

Abstract
A new method is described for the detection of disfavored reaction pathways. The approach combines organic synthesis, to independently prepare reactant and product, with the low detection limits of confocally adjusted fluorescence correlation spectroscopy. Selective detection of disfavored products is achieved by designing a system in which the analyte displays a unique absorption and emission of light. This was accomplished through application of a substance-selective intramolecular charge transfer. The power of this technique was demonstrated by monitoring the progress of the thermal retro-[2+2]-cycloaddition. In conjunction with concurrent 1H-NMR monitoring, the relative abundance of major and disfavored reaction products can be determined and used to calculate the energetics of processes disfavored by more than 5 kcal/mol.
Keywords: fluorescence correlation spectroscopy, retrocycloaddition, energetically disfavored reactions, regioselectivity
The development of new strategies for the enhancement of disfavored processes requires an efficient means to specifically characterize a minuscule amount of one compound in a gross excess of similar substances (1). At an ambient 10 kcal/mol (1 kcal = 4.18 kJ) of disfavoring, this would correspond to detection of one specific molecule in the presence of 17 million others. Therefore, an effective screen must be capable of rapidly detecting select species at low concentrations, ideally at the molecular level. Recently, Eigen and Rigler (2) described a method to detect single molecules by monitoring the time-dependent autocorrelated fluorescence of a molecule of rhodamine-labeled DNA diffusing through a stationary beam of light. To reach the single-molecule level, a volume element of 0.2 fl was generated by the placement of a confocally imaged pinhole within the beam of a diffraction-limited laser, allowing the detection of fluorophores at concentrations as low as 10−18 M (3). When joined with a means for selective excitement and/or fluorescence, this technique provides a powerful tool to specifically quantify the low level of a minor product within a reaction array. This paper describes an application of this method for the detection an unfavorable product of the thermal [2+2]-cycloreversion.
In the context of the Woodward and Hoffmann rules, the thermal retro-[2+2]-cycloaddition is symmetry forbidden and therefore proceeds through an intermediate whose character lies between that of a diradical and zwitterion (4–6). As shown in Scheme I ![]() , 3-methylenecyclohexene is thermally stable; however, its photochemically generated spirocyclobutanes readily undergo thermal reversal (7). For the most part, the regiochemical outcome of methyl- and vinyl-substituted cyclobutane fragmentation can be described by the stability of its intrinsic diradical intermediate(s) (8, 9). From this, one can estimate that cleavage of 1,2-diphenylcyclobutane, as shown in Scheme II
, 3-methylenecyclohexene is thermally stable; however, its photochemically generated spirocyclobutanes readily undergo thermal reversal (7). For the most part, the regiochemical outcome of methyl- and vinyl-substituted cyclobutane fragmentation can be described by the stability of its intrinsic diradical intermediate(s) (8, 9). From this, one can estimate that cleavage of 1,2-diphenylcyclobutane, as shown in Scheme II  , would favor the production of styrene by approximately the radical-stabilization energy imparted by addition of phenyl group, typically 12 kcal/mol (10). At room temperature, this corresponds the production of one molecule of stilbene in the presence of 500 million molecules of styrene. Experimentally, cis-1,2-diphenylcyclobutane undergoes a first-order fragmentation to styrene, activation energy Ea = 35.8 kcal/mol, and rearrangement to trans-1,2-diphenylcyclobutane, Ea = 35.8 kcal/mol, at 200°C in tetrachloroethylene. The corresponding trans isomer likewise fragments at slightly elevated temperature to again provide styrene (11). In both instances, stilbene was not detected. This report describes the design of a system to detect and quantify the relative population of stilbene. In doing so, one needs a means to dramatically differentiate between stilbene and styrene. Chemically, this can accomplished by modifying the system such that the styrene product absorbs and/or fluoresces within a unique region of the visible spectrum. One way to accomplish this is to take advantage of intramolecular charge transfer. The extent of this effect can be seen in the comparison of trans,trans-1-(4-nitrophenyl)-4-(4-N,N-dimethylamino)butadiene (NND), absorption λmax = 418 nm in heptane and fluorescence λmax = 602 nm in benzene (12), to trans,trans-1,4-diphenylbutadiene, absorption λmax = 313 nm in hexane, fluorescence λmax = 412, 455, and 510 nm in benzene (13). Developed upon these principles, this report exposes a new means to quantify the energy of reactions which are not observed with the more traditional spectroscopic techniques, such as NMR, IR, UV–visible, and fluorescence spectroscopy.
, would favor the production of styrene by approximately the radical-stabilization energy imparted by addition of phenyl group, typically 12 kcal/mol (10). At room temperature, this corresponds the production of one molecule of stilbene in the presence of 500 million molecules of styrene. Experimentally, cis-1,2-diphenylcyclobutane undergoes a first-order fragmentation to styrene, activation energy Ea = 35.8 kcal/mol, and rearrangement to trans-1,2-diphenylcyclobutane, Ea = 35.8 kcal/mol, at 200°C in tetrachloroethylene. The corresponding trans isomer likewise fragments at slightly elevated temperature to again provide styrene (11). In both instances, stilbene was not detected. This report describes the design of a system to detect and quantify the relative population of stilbene. In doing so, one needs a means to dramatically differentiate between stilbene and styrene. Chemically, this can accomplished by modifying the system such that the styrene product absorbs and/or fluoresces within a unique region of the visible spectrum. One way to accomplish this is to take advantage of intramolecular charge transfer. The extent of this effect can be seen in the comparison of trans,trans-1-(4-nitrophenyl)-4-(4-N,N-dimethylamino)butadiene (NND), absorption λmax = 418 nm in heptane and fluorescence λmax = 602 nm in benzene (12), to trans,trans-1,4-diphenylbutadiene, absorption λmax = 313 nm in hexane, fluorescence λmax = 412, 455, and 510 nm in benzene (13). Developed upon these principles, this report exposes a new means to quantify the energy of reactions which are not observed with the more traditional spectroscopic techniques, such as NMR, IR, UV–visible, and fluorescence spectroscopy.
MATERIALS AND METHODS
Unless otherwise noted, all reagents and chemicals were purchased from commercial sources and were used without further purification. Reactions were conducted under an argon atmosphere in rigorously dried glassware and magnetically stirred with a Teflon-coated stirbar. Anhydrous solvents were freshly distilled as follows: tetrahydrofuran (THF) from sodium benzophenone ketyl and methylene chloride from calcium hydride. Materials to be allowed to react under anhydrous conditions were dried extensively with toluene azeotrope prior to use. TLC on Merck silica gel DC 60 plates was routinely used to monitor reactions and stained with iodine absorbed on silica gel. Rf values were collected from runs in 33:67 (vol/vol) ethyl acetate/hexane. Melting points were measured on a Büchi 520 apparatus and are uncorrected. IR spectra were collected on a Perkin–Elmer Paragon 1000 PC Fourier-transform IR spectrometer. Samples were prepared on sodium chloride plates, neat or in a chloroform smear. UV–visible and fluorescence spectra were measured on a Perkin–Elmer Lambda 17 UV-Vis spectrometer and a Perkin–Elmer LS-5B luminescence spectrometer, respectively. 1H-NMR and 13C-NMR spectra were obtained on a Bruker MSL300 at 300 MHz and 75 MHz, respectively. Chemical shifts (δ) are given in ppm and coupling constants (J) in Hz. Microanalyses were obtained from Beller Microanalytisches Labor (Göttingen, Germany). Standard flash chromatography was performed on Merck 9395 silica gel using a gradient from hexane to the solvent listed. All materials for fluorescence correlation spectroscopy (FCS) studies were purified by repetitive column chromatography using spectral grade solvents on Merck type 7754 silica gel, which was previously soaked in and washed with spectral grade chloroform to remove any possible fluorescent contaminants. Samples were further recrystallized from spectral grade solvents where possible. Reactions are summarized in Scheme III, shown in Fig. 1.
Figure 1.

Reaction a: allyltriphenylphosphonium bromide, n-butyllithium, THF, 0°C to room temperature (RT), 3 h, 94% yield of a 1:1 mixture. Reaction b: NaBH4, MeOH/THF/Et2O (5:5:1), 0°C to RT, 97% yield. Reaction c: CBr4, PPh3, 4-Å molecular sieves, CH2Cl2, 0°C to RT, 86% yield. Reaction d: P(OEt)3, dimethylformamide (DMF), 155°C, 1.5 h, 99% yield. Reaction e: (i) sodium hexamethyldisilazide (NaHMDS) (2 eq), DMF, THF, 0°C to RT, 1 h; (ii) N,N-dimethylaminocinnamaldehyde, THF, −20°C to RT, 6 h, 55% yield. Reaction f: (i) diisobutylaluminum hydride (DIBAL-H) (3.5 eq), PhCH3, −78°C, 45 min; (ii) quench with 10% HOAc/H2O, −78°C to RT, 1 h, 68% yield of a 1:1 mixture. Reaction g: (i) 8, NaHMDS, DMF, THF, 0°C to RT, 1 h; (ii) add 10 in THF, 6 h, 0°C to RT, 4 h, 89% yield.
Synthesis of Cyclobutane 1.
Styrene 3 (5.48 g, 28.7 mmol), synthesis below, dissolved in 37 ml of acrylonitrile (572 mmol) and 1.8 liters of spectral-grade toluene was deoxygenated by bubbling with argon for 15 min and then irradiated with a high-pressure mercury lamp (150 W) in a Duran glass vessel (>310 nm). Ambient temperature was maintained by submerging the light source into a quartz cavity, cooled with continuous water flow, and harvesting the light outside. After irradiation for 148 h, the volatile components were removed by rotary evaporation and pure material was obtained by flash chromatography (20% ethyl acetate/hexane), yielding 640 mg (18%) of 9: Rf = 0.45; 1H (CDCl3) (D = 2H): δ 7.10 (d, J = 8.6 Hz, 2H), 6.70 (d, J = 8.6 Hz, 2H), 3.75 (q, J = 8.5 Hz, 1H), 3.48 (p, J = 3.8 Hz, 1H), 2.83 (s, 6H), 2.47 (m, 1H), 2.38 (m, 1H), 2.26 (m, 1H), 2.09 (m, 1H); 13C (CDCl3): 23.1, 26.1, 31.1, 40.5, 41.7, 122–7, 127.9; IR (CHCl3): 3084, 2887, 2801, 1614, 1520, 1444, 1408, 1353, 1222, 1166, 1062, 989, 947, 887, 821, 536, 502 cm−1. Elemental analysis (C13H16N2): calculated, C 77.97, H 8.05, N 13.99; found, C 78.06, H 8.13, N 13.85.
At −78°C, 1.13 ml of a 1 M solution of DIBAL-H in toluene (1.5 eq) was added over 15 min to nitrile 9 (150.2 mg, 0.75 mmol) dissolved in 5 ml of toluene. After 45 min at −78°C, the reaction was quenched with 1 ml of 10% aqueous HOAc and warmed to room temperature. The resulting solution was partitioned between 40 ml of CH2Cl2 and 15 ml of saturated sodium bicarbonate and extracted. The aqueous phase was further extracted with three 40-ml portions of CH2Cl2. The combined organic layers were dried with Na2SO4, concentrated, and directly submitted to flash chromatography (25% ethyl acetate/hexane) yielding 104.5 mg (68%) as a 1:1 mixture of aldehydes 14: Rf = 0.49; 1H (CDCl3): trans isomer: δ 9.75 (d, J = 1.9 Hz, 1H), 7.10 (d, J = 8.6 Hz, 2H), 6.68 (d, J = 8.6 Hz, 2H), 3.65 (m, 1H), 3.23 (m, 2H), 2.38–2.21 (m, 4H); cis isomer: δ 9.50 (d, J = 2.7 Hz, 1H), 7.05 (d, J = 8.3 Hz, 2H), 6.66 (d, J = 8.3 Hz, 2H), 3.99 (q, J = 8.7 Hz, 1H), 3.43 (m, 1H), 2.38–2.21 (m, 3H), 2.12 (m, 1H); IR (CHCl3): 2917, 1723, 1614, 1523, 1351, 818, 717 cm−1.
A solution of bromide 7 (62.1 mg, 0.268 mmol), synthesis below, and triethylphosphite (53.2 μl, 0.314 mmol) was heated at 150°C for 1.5 h in 1 ml of DMF. This solution was gradually cooled to 0°C and fresh sodium bis(trimethylsilyl)amide (0.54 ml, 1.0 M in THF) was added. The appropriate deprotonation was verified by the fact that a deep magenta color was produced upon exceeding the first equivalent of base. After 30 min at 0°C, the solution was warmed to room temperature and kept there for 20 min. Aldehydes 10 (12.4 mg, 0.061 mmol) were introduced in 3 ml of THF to the above solution at 0°C. After 3 h at room temperature, 0.5 ml of water was added and the pH was adjusted to 7 with 5% aqueous HCl. Crude product was obtained by repetitive extraction with CH2Cl2, dried with Na2SO4, and concentrated. Flash chromatography (10% ethyl acetate/hexane) afforded 24.7 mg (89%) of a 4:1 mixture of trans and cis isomers (at the ring junction). The pure trans isomer 1 was obtained by repetitive flash chromatography on Merck 7754 silica gel with spectral-grade 10% ethyl acetate/hexane or by thermolysis at 120°C in p-xylenes. Rf = 0.52; 13C (CDCl3): trans isomer: δ 24.6, 26.3, 40.7, 43.9, 47.1, 112.9, 116.4, 118.0, 125.3, 126.7, 127.2, 128.2, 132.0, 145.5, 147.5, 155.7; cis isomer: δ 24.6, 26.3, 40.7, 43.9, 47.1, 112.9, 116.4, 118.0, 125.3, 126.7, 127.2, 128.2, 132.0, 145.5, 147.5, 155.7. IR (neat): 3444, 2924, 1618, 1594, 1523, 1478, 1437, 1272, 1192, 1167, 818, 753 cm−1. High-resolution chemical ionization MS: calculated, 338.1630; found, 338.1639.
Synthesis of 2.
Sodium bis(trimethylsilyl)amide (1.27 ml, 1.0 M in THF) was added to a solution of phosphate 8 (143.6 mg, 0.453 mmol), synthesis below, in 0.8 ml of DMF at 0°C. Thirty minutes later, the solution was warmed to room temperature and kept there for 20 min. At this point, the contents were recooled to −20°C and allowed to react with a solution of N,N-dimethylaminocinnamaldehyde (74.2 mg, 0.430 mmol) in 4 ml of THF. After 8 h at ambient temperature, 15 ml of ice-cold brine was added. The pH was adjusted to 7 with dilute HCl and the crude product was obtained by repetitive extraction with 10% THF in CH2Cl2, drying with Na2SO4, and concentration. Flash chromatography (33% CHCl3/hexane) and recrystallization from 10:1 heptane/THF provided 302.7 mg (55%) of 2: mp 216.3–217.2°C; Rf = 0.43; (CDCl3): δ 10.74 (s, 1H), 7.99 (d, J = 8.7 Hz, 1H), 7.24 (d, J = 8.7 Hz, 2H), 7.08 (dd, J = 7.8, 15.5 Hz, 1H), 7.03 (dd, J = 7.8, 16.9 Hz, 1H), 7.02 (d, J = 16.9 Hz, 1H), 6.75 (d, J = 7.8 Hz, 2H), 6.74 (s, 1H), 6.65 (d, J = 8.7 Hz, 2H), 6.47 (d, J = 15.4 Hz, 1H), 2.98 (s, 6H); 13C-NMR (dimethyl-d6 sulfoxide): δ 30.6, 112.3, 115.9, 117.0, 124.1, 124.6, 125.8, 127.4, 128.0, 133.9, 135.2, 136.7, 145.6, 150.6, 153.6; IR (trace CHCl3): 3850, 3741, 2357, 2169, 1574, 1470, 1219, 962, 944, 772, 674 cm−1. Elemental analysis (C18H18N2O3): calculated, C 69.66, H 5.85, N 9.03; found, C 69.72, H 5.97, N 9.06.
4-Vinyl-N,N-dimethylbenzylamine (3).
n-Butyllithium (5.24 ml, 2.49 M in hexane, 13.1 mmol) was added to a solution of methyltriphenylphosphonium bromide (4.78 g, 13.4 mmol) in 15 ml of THF (15 ml) at 0°C. After 30 min at room temperature, the solution was recooled to −20°C and p-dimethylaminobenzaldehyde (1.0 g, 6.70 mmol) was introduced in 10 ml of THF. The solution was gradually warmed to room temperature over 2 h. At this point, the contents were poured on 10 ml of water and 0.5 ml of brine, extracted with CH2Cl2, dried with Na2SO4, and concentrated. Flash chromatography (20% ethyl acetate/hexane) afforded 921.7 mg (94%) of 3: Rf = 0.65; 1H (CDCl3): δ 7.28 (d, J = 8.7 Hz, 2H), 6.67 (d, J = 8.7 Hz, 2H), 6.62 (dd, J = 10.8, 17.7 Hz, 1H), 5.52 (dd, J = 1.1, 17.6 Hz, 1H), 5.00 (d, J = 1.1, 10.8 Hz, 1H), 2.94 (s, 6H); 13C (CDCl3): δ 40.0, 109.0, 112.3, 126.8, 136.5, 150.3; IR (neat): 3084, 2887, 2801, 1614, 1520, 1444, 1408, 1353, 1222, 1166, 1062, 989, 947, 887, 821, 536, 502 cm−1.
Butadiene 4.
A solution of n-butyllithium (2.70 ml, 2.50 M in hexane, 6.77 mmol) was added at 0°C to allyltriphenylphosphonium bromide (4.58 g, 11.98 mmol) in 15 ml of THF. After slow warming to room temperature, aldehyde 5 (1.0 g, 5.98 mmol) in 10 ml of THF was added dropwise to the above ylide cooled to −30°C. The reaction was complete within 2 h at ambient temperature. At this point, the reaction was quenched with 20 ml of water and the pH was adjusted to 7 with dilute HCl. The product was extracted with CH2Cl2, dried, and concentrated. Flash chromatography (20% ethyl acetate/hexane) yielded 1.07 g (94%) of a 1:1 mixture of 4 and its corresponding cis isomer. Pure 4 was obtained by repetitive column chromatography on silica gel (Merck 9385) doped with approximately 5% silver nitrate (using 10% ethyl acetate/hexane): Rf = 0.44; 13C (CDCl3) on 1:1 mixture: δ 116.6, 117.8, 119.3, 120.9, 121.2, 122.4, 124.8, 125.2, 127.4, 128.2, 128.3, 128.4, 130.0, 132.0, 133.4, 133.6, 134.5, 134.8, 136.1, 146.6, 146.9, 154.9, 155.3. Elemental analysis (C10H9NO3): calculated, C 62.82, H 4.74, N 7.33; found, C 62.83, H 4.85, N 7.36.
Phosphate 8.
The synthesis of phosphate 8 was accomplished in three operations from commercially available aldehyde 5. The functional conversion began by reducing aldehyde 5 (3.74 g, 22.4 mmol) with NaBH4 (3.46 g, 91.5 mmol) in 77 ml of a 5:5:1 mix of methanol, diethyl ether, and THF. This was accomplished by adding NaBH4 to the above solution of 5 at 0°C over 30 min. After 2 h at ambient temperature, the reaction was quenched with 10% HCl (until the pH was 6), poured on 80 ml of brine, extracted with CH2Cl2, dried with Na2SO4, and concentrated. The crude product was used directly for the next step. Pure material was obtained through flash chromatography (50% ethyl acetate/hexane), yielding 3.67 g (97%) of 6: mp 79.7–81.3°C; Rf = 0.37; 1H (CDCl3): δ 10.60 (s, 1H), 8.06 (d, J = 8.8 Hz, 1H), 7.14 (s, 1H), 6.94 (d, J = 8.8 Hz, 1H), 4.73 (d, J = 5.5 Hz, 2H), 1.97 (t, J = 5.5 Hz, 1H); 13C (CDCl3): δ 63.7, 116.9, 117.9, 125.2, 151.7, 155.3; IR (CHCl3): 3459, 2356, 1620, 1579, 1520, 1475, 1252, 1154, 1025, 822, 741, 689 cm−1. Elemental analysis (C7H7NO4): calculated, C 49.71, H 4.17, N 8.28; found, C 50.13, H 4.30, N 8.20.
Carbon tetrabromide (2.86 g, 8.63 mmol) was added in portions over 30 min to a solution of 6 (≈1.22 g, ≈7.22 mol), 1.2 g of powdered 4-Å molecular sieves, and PPh3 (2.45 g, 9.35 mmol) in 25 ml of dry CH2Cl2 at 0°C. The reaction mixture was warmed to room temperature over 1.5 h and allowed to stand for an additional 1 h. At this point, the mixture was poured on 40 ml of water, extracted with CH2Cl2, dried with Na2SO4, and concentrated. Pure material was obtained by flash chromatography (33% ethyl acetate/hexane) yielding 1.42 g (86%) of 7: mp 68.9–70.2°C; Rf = 0.58; 1H (CDCl3): δ 10.58 (s, 1H), 8.07 (d, J = 8.6 Hz, 1H), 7.16 (d, J = 1.9 Hz, 1H), 6.99 (dd, J = 1.9, 8.8 Hz, 1H), 4.39 (s, 2H); 13C (CDCl3): δ 30.6, 120.1, 120.8, 125.7, 147.8, 155.2; IR (CHCl3): 2354, 1622, 1584, 1479, 1328, 1258 cm−1. Elemental analysis (C7H6NO3Br): calculated, C 36.23, H 2.61, N 6.04; found, C 36.10, H 2.69, N 5.92.
A mixture of 7 (0.91 g, 3.98 mmol) and triethylphosphite (0.87 ml, 5.09 mmol) in 2.5 ml of dry DMF (2.5 ml) was refluxed at 155°C for 1.8 h. Pure material was obtained through flash chromatography (25% ethyl acetate/hexane), yielding 1.14 g (99%) of 8: mp 59.3–62.1°C, Rf = 0.09; 1H (CDCl3): δ 10.56 (s, 1H), 8.01 (d, J = 8.7 Hz, 1H), 7.04 (dd, J = 2.2 Hz, JH-P = 2.2 Hz, 1H), 6.91 (ddd, J = 2.2, 8.7 Hz, JH-P = 2.2 Hz, 1H), 4.04 (qd J = 7.1 Hz, JH-P = 8.1 Hz, 2H), 3.04 (d, JH-P = 22.5 Hz, 2H), 1.25 (q, J = 7.1 Hz, 3H); 13C (CDCl3): δ 16.2 (d, JC-P = 5.3 Hz), 34.2 (d, JC-P = 136.7 Hz), 62.4 (d, JC-P = 6.5 Hz), 120.7 (d, JC-P = 8.3 Hz), 121.8 (d, JC-P = 6.0 Hz), 125.0 (d, JC-P = 2.4 Hz), 143.2 (d, JC-P = 8.8 Hz), 154.9 (d, JC-P = 3.8 Hz); IR (CHCl3): 3850, 3444, 2985, 1623, 1587, 1520, 1480, 1443, 1332, 1252, 1164, 1051, 1025, 968, 843, 761, 654, 605, 518 cm−1. Elemental analysis (C11H16NO6P): calculated, C 45.68, H 5.58, N 4.84; found, C 45.60, H 5.74, N 4.83.
Fragmentation of Cyclobutane 1.
Spectral-grade solvents were purchased from Aldrich and redistilled until baseline of less than 4 kHz was detected in the FCS spectrometer. All glassware was washed with 3% hydrogen peroxide, spectral-grade water, chloroform, and heptane at 60°C and dried at 140°C for 8 h. The thermolysis was conducted in a 15-ml Ace borosilicate thick-walled pressure tube (Aldrich Z18, 109–9) which was soaked in 30% ammonium hydroxide for 18 h, washed with spectral-grade water until the pH became neutral, washed with chloroform and warm heptane, and oven dried at 140°C for 24 h. Blanks were obtained prior to every transfer and checked by FCS for possible contamination. Serial dilution was performed in 10-ml volumetric flasks and used immediately. FCS measurements were obtained in gold vials, readily cleaned by soaking in warm heptane, which were mounted in silver carriers, as used by Eigen and Rigler (2). Samples were added by means of disposable glass pipettes.
A solution of 1 (20.8 mg, 0.062 mmol) in 9 ml of p-xylene was placed in a pressure tube and deoxygenated with a rapid bubbling of argon for 5 min. The system was rapidly closed and completely immersed in a silicon oil bath maintained at 150°C (±2°C) so that only the uppermost portion of flask remained in contact with the atmosphere. The reaction was periodically monitored by rapid cooling to ambient temperature, removing exactly 1.5 ml, degassing, resealing, and submerging into an oil bath. The samples were placed in 5-ml volumetric flasks and evaporated at 0.5 mmHg (partial evaporation of styrene 3 occurred under these conditions). Each sample was diluted to 5 ml with heptane and then further diluted by four repetitive serial dilutions with heptane, providing an array of concentrations of initial 1 varying between 2 × 10−3 and 2 × 10−8. At the same time, reference standards of 1, 3, and 4 were prepared by dissolving the substance in 1 ml of CHCl3 and diluting with 9 ml of heptane. Again, the samples were further diluted by repetitive 10-fold dilution with heptane, providing an array of concentrations between 10−3 and 10−13 M. Beginning with the lowest concentration, samples were placed in a gold vial and measured for possible autocorrelation through a coverslip-protected objective. The FCS spectrometer used was that photographed and described by Eigen and Rigler (2). A dual detector system was used such that fluorescence could be concurrently measured through both Omega optics 530 and 680 bandpass 45 filters. All samples were excited at 457 nm with a power of 0.5 mW.
RESULTS AND DISCUSSION
The intial plan was to develop a reaction manifold that produced the chromophore NND in a highly disfavored sense. However, upon investigation, the low quantum efficiency of NND, 0.093 in heptane (12), prevented its detection in the FCS spectrometer below 10−7 M. At 10 kcal/mol, this detection limit would require at least molar level of the major product at room temperature. On the other hand, addition of a hydrogen bonding group could enhance the quantum efficiency by stabilization of the charge-transfer resonance form of NND. Therefore, dye 2 was synthesized as a possible minor product from 1 (refer to Scheme III). The high trans-stereoselectivity observed in the Wadsworth–Horner–Emmons modified Wittig reaction is well documented for the construction of trans-olefins (14). Its application can readily be extended for the construction of cyclobutane 1 and dye 2. Introduction of a double bond adjacent to the 3-hydroxy-4-nitrophenyl group of 2 appeared to be more efficient than next to the N,N-dimethylaniline group, since the corresponding phosphate 8 was readily prepared and could be used for the synthesis of both 1 and 2, and the Wittig counterpart for the synthesis of 2, N,N-dimethylaminocinnamaldehyde, was commercially available. Phosphate 8 was obtained in three steps from commercially available 3-hydroxy-4-nitrobenzaldehyde (5) in an overall yield of 78%. The synthesis began with the reduction of 5 with sodium borohydride to furnish alcohol 6. This was then transformed to bromide 7 by using the method of Appel (15). Subsequently, conversion to 8 was achieved by Michaelis–Arbuzov displacement with triethylphosphite in refluxing DMF (16). The dianion of 8 could be generated readily by the addition 2 eq of sodium bis(trimethylsilyl)amide. Reaction with N,N-dimethylaminocinnamaldehyde afforded 2 with a purity greater than 98% (by GC) after two recrystallizations from a heptane/THF mixture. When 2 was excited at 450 nm, the fluorescence quantum yield was 0.42 in heptane, as compared 0.097 for NND. Furthermore, acetylation of the phenolic group also lowered the quantum yield to 0.19 in heptane. This suggests that this quantum enhancement arises from hydrogen bond stabilization of the zwitterion resonance form (Scheme IV). 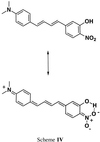 Unlike NND, analog 2 readily autocorrelated in the FCS spectrometer at concentrations of 10−12 M.
Unlike NND, analog 2 readily autocorrelated in the FCS spectrometer at concentrations of 10−12 M.
The construction of cyclobutane 1 began with photolysis of styrene 3, which in turn was prepared by olefination of N,N-dimethylaminobenzaldehyde. Samples of styrene 3 in the presence of a 20-fold excess of acrylonitrile were irradiated with a 150-W high-pressure mercury lamp for 144 h. Although only an unoptimized 18% yield of 9 was recovered, the rapid entry of this route supported its use. Additionally, a 4% yield of the trans isomer of 9 was obtained. Assignment of the isomers was readily verified by the combination of conformational and NMR analysis. In contrast to the trans isomer, in which both nitrile and aryl substituents occupy pseudo-equatorial positions, the lowest-energy conformation of the cis isomer favors a pseudo-axially oriented nitrile. This selectively permits the corresponding pseudo-equatorial positioned hydrogen adjacent to nitrile group to experience w-coupling across the cyclobutane ring. This coupling was observed in the 1H correlated spectroscopy spectrum of only the major isomer, assigned 9. Conversion to aldehydes 10 required for Wittig coupling was accomplished in one flask by reduction with DIBAL-H at −78°C. Potential over-reduction and rearrangement was avoided by quenching the metalloimine intermediate at −78°C with 10% aqueous acetic acid. Under these conditions, an equivalent mixture of cis and trans aldehydes 10 was obtained. The presence of this and preceding isomerizations had no consequence, since condensation of 10 with the dianion of phosphate 8 provided a 4:1 mixture of 1 and its cis isomer 11. Furthermore, this mixture was isomerized to pure 1, without fragmentation, upon heating to 120°C. The structural assignment of these isomers was verified by the same analysis as used for nitrile 9. Only the minor isomer 11 experienced this additional w-coupling.
Although the absorption and fluorescence spectra suggested that 1, 3, and 4 would not interfere with selective irradiation and fluorescence of 2, reference analyses were measured for verification (Table 1). Autocorrelation was not detected in samples of 1, 3, and 4 at concentrations above 10−4 M (see Fig. 2). At isometric concentrations, samples of 1, 2, 3, and 4 in heptane fluoresced with relative intensities of fluctuation of 520:107:10:150. Under the identical conditions used for samples of 1, 3, and 4, dye 2 readily autocorrelated between 10−7 and 10−12 M. Thermolysis of cyclobutane 1 (6.8 mM in p-xylene) at 150°C (±2°C) induced fragmentation to 3 + 4 and rearrangement to 12, the latter by virtue of a vinylcyclobutane rearrangement (17–20). The progress of the reaction was monitored by periodic sampling (Fig. 3). Significant autocorrelation was detected as early as 100 min in a sample which was diluted to an initial 2 × 10−6 M in 1. This was verified to arise from 2 by the fact that it autocorrelated increasingly proportional with concentration. NMR analysis showed that a sample taken after 126 min contained approximately 5 × 10−7 M 3/4 (see Fig. 4). The concentration of 2 at this interval was estimated at 2.3 × 10−11 M by standardization with known concentrations of 2. On the basis of this evidence, the thermolysis of 1 favors fragmentation to 3/4 over 2 by 1.6 (±0.2) × 104. Repetition of this calculation at other time intervals deviated within 10%. At 150°C, this corresponds to an 8.7 (±0.2) kcal/mol disfavoring of the production of 2.
Table 1.
Absorption and fluorescence maxima for compounds 1–4
| Substance | Absorption λmax, nm | Fluorescence λmax, nm |
|---|---|---|
| 1 | 206, 257, 320 | 351, 394 |
| 2 | 318, 455 | 525, 563 |
| 3 | 291 | 355 |
| 4 | 209, 338 | 431 |
Figure 2.

Autocorrelation functions for samples of 1–4 in heptane. Samples were irradiated at 457 nm (0.5 mW) and collected through an Omega Optics 530 bandpass 45 filter. For each sample, the upper trace indicates the frequency of emitted photons of light (kHz). The lower trace provides the autocorrelation function, where the number of particles inside the volume element is given by 1/g2.
Figure 3.

1H-NMR spectral monitor (300 MHz in CDCl3) of the thermolysis of trans-cyclobutane 1 at 150 (±0.2)°C. Notations above the spectra provide peak assignments and are given as follows: ∗, cyclobutane 1; †, styrene 3; O, diene 4; and X, isomers of 12.
Figure 4.

Representative autocorrelation function of the thermolysis of 1 in heptane. This sample was obtained after 2.1 h at 150 (±0.2)°C. It was irradiated at 457 nm (0.5 mW) and collected through an Omega Optics 530 bandpass 45 filter (upper traces) and 680 bandpass 45 filter (lower traces).
This model demonstrated monitoring of the highly disfavored fragmentation of 1 to 2. Although this application requires the production of only 2, this study illustrates a new technique which permits detection of highly energetically disfavored products, opening a new frontier for the verification and calculation of chemical energetics. We are currently adapting this method for general mechanistic application as well as for the detection and quantification of short-lived intermediates.
Acknowledgments
The concept of this work was devised by Richard Lerner of the Scripps Research Institute. Special thanks is given to Manfred Eigen for his gracious support while J.J.L. was at the Max Planck Institute for Biophysical Chemistry (MPI-BPC) in Göttingen, Germany. The bulk of the synthetic work was carried out in the laboratories of Hansjorg Eibl (MPI-BPC). Photolysis as well as the 1H correlated spectroscopy experiments were run at the Georg-August Universität with the generous assistance of Lutz Tietze and Reinhard Machineck, respectively.
ABBREVIATIONS
- FCS
fluorescence correlation spectroscopy
- THF
tetrahydrofuran
- DIBAL-H
diisobutylaluminum hydride
- DMF
N,N-dimethylformamide
- NND
trans,trans-1-(4-nitrophenyl)-4-(4-N,N-dimethylamino)butadiene
References
- 1.Schultz P G, Lerner R A. Science. 1995;269:1835–1842. doi: 10.1126/science.7569920. [DOI] [PubMed] [Google Scholar]
- 2.Eigen M, Rigler R. Proc Natl Acad Sci USA. 1994;91:5740–5747. doi: 10.1073/pnas.91.13.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rigler, R. (1995) J. Biotechnol. 41, 86, 177–186. [DOI] [PubMed]
- 4.Woodward R B, Hoffmann R. Angew Chem Int Ed Engl. 1969;8:781–852. [Google Scholar]
- 5.Schaumann E, Ketcham R. Angew Chem Int Ed Engl. 1982;21:225–314. [Google Scholar]
- 6.von E. Doering W, Roth W R, Breuckmann R, Figge L, Lennartz H-W, Fessner W-D, Prinzbach H. Chem Ber. 1988;121:1–9. [Google Scholar]
- 7.von E. Doering W, Belfield K D, He J. J Am Chem Soc. 1993;115:5414–5421. [Google Scholar]
- 8.Gerberich H R, Walters W D. J Am Chem Soc. 1961;83:4884–4888. [Google Scholar]
- 9.Trecker D J, Henry J P. J Am Chem Soc. 1964;86:902–905. [Google Scholar]
- 10.Wayner D D M, Griller D. Adv Free Radical Chem. 1990;1:159–192. [Google Scholar]
- 11.Jones G, II, Chow V L. J Org Chem. 1974;39:1447–1448. [Google Scholar]
- 12.Shin D M, Whitten D G. J Phys Chem. 1988;92:2945–2956. [Google Scholar]
- 13.Hirschberg Y, Bergmann E, Bergmann F. J Am Chem Soc. 1950;72:5116–5117. [Google Scholar]
- 14.Wadsworth W S., Jr Org React. 1977;25:73–253. [Google Scholar]
- 15.Appel R. Angew Chem Int Ed Engl. 1975;14:801–811. [Google Scholar]
- 16.Arbuzov B A. Pure Appl Chem. 1964;9:307–335. [Google Scholar]
- 17.Overberger C G, Borchert A E. J Am Chem Soc. 1960;82:1007–1008. [Google Scholar]
- 18.Frey H M. Adv Phys Org Chem. 1966;4:170–183. [Google Scholar]
- 19.Berson J A, Nelson G L. Acc Chem Res. 1968;1:152–160. [Google Scholar]
- 20.Ellis R J, Frey H M. Trans Faraday Soc. 1963;59:2076–2079. [Google Scholar]