.C. Neidhardt, editor. ASM Press. Washington, DC, USA. 2708–2720.
- 2.Pupo GM, Karaolis DKR, Lan RT, Reeves PR. Evolutionary relationships among pathogenic and nonpathogenic Escherichia coli strains inferred from multilocus enzyme electrophoresis and mdh sequence studies. Infect Immun. 1997;65:2685–2692. doi: 10.1128/iai.65.7.2685-2692.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blattner FR, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 4.Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci USA. 1998;95:9413–9417. doi: 10.1073/pnas.95.16.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perna NT, et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001;409:529–533. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- 6.Milkman R. Recombination and population structure in Escherichia coli. Genetics. 1997;146:745–750. doi: 10.1093/genetics/146.3.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reid SD, Herbelin CJ, Bumbaugh AC, Selander RK, Whittam TS. Parallel evolution of virulence in pathogenic Escherichia coli. Nature. 2000;406:64–67. doi: 10.1038/35017546. [DOI] [PubMed] [Google Scholar]
- 8.Martinez MB, Whittam TS, McGraw EA, Rodrigues J, Trabulsi LR. Clonal relationship among invasive and non-invasive strains of enteroinvasive Escherichia coli serogroups. FEMS Microbiol Lett. 1999;172:145–151. doi: 10.1111/j.1574-6968.1999.tb13462.x. [DOI] [PubMed] [Google Scholar]
- 9.Picard B, et al. The link between phylogeny and virulence in Escherichia coli extraintestinal infection. Infect Immun. 1999;67:546–553. doi: 10.1128/iai.67.2.546-553.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bingen E, et al. Phylogenetic analysis of Escherichia coli strains causing neonatal meningitis suggests horizontal gene transfer from a predominant pool of highly virulent B2 group strains. J Infect Dis. 1998;177:642–650. doi: 10.1086/514217. [DOI] [PubMed] [Google Scholar]
- 11.Nataro JP, Kaper JB. Diarrheagenic Escherichia coli. Clin Microbiol Rev. 1998;11:142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donnenberg, M.S., and Welch, R.A. 1996. Virulence determinants of uropathogenic Escherichia coli. In Urinary tract infections: molecular pathogenesis and clinical management. H.L.T. Mobley and J.W. Warren, editors. ASM Press. Washington, DC, USA. 135–174.
- 13.Donnenberg MS. Interactions between enteropathogenic Escherichia coli and epithelial cells. Clin Infect Dis. 1999;28:451–455. doi: 10.1086/515159. [DOI] [PubMed] [Google Scholar]
- 14.Anantha RP, Stone KD, Donnenberg MS. Effects of bfp mutations on biogenesis of functional enteropathogenic Escherichia coli type IV pili. J Bacteriol. 2000;182:2498–2506. doi: 10.1128/jb.182.9.2498-2506.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anantha RP, Stone KD, Donnenberg MS. The role of BfpF, a member of the PilT family of putative nucleotide-binding proteins, in type IV pilus biogenesis and in interactions between enteropathogenic Escherichia coli and host cells. Infect Immun. 1998;66:122–131. doi: 10.1128/iai.66.1.122-131.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knutton S, Shaw RK, Anantha RP, Donnenberg MS, Zorgani AA. The type IV bundle-forming pilus of enteropathogenic Escherichia coli undergoes dramatic alterations in structure associated with bacterial adherence, aggregation and dispersal. Mol Microbiol. 1999;33:499–509. doi: 10.1046/j.1365-2958.1999.01495.x. [DOI] [PubMed] [Google Scholar]
- 17.Bieber D, et al. Type IV pili, transient bacterial aggregates, and virulence of enteropathogenic Escherichia coli. Science. 1998;280:2114–2118. doi: 10.1126/science.280.5372.2114. [DOI] [PubMed] [Google Scholar]
- 18.Frankel G, et al. Enteropathogenic and enterohaemorrhagic Escherichia coli: more subversive elements. Mol Microbiol. 1998;30:911–921. doi: 10.1046/j.1365-2958.1998.01144.x. [DOI] [PubMed] [Google Scholar]
- 19.Lee CA. Type III secretion systems: machines to deliver bacterial proteins into eukaryotic cells? Trends Microbiol. 1997;5:148–156. doi: 10.1016/S0966-842X(97)01029-9. [DOI] [PubMed] [Google Scholar]
- 20.DeVinney R, Knoechel DG, Finlay BB. Enteropathogenic Escherichia coli: cellular harassment. Curr Opin Microbiol. 1999;2:83–88. doi: 10.1016/s1369-5274(99)80014-9. [DOI] [PubMed] [Google Scholar]
- 21.Kenny B. Phosphorylation of tyrosine 474 of the enteropathogenic Escherichia coli (EPEC) Tir receptor molecule is essential for actin nucleating activity and is preceded by additional host modifications. Mol Microbiol. 1999;31:1229–1241. doi: 10.1046/j.1365-2958.1999.01265.x. [DOI] [PubMed] [Google Scholar]
- 22.Luo Y, et al. Crystal structure of enteropathogenic Escherichia coli intimin-receptor complex. Nature. 2000;405:1073–1077. doi: 10.1038/35016618. [DOI] [PubMed] [Google Scholar]
- 23.Wachter C, Beinke C, Mattes M, Schmidt MA. Insertion of EspD into epithelial target cell membranes by infecting enteropathogenic Escherichia coli. Mol Microbiol. 1999;31:1695–1707. doi: 10.1046/j.1365-2958.1999.01303.x. [DOI] [PubMed] [Google Scholar]
- 24.Taylor KA, Luther PW, Donnenberg MS. Expression of the EspB protein of enteropathogenic Escherichia coli within HeLa cells affects stress fibers and cellular morphology. Infect Immun. 1999;67:120–125. doi: 10.1128/iai.67.1.120-125.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalman D, et al. Enteropathogenic E. coli acts through WASP and Arp2/3 complex to form actin pedestals. Nat Cell Biol. 1999;1:389–391. doi: 10.1038/14087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McNamara BP, Donnenberg MS. A novel proline-rich protein, EspF, is secreted from enteropathogenic Escherichia coli via the type III export pathway. FEMS Microbiol Lett. 1998;166:71–78. doi: 10.1111/j.1574-6968.1998.tb13185.x. [DOI] [PubMed] [Google Scholar]
- 27.McNamara BP, et al. Translocated EspF protein from enteropathogenic Escherichia coli disrupts host intestinal barrier function. J Clin Invest. 2001;107:621–629. doi: 10.1172/JCI11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crane, J.K., McNamara, B.P., and Donnenberg, M.S. 2001. Role of EspF in host cell death induced by enteropathogenic Escherichia coli.Cellular Microbiology. In press. [DOI] [PubMed]
- 29.Klapproth J-M, et al. A large toxin from pathogenic Escherichia coli strains that inhibits lymphocyte activation. Infect Immun. 2000;68:2148–2155. doi: 10.1128/iai.68.4.2148-2155.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whittam TS, McGraw EA. Clonal analysis of EPEC serogroups. Revista de Microbiologia. 1996;27(Suppl. 1):7–16. [Google Scholar]
- 31.Campos LC, Whittam TS, Gomes TAT, Andrade JRC, Trabulsi LR. Escherichia coli serogroup O111 includes several clones of diarrheagenic strains with different virulence properties. Infect Immun. 1994;62:3282–3288. doi: 10.1128/iai.62.8.3282-3288.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tobe T, et al. Complete DNA sequence and structural analysis of the enteropathogenic Escherichia coli adherence factor plasmid. Infect Immun. 1999;67:5455–5462. doi: 10.1128/iai.67.10.5455-5462.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blank TE, Zhong H, Bell AL, Whittam TS, Donnenberg MS. Molecular variation among type IV pilin (bfpA) genes from diverse enteropathogenic Escherichia coli strains. Infect Immun. 2000;68:7028–7038. doi: 10.1128/iai.68.12.7028-7038.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyce TG, Swerdlow DL, Griffin PM. Current concepts: Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med. 1995;333:364–368. doi: 10.1056/NEJM199508103330608. [DOI] [PubMed] [Google Scholar]
- 35.Tarr PI. Escherichia coli 0157:H7: clinical, diagnostic, and epidemiological aspects of human infection. Clin Infect Dis. 1995;20:1–10. doi: 10.1093/clinids/20.1.1. [DOI] [PubMed] [Google Scholar]
- 36.Griffin PM, Tauxe RV. The epidemiology of infections caused by Escherichia coli O157:H7, other enterohemorrhagic E. coli, and the associated hemolytic uremic syndrome. Epidemiol Rev. 1991;13:60–98. doi: 10.1093/oxfordjournals.epirev.a036079. [DOI] [PubMed] [Google Scholar]
- 37.Burland V, et al. The complete DNA sequence and analysis of the large virulence plasmid of Escherichia coli O157:H7. Nucleic Acids Res. 1998;26:4196–4204. doi: 10.1093/nar/26.18.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makino K, et al. Complete nucleotide sequences of 93-kb and 3.3-kb plasmids of an enterohemorrhagic Escherichia coli O157:H7 derived from Sakai outbreak. DNA Res. 1998;5:1–9. doi: 10.1093/dnares/5.1.1. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida T, et al. Primary cultures of human endothelial cells are susceptible to low doses of Shiga toxins and undergo apoptosis. J Infect Dis. 1999;180:2048–2052. doi: 10.1086/315116. [DOI] [PubMed] [Google Scholar]
- 40.Elliott SJ, Yu J, Kaper JB. The cloned locus of enterocyte effacement from enterohemorrhagic Escherichia coli O157:H7 is unable to confer the attaching and effacing phenotype upon E-coli K-12. Infect Immun. 1999;67:4260–4263. doi: 10.1128/iai.67.8.4260-4263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pelayo JS, et al. Virulence properties of atypical EPEC strains. J Med Microbiol. 1999;48:41–49. doi: 10.1099/00222615-48-1-41. [DOI] [PubMed] [Google Scholar]
- 42.Feng P, Lampel KA, Karch H, Whittam TS. Genotypic and phenotypic changes in the emergence of Escherichia coli O157:H7. J Infect Dis. 1998;177:1750–1753. doi: 10.1086/517438. [DOI] [PubMed] [Google Scholar]
- 43.McGraw EA, Li J, Selander RK, Whittam TS. Molecular evolution and mosaic structure of α, β, and γ intimins of pathogenic Escherichia coli. Mol Biol Evol. 1999;16:12–22. doi: 10.1093/oxfordjournals.molbev.a026032. [DOI] [PubMed] [Google Scholar]
- 44.Wagner PL, Acheson DW, Waldor MK. Isogenic lysogens of diverse shiga toxin 2-encoding bacteriophages produce markedly different amounts of Shiga toxin. Infect Immun. 1999;67:6710–6714. doi: 10.1128/iai.67.12.6710-6714.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karch H, et al. A genomic island, termed high-pathogenicity island, is present in certain non-O157 Shiga toxin-producing Escherichia coli clonal lineages. Infect Immun. 1999;67:5994–6001. doi: 10.1128/iai.67.11.5994-6001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang WL, et al. Molecular characteristics and epidemiological significance of Shiga toxin-producing Escherichia coli O26 strains. J Clin Microbiol. 2000;38:2134–2140. doi: 10.1128/jcm.38.6.2134-2140.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]