

Abstract
Alkylation of DNA by 7r,8t-dihydroxy,9t,10t-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene (anti-BPDE) forms mainly trans adducts (with respect to the C-9/10 positions). We recently described a halide-catalyzed pathway that preferentially generates cis adducts and now report that the trans chlorohydrin of anti-BPDE (trans-BPDCH) is an intermediate in the chloride-catalyzed reaction. trans-BPDCH was synthesized, and both it and anti-BPDE were reacted with deoxyadenosine as a model DNA nucleophile. The stereochemistry and yields of deoxyadenosine adducts were determined as a function of chloride concentration. In the absence of salt, the fraction of cis adducts obtained from anti-BPDE and trans-BPDCH are 0.33 and 0.67, respectively. Adding sodium chloride increases the fraction of cis adducts (and consequently decreases the fraction of trans adducts), with the midpoint of the increase for both substrates at approximately 35–40 mM chloride. The chloride-dependent curves for BPDE and BPDCH converge at 1 M chloride, where the fraction of cis adducts is 0.88. Chloride also increases the total yield of cis adducts with either substrate, whereas the yield of trans adducts from the chlorohydrin is not significantly changed. These results support a mechanism by which chloride ion undergoes nucleophilic addition to the benzylic C-10 position of anti-BPDE. This generates a trans halohydrin that alkylates DNA with inversion of configuration to form a cis adduct. This pathway may have biological significance because chlorohydrins could form in serum or in cells with relatively high intracellular concentrations of chloride.
Keywords: halide ion, carcinogen, adduct stereochemistry, bay region epoxides
Polycyclic aromatic hydrocarbons (PAHs) are widespread environmental contaminants, and a number of these hydrocarbons, including benzo[a]pyrene, are potent carcinogens. Benzo[a]pyrene is cytotoxic and mutagenic, and induces cell transformation in culture (1, 2). Although benzo[a]pyrene is a weak complete carcinogen, it is an initiator and interacts strongly with DNA, causing mutations and chromosomal damage (2).
Benzo[a]pyrene is metabolically activated by a series of reactions to form several isomeric bay region diol epoxides (3). The most mutagenic and carcinogenic of these is (7R,8S)-dihydroxy-(9S,10R)-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene [(+)-anti-BPDE]; this isomer is also the one formed in highest amounts in vivo (3–5). BPDE and other activated PAH carcinogens probably initiate tumors by alkylation of DNA. The primary alkylation site of (+)-anti-BPDE is the exocyclic amino group of deoxyguanosine (3, 6–10). Minor adducts are formed by alkylation of the exocyclic amino group of deoxyadenosine (dAdo) and, to a lesser extent, that of deoxycytidine (9, 11, 12). In addition, BPDE forms an unstable adduct at the N-7 position of deoxyguanosine, which results in depurination (13). Another pathway of PAH activation is by metabolic conversion to radical cations (14), which alkylate DNA to form adducts that are also lost by depurination.
The formation of adducts between bay region diol epoxides and DNA has been extensively studied (15–19). An early report (20) on the structure of the major anti-BPDE adduct isolated from DNA indicated that the substituents at the C-9 and C-10 positions were trans (a trans adduct). Subsequent work has shown that at low buffer concentration, and without additional salt, 98% of the exocyclic amino adducts formed between racemic anti-BPDE and DNA are trans (10, 12, 21). Based on this type of evidence, trans adducts have been the focus of attention in carcinogenesis studies.
Only a small percentage of the epoxide reacts to form adducts with duplex DNA, whereas the remainder undergoes hydrolysis to form 7r,8t,9t,10c-tetrahydroxy-7,8,9,10-tetrahydrobenzo[a]pyrene (trans-tetrol) and 7r,8t,9t,10t-tetrahydroxy-7,8,9,10-tetrahydrobenzo[a]pyrene (cis-tetrol). The stereochemistry of hydolysis products, under low salt conditions, is also predominantly trans (16). DNA catalyzes hydrolysis of BPDE, and the rate-determining step both in this reaction and in alkylation is the formation of a carbocation intermediate (16, 18, 22). The carbocation directly alkylates nucleophilic groups in DNA.
Recently, we reported (21, 23, 24) that halide ions catalyze BPDE reactions by a new pathway, which is distinguished from the SN1 (carbocation) pathway by a much higher yield of cis products. Chloride, bromide, and iodide ions catalyze the formation of both cis hydrolysis products and cis adducts (cis refers to the stereochemistry of the c-9/10 positions; see Fig. 6 for structures of cis and trans adducts). Catalytic efficiency depends on the nucleophilicity of the halide, and we postulated that this reaction is mediated by a trans halohydrin. In this paper, we demonstrate that a trans chlorohydrin, 7r,8t,9t-trihydroxy-10c-chloro-7,8,9,10-tetrahydrobenzo[a]pyrene (trans-BPDCH) is an intermediate in chloride-catalyzed cis adduct formation between anti-BPDE and dAdo and that chloride ion increases the proportion and yield of cis adducts with either BPDE or BPDCH.
Figure 6.

The carbocation and chlorohydrin activation routes for the preferential formation of trans and cis adducts, respectively. The upper pathway depicts the acid-catalyzed formation of a C-10 carbocation intermediate, which generates primarily trans adducts with duplex DNA. Substantial amounts of cis products are also formed through this pathway by reaction with DNA monomers. The lower pathway depicts the mechanism we propose for the chloride ion-catalyzed formation of a cis adduct. The trans chlorohydrin would form exclusively cis adducts by an SN2 inversion of configuration. However, it can also lose chloride and generate trans products through a carbocation intermediate.
MATERIALS AND METHODS
Chemicals and Supplies.
Racemic anti-BPDE was purchased from Chemsyn Science Laboratories (Lenexa, KS) or prepared as described (25). Reverse-phase C18 Sep-Pak cartridges were obtained from Millipore.
Synthesis of trans-BPDCH.
The chlorohydrin was synthesized by treatment of (±)-anti-BPDE with lithium chloride in tetrahydrofuran (THF) containing acetic acid (Fig. 1), at room temperature or below (26). The chlorohydrin was purified by normal-phase HPLC before use, using a 47% THF/hexane mobile phase. The stereochemistry of the trans-BPDCH was established by NMR spectroscopy and complete conversion of the synthetic product to the parent epoxide under basic conditions (this reaction occurs to an appreciable extent only at a pH ≥ 12). cis-BPDCH was not detected in these preparations.
Figure 1.

Scheme for the synthesis of the bay region trans chlorohydrin from racemic anti-BPDE [only the (+)-enantiomer of BPDE is shown]. Trans refers to the stereochemistry of the C-9 and C-10 substituents of the hydrocarbon.
Formation of Adducts Between BPDE or the Trans Chlorohydrin and dAdo.
Adducts between (±)-anti-BPDE or (±)-trans-BPDCH and dAdo were generated in 1-ml samples containing 10 mM sodium cacodylate buffer, pH 7.0, 5% THF (vol/vol), and varying concentrations of sodium chloride or sodium nitrate, at a temperature of 22.6 ± 0.2°C. The data for Fig. 2 were obtained using 30 mM dAdo and 40 μM BPDE or 75 μM BPDCH. The data for Fig. 3 were obtained using 10 mM dAdo and 40 μM BPDE or BPDCH. Samples were allowed to react for a minimum of 2 hr.
Figure 2.

Reverse-phase HPLC chromatograms of dAdo adducts formed by alkylation with (±)-anti-BPDE or its racemic chlorohydrin derivative (trans-BPDCH). (A) Adducts formed with the epoxide in the absence of chloride. (B) Adducts formed with the epoxide in the presence of 175 mM NaCl. (C) Adducts formed with trans-BPDCH in the absence of chloride. (D) Co-injection of the samples used in A and C. Peaks in A are labeled with the enantiomer of BPDE from which the adduct was derived [(+), RSSR; (−), SRRS] and with the epoxide ring opening geometry (c = cis; t = trans). Adducts were formed at room temperature in 10 mM sodium cacodylate buffer (pH 7.0) containing 5% THF (vol/vol), and analyzed by reverse-phase HPLC using a 1:1 methanol/water mobile phase and fluorescence detection.
Figure 3.

The fraction of cis dAdo adducts formed by (+)-anti-BPDE or the corresponding enantiomer of trans-BPDCH as a function of sodium chloride or sodium nitrate concentration (fraction cis + trans adducts = total adducts). Reaction conditions and adduct analysis were as described in Materials and Methods. The curves were obtained by fitting the data points to Eq. 1 using nonlinear regression as described in the text; see Table 1 for curve parameters.
Isolation of Adducts.
After reaction, samples were reduced in volume (0.6 ml or less) to remove THF and were diluted to 5 ml with water. They were loaded onto a C18 Sep-Pak cartridge and washed with 20 ml of 10% methanol/water (vol/vol) to remove most of the unmodified dAdo. After this step, illumination of the cartridge with 366-nm UV light revealed a fluorescent band of adsorbed tetrols and adducts at the top of the cartridge, indicating that no significant loss of these components had occurred. The adducts (as well as the BPDE or BPDCH hydrolysis products) were then eluted from the Sep-Pak with 2.0 ml of methanol (control experiments indicated that this procedure elutes >99% of the adsorbed adducts). These Sep-Pak eluates were evaporated, and the residue from each sample was taken up in 0.6 ml of 50% methanol/water (vol/vol). The samples were then filtered using 0.2-μm Nylon-66 membranes and stored at −20°C.
Individual adducts were resolved by reverse-phase HPLC using a C18 column (Microsorb 0.46 × 25 cm, 5 μm particle, 100 Å pore; Rainin Instruments) and an isocratic methanol/water (vol/vol) mobile phase, with a flow rate of 0.8 ml/min. The proportion of methanol in the eluant was 50% for the chromatograms in Fig. 2, and 53% for the data in Figs. 3, 4, 5 and Table 1. Detection was by fluorescence, with 245-nm excitation and >320-nm emission (see Fig. 2) or by absorbance at 245 nm (see Figs. 3, 4, 5 and Table 1). The chromatograms were transferred to a Macintosh computer using a Rainin Control/Interface Module, and the peak areas were integrated using Rainin macintegrator software. The stereochemistry data in Fig. 3 and Table 1 are based on the adducts derived from the more carcinogenic (+)-enantiomer of anti-BPDE (RSSR) or the corresponding enantiomer of trans-BPDCH (RSSS) because they are more easily resolved than the products of the SRRS (BPDE) or SRRR (chlorohydrin) enantiomers. Under all conditions, the fraction of cis adducts was similar for both enantiomers of a given compound.
Figure 4.

The yield of cis and trans dAdo adduct formation by anti-BPDE or trans-BPDCH as a function of sodium chloride concentration. The amount of adduct formed was divided by the amount of epoxide or chlorohydrin initially added. The normalized yield is independent of hydrocarbon concentration as long as dAdo is present in large excess. Reaction conditions, adduct analysis, and the calculation of the yield were as described in Materials and Methods.
Figure 5.

The fraction of cis hydrolysis products (tetrols) formed by anti-BPDE or trans-BPDCH as a function of sodium chloride concentration (fraction cis + trans tetrols = total tetrols). Reaction conditions and product analysis were as described in Materials and Methods. The curves were obtained by fitting the data points to Eq. 1 using nonlinear regression as described in the text; see Table 1 for curve parameters.
Table 1.
The effect of chloride concentration on Eq. 1 parameters for cis product formation
To determine the yield of adducts as a fraction of the initial hydrocarbon, total tetrol peak areas were plotted as a function of total adduct peak areas for both BPDE and BPDCH. The two plots give parallel lines whose slope is the ratio of the extinction coefficient of tetrol to that of the adducts. This analysis is valid because there are no other significant products from the dAdo alkylation reactions.
MS Characterization of Adducts.
The fourth-eluting dAdo adduct peak derived from the chlorohydrin (corresponding to the cis adduct of (+)-anti-BPDE) was isolated by HPLC, and its mass spectrum was obtained as described (27). The underivatized adduct was analyzed by liquid secondary ion MS in a negative mode.
Analysis of Adduct Stereochemistry as a Function of the Salt Concentration.
Nonlinear regression was used to fit the salt concentration dependence data to the equation
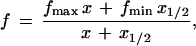 |
1 |
in which x is the salt concentration, f is the fraction of the adducts that are cis, fmin is the minimum fraction cis at [salt] = 0, fmax is the limiting maximum fraction cis obtained as [salt] → ∞, and x1/2 is the value of x corresponding to the median value of f, which is (fmin + fmax)/2. Eq. 1 describes a rectangular hyperbola; when plotted versus log x, such a curve will have an inflection point at x = x1/2. In general, the halide concentration dependence of the product fraction for a reaction with two products should follow an equation of this form when the products are formed by kinetically independent halide-catalyzed and non-halide-catalyzed pathways, and the halide kinetic effect is first order. In our experimental system, these conditions should be fulfilled as long as reversion of the chlorohydrin to the epoxide is negligible, which is the case at neutral pH. In the expression for product fraction, x1/2 represents the ratio of the rate constant of the non-halide-catalyzed reaction(s) to that of the halide-catalyzed reaction(s).
RESULTS
Deoxyadenosine Adducts from BPDE and the Chlorohydrin.
An HPLC chromatogram of adducts formed by (±)-anti-BPDE and dAdo in the absence of chloride ion is shown in Fig. 2A. The four diastereomeric adducts result from trans and cis epoxide ring opening by the two enantiomers of BPDE. The structures of these adducts were determined by comparison to authentic standards prepared as described (9, 27). An HPLC chromatogram of the four adducts generated between (±)-trans-BPDCH and dAdo under the same conditions is shown in Fig. 2C.
Structures of BPDCH–dAdo Adducts.
BPDE alkylates dAdo in DNA with the formation of an adduct in which the C-10 position of the hydrocarbon is linked to the exocyclic amino group of the base (3, 9, 28, 29). The mass spectrum (liquid secondary ion MS, negative mode) of the cis dAdo adduct derived from the (+)-enantiomer of BPDCH was consistent with this structure. The spectrum of this adduct exhibited an ion at m/z 552 (M-1) and an ion at m/z 436 (M-117) corresponding to loss of a deoxyribose fragment.
HPLC co-chromatography of the four BPDCH–dAdo adducts with the four BPDE–dAdo adducts was carried out using mobile phases varying from 50% to 55% methanol (a range over which adduct retentions varied more than 2-fold). In each case, the corresponding adducts coeluted, and only four peaks were obtained (Fig. 2D). These results indicate that the structures of the BPDCH adducts are identical to those formed from BPDE.
Stereochemistry of BPDE–dAdo and BPDCH–dAdo Adducts.
In the absence of salt, anti-BPDE exhibits a moderate preference for trans adduct formation with dAdo. This is illustrated by the chromatogram in Fig. 2A, where the adducts are 69% trans and 31% cis. Chloride catalysis resulting from the addition of 175 mM NaCl reverses the stereochemical preference, yielding adducts that are 74% cis (Fig. 2B). The distribution of anti-BPDE adducts formed in the presence of 175 mM NaCl is similar to that of chlorohydrin adducts generated in the absence of chloride ion, where a value of 68% cis was obtained (Fig. 2C).
Dependence of Adduct Stereochemistry on Salt Concentration.
The stereochemistry of dAdo adducts formed by reaction with BPDE or BPDCH depends on chloride concentration (Fig. 3). To permit examination of ionic strength or other effects separately from the chloride effect, we also generated adduct samples using sodium nitrate instead of sodium chloride. The curves were obtained by using nonlinear regression to fit the data to Eq. 1. The parameter values for the curves in Fig. 3 are given in Table 1.
In the absence of chloride, the proportion of cis adducts produced by trans-BPDCH is roughly twice that produced by anti-BPDE. As chloride concentration increases, the fraction of cis products increases in both cases, but more rapidly for BPDE, and both curves converge at 1 M chloride, where the fraction of cis adducts is 0.88. Sodium nitrate has only a small effect on the stereochemistry of the BPDE–dAdo reaction and almost no effect on the BPDCH reaction.
Salt Effects on Yields of BPDE–dAdo and BPDCH–dAdo Adducts.
The yields of cis and trans dAdo adducts formed from BPDE and BPDCH as functions of sodium chloride concentration are shown in Fig. 4. Yields were normalized to the amount of epoxide or chlorohydrin added to the assay as described in the legend to Fig. 4. The absolute levels of cis adducts increase with higher chloride concentration in both cases. The amount of cis adducts increases 3.8-fold with BPDCH, and 7-fold with BPDE, as the chloride concentration increases from 0 to 2.56 M. The midpoints of these increases occur at approximately 160 mM chloride. In contrast, the yield of trans adducts either remains constant (in the case of BPDCH) or decreases slightly (in the case of BPDE) over the range of chloride concentration tested. Sodium nitrate has a much smaller effect on the yields of these reactions (data not shown).
Dependence of Tetrol Stereochemistry on Salt Concentration.
The stereochemistry of tetrols derived from hydrolysis of BPDE and BPDCH in dAdo adduct assays also depends on salt concentration (Fig. 5). The curves were obtained by fitting the data to Eq. 1. In the absence of chloride, the fraction of cis-tetrol produced by trans-BPDCH is slightly higher than that produced by anti-BPDE (Table 1). A small difference is maintained over the range of chloride concentrations tested. The highest level of chloride tested raised the proportion of cis-tetrol by a factor of roughly 2.5 in both cases. The midpoints of the change in stereochemistry were in the 100–140 mM chloride range. As we reported earlier (21), sodium nitrate has only a small effect on the stereochemistry of tetrols formed by hydrolysis of BPDE; for BPDCH, this effect is smaller yet (data not shown).
DISCUSSION
Previously, we reported that chloride catalyzes BPDE alkylation of nucleic acids, resulting in an increase in the fraction of cis adducts (21, 23). In this paper, we demonstrate that trans-BPDCH alkylates dAdo to form the same products as anti-BPDE but produces a larger proportion of those with cis stereochemistry. These and other results reported here strongly support the intermediate formation of a trans chlorohydrin in the chloride-catalyzed alkylation pathway of BPDE. Similar results have been obtained with a number of other nucleophiles, including DNA. The mechanisms of adduct formation by the acid-catalyzed and the proposed chloride-catalyzed pathways are depicted in Fig. 6.
The SN1 pathway for the hydrolysis of anti-BPDE yields predominantly trans-tetrol. To account for this behavior Jerina and coworkers (30, 31) proposed that a more abundant (low energy) conformer with a pseudo-axial C-9 hydroxy group preferentially forms trans-tetrol, whereas a less abundant (high energy) conformer with a pseudo-equatorial C-9 hydroxy group generates cis-tetrol. Dipple and coworkers (10) have used analogous arguments to explain the almost exclusive trans stereochemistry of DNA adducts. It was suggested that the low energy conformer could readily alkylate sites in duplex DNA, whereas the bulkier high energy conformer was excluded. The inherently higher reactivity of the high energy conformer was used to explain the higher levels of cis adduct formation with nucleic acid monomers, where steric constraints would not be stringent compared with DNA.
We have investigated the effect of temperature on the product stereochemistry of the BPDE hydrolysis and alkylation reactions (24). Increasing temperature increases the fraction of cis products obtained from both reactions. The temperature effect was significantly reduced in the presence of halides, suggesting it mainly influenced the carbocation pathway. These results support the view that the halide-catalyzed route is distinct from the SN1 pathway.
Indeed, the NMR spectrum of the BPDCH indicates that the C-9 hydroxy group in this derivative is pseudo-axial, and as proposed by Cheng et al. (10), a substrate with this conformation should lead primarily to trans products and not the predominantly cis products we obtain. This observation rules out the possibility that the ground state conformation of BPDCH is responsible for the stereochemical outcome of these halide-catalyzed reactions.
The proposed mechanism of the halide-catalyzed reaction is nucleophilic substitution of the C-10 position in anti-BPDE by halide to form a trans halohydrin, followed by SN2 displacement of the halide by a nucleophile to form a cis product (21, 23, 24). Kinetic evidence indicates that whereas chloride substitution is predominantly SN1, iodide is mainly SN2, and bromide is intermediate between SN1 and SN2 (21).
Reactions carried out with synthetic trans-BPDCH provide clear evidence in support of its intermediate formation in the halide-catalyzed reactions. Alkylations by the bay region diol epoxide and the corresponding chlorohydrin form the same exocyclic amino group adducts with DNA nucleophiles. trans-BPDCH also reacts to form a greater proportion of cis hydrolysis and alkylation products than anti-BPDE in the absence of chloride. In the presence of chloride, the fraction of cis adducts produced with BPDE increases markedly, whereas a more modest increase is obtained with trans-BPDCH (whose cis fraction in the absence of chloride is already elevated compared with the epoxide). At very high chloride concentrations, conversion of the carbocation into the chlorohydrin should be nearly complete, and alkylation reactions by anti-BPDE and trans-BPDCH should give the same product stereochemistry. Our finding that the proportion of cis dAdo adducts produced by the two compounds was essentially the same at or above 1 M chloride strongly supports the proposed mechanism. Finally, a BPDE sample hydrolyzed in the presence of high concentrations of iodide exhibits a transient red-shifted absorbance spectrum identical to that of the bay region iodohydrin of BPDE, providing direct evidence that halohydrins form under these conditions (unpublished work).
The second step in the proposed halohydrin pathway is SN2 displacement of the halide by nucleophiles to form cis products. As required for an SN2 reaction, trans-BPDCH undergoes nucleophilic substitution with inversion of configuration. SN2 reactions are favored by strong nucleophiles and, as expected, the proportion of cis products obtained by reaction with a strong nucleophile (dAdo) is higher than with a weak nucleophile (water). This is demonstrated by comparison of the data in Figs. 3 and 5. In addition, the SN2/SN1 ratio of rate constants (reciprocal of x1/2) calculated from this data is three times higher for alkylation than for hydrolysis.
In addition to nucleophilic displacement, the chlorohydrin can also react through the SN1 pathway by loss of halide and generation of a carbocation, which leads to trans product formation. As expected, adding chloride suppresses the SN1 pathway by shifting the equilibrium from the carbocation toward the chlorohydrin. Even at the high concentrations of chloride tested here, the rate of the SN1 pathway is sufficient to capture halohydrin because some trans products are still observed.
Sodium nitrate, a non-nucleophilic salt, causes small increases in adduct yields and proportions of cis products, particularly with the epoxide as substrate. Similar effects by this and other nonhalide salts on the product ratio and kinetics of BPDE hydrolysis were reported previously (21). The mechanism of this effect is not known, but it clearly differs from the mechanism of the halide effect. Consideration of the differences in the magnitudes of the effects of various salts is instructive. At 500 mM, the three nonhalide salts tested all increase the BPDE hydrolysis cis/trans product ratio by about 45%. A comparable effect would be obtained from 8.2 mM sodium chloride, 1.1 mM potassium bromide, or 0.17 mM potassium iodide. The potencies of the halide effects are several orders of magnitude greater than those of the nonhalide salt effects and vary widely according to nucleophilicity.
The binding studies reported here have been carried out on dAdo, a minor target of BPDE alkylation in DNA. However, we have obtained qualitatively similar results with deoxyguanosine. DNA adducts with a cis configuration at deoxyguanosine are quasi-intercalated and disrupt the duplex of DNA (32–34). It is possible that the disruption caused by cis adducts is mutagenic. The relative mutagenic and carcinogenic activities of cis and trans adducts are not yet known, but under some conditions, cis adducts might interfere with DNA processing to a greater degree than trans adducts, which are mostly external and nondisruptive (35, 36). In the presence of chloride, the shift in stereochemistry from trans to cis adducts (or the larger total quantity of cis adducts formed) could lead to an increase in initiation of tumorigenesis. Thus, the chlorohydrin pathway could contribute to the carcinogenic properties of bay region epoxides if it operates in vivo. This would require an environment with a chloride concentration high enough to permit significant conversion of the epoxide to the chlorohydrin.
One possibility would be in serum or other extracellular fluids, where the concentration of chloride ion is near 150 mM. Whether chlorohydrins or other halohydrins would undergo cellular uptake and survive transit to a cell nucleus is not known. Our preliminary evidence indicates that the trans-BPDCH is considerably less stable than BPDE, at least in dilute aqueous buffer.
The concentration of chloride ion in typical cells is only 5–15 mM, and at this level, a significant degree of conversion of BPDE to BPDCH appears unlikely. However, the natural targets of PAH carcinogens are cells in the epithelia of lung, skin, esophagus, mouth, and breast. Cells in these tissues have higher intracellular chloride ion concentration than cells in non-epithelial tissues. Many types of epithelial cells contain 30–50 mM chloride, whereas secretory epithelial cells contain 60–90 mM chloride (37–39). The degree to which the BPDCH would form and alkylate intracellular targets remains to be determined. However, the chloride-catalyzed pathway could operate at a level high enough to influence stereochemistry and levels of adducts in epithelial cells, and therefore, might influence PAH mutagenicity and tumorigenicity, as well as tissue susceptibility.
Acknowledgments
This paper is dedicated to Melvin Calvin. We thank the University of California, San Francisco, National Institutes of Health Mass Spectrometry Facility for analyses. This work was supported, in part, by grants from the National Cancer Institute (CA40598) and the National Institute on Environmental Health Sciences (ES06869).
ABBREVIATIONS
- PAH
polycyclic aromatic hydrocarbon
- (+)-anti-BPDE
(7R,8S)-dihydroxy-(9S,10R)-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene
- cis-tetrol
racemic 7r,8t,9t,10t-tetrahydroxy-7,8,9,10-tetrahydrobenzo[a]pyrene
- trans-tetrol
racemic 7r,8t,9t,10c-tetrahydroxy-7,8,9,10-tetrahydrobenzo[a]pyrene
- trans-BPDCH
racemic 7r,8t,9t-trihydroxy-10c-chloro-7,8,9,10-tetrahydrobenzo[a]pyrene
- dAdo
deoxyadenosine
- THF
tetrahydrofuran
References
- 1.Landolph J R, Heidelberger C. Proc Natl Acad Sci USA. 1979;76:930–934. doi: 10.1073/pnas.76.2.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gehly E B, Landolph J R, Heidelberger C, Nagasawa H, Little J B. Cancer Res. 1982;42:1866–1875. [PubMed] [Google Scholar]
- 3.Thakker D H, Yagi H, Levin W, Wood A W, Conney A H, Jerina D M. In: Bioactivation of Foreign Compounds. Anders M W, editor. New York: Academic; 1985. pp. 177–242. [Google Scholar]
- 4.Wood A W, Chang R L, Levin W, Yagi H, Thakker D R, Jerina D M, Conney A H. Biochem Biophys Res Commun. 1977;77:1389–1396. doi: 10.1016/s0006-291x(77)80133-2. [DOI] [PubMed] [Google Scholar]
- 5.Buening M K, Wislocki P G, Levin W, Yagi H, Thakker D R, Akagi H, Koreeda M, Jerina D M, Conney A H. Proc Natl Acad Sci USA. 1978;75:5358–5361. doi: 10.1073/pnas.75.11.5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koreeda M, Moore P D, Yagi H, Yeh H J, Jerina D M. J Am Chem Soc. 1976;98:6720–6722. doi: 10.1021/ja00437a061. [DOI] [PubMed] [Google Scholar]
- 7.King H W S, Osborne M R, Beland F A, Harvey R G, Brookes P. Proc Natl Acad Sci USA. 1976;73:2679–2681. doi: 10.1073/pnas.73.8.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeffrey A M, Jennette K W, Blobstein S H, Weinstein I B, Beland F A, Harvey R G, Kasai H, Miura I, Nakanishi K. J Am Chem Soc. 1976;98:5714–5715. doi: 10.1021/ja00434a060. [DOI] [PubMed] [Google Scholar]
- 9.Straub K M, Meehan T, Burlingame A L, Calvin M. Proc Natl Acad Sci USA. 1977;74:5285–5289. doi: 10.1073/pnas.74.12.5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng S C, Hilton B D, Roman J M, Dipple A. Chem Res Toxicol. 1989;2:334–340. doi: 10.1021/tx00011a011. [DOI] [PubMed] [Google Scholar]
- 11.Meehan T, Straub K, Calvin M. Nature (London) 1977;269:725–727. doi: 10.1038/269725a0. [DOI] [PubMed] [Google Scholar]
- 12.Sayer J M, Chadha A, Agarwal S K, Yeh H J, Yagi H, Jerina D M. J Org Chem. 1991;56:20–29. [Google Scholar]
- 13.Osborne M, Merrifield K. Chem Biol Interact. 1985;53:183–195. doi: 10.1016/s0009-2797(85)80095-8. [DOI] [PubMed] [Google Scholar]
- 14.Cavalieri E L, Rogan E G. Xenobiotica. 1995;25:677–688. doi: 10.3109/00498259509061885. [DOI] [PubMed] [Google Scholar]
- 15.Geacintov N E. In: Polycyclic Aromatic Hydrocarbons: Structure Activity Relationships. Yang S K, Silverman B D, editors. Vol. 2. Boca Raton, FL: CRC; 1988. pp. 181–206. [Google Scholar]
- 16.Harvey R G, Geacintov N E. Acc Chem Res. 1988;21:66–73. [Google Scholar]
- 17.Gräslund A, Jernström B. Q Rev Biophys. 1989;22:1–37. [PubMed] [Google Scholar]
- 18.Jerina D M, Chadha A, Cheh A M, Schurdak M E, Wood A W, Sayer J M. In: Biological Reactive Intermediates IV: Molecular and Cellular Effects and Their Impact on Human Health. Witmer C M, Snyder R, Jollow D J, Kalf G F, Kocsis J J, Sipes I G, editors. New York: Plenum; 1991. pp. 533–553. [Google Scholar]
- 19.Dipple A. In: DNA Adducts: Identification and Biological Significance. Hemminki K, Dipple A, Shuker D E G, Kadlubar F F, Segerbäck D, Bartsch H, editors. Lyon, France: International Agency for Research on Cancer; 1994. pp. 107–129. [Google Scholar]
- 20.Jeffrey A M, Weinstein I B, Jennette K W, Grzeskowiak K, Nakanishi K, Harvey R G, Autrup H, Harris C. Nature (London) 1977;269:348–350. doi: 10.1038/269348a0. [DOI] [PubMed] [Google Scholar]
- 21.Wolfe A R, Meehan T. Chem Res Toxicol. 1994;7:110–119. doi: 10.1021/tx00037a016. [DOI] [PubMed] [Google Scholar]
- 22.Meehan T, Bond D M. Proc Natl Acad Sci USA. 1984;81:2635–2639. doi: 10.1073/pnas.81.9.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolfe A R, Yamamoto J, Meehan T. Proc Natl Acad Sci USA. 1994;91:1371–1375. doi: 10.1073/pnas.91.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolfe A R, Song Q, Meehan T. Polycyclic Aromatic Compounds. 1996;10:203–210. [Google Scholar]
- 25.Harvey R G, Fu P P. In: Polycyclic Hydrocarbons and Cancer. Gelboin H V, T’so P O P, editors. Vol. 1. New York: Academic; 1978. pp. 133–165. [Google Scholar]
- 26.Bajwa J S, Anderson R C. Tetrahedron Lett. 1991;32:3021–3024. [Google Scholar]
- 27.Yamamoto J, Subramaniam R, Wolfe A R, Meehan T. Biochemistry. 1990;29:3966–3972. doi: 10.1021/bi00468a025. [DOI] [PubMed] [Google Scholar]
- 28.Jeffrey A M, Grzeskowiak K, Weinstein I B, Nakanishi K, Roller P, Harvey R G. Science. 1979;206:1309–1311. doi: 10.1126/science.316186. [DOI] [PubMed] [Google Scholar]
- 29.Jernström B, Gräslund A. Biophys Chem. 1994;49:185–199. doi: 10.1016/0301-4622(93)e0087-l. [DOI] [PubMed] [Google Scholar]
- 30.Sayer J M, Yagi H, Silverton J V, Friedman S L, Whalen D L, Jerina D M. J Am Chem Soc. 1982;104:1972–1978. [Google Scholar]
- 31.Sayer J M, Whalen D L, Friedman S L, Paik A, Yagi H, Vyas K P, Jerina D M. J Am Chem Soc. 1984;106:226–233. [Google Scholar]
- 32.Cosman M, de los Santos C, Fiala R, Hingerty B E, Ibanez V, Luna E, Harvey R, Geacintov N E, Broyde S, Patel D J. Biochemistry. 1993;32:4145–4155. doi: 10.1021/bi00067a001. [DOI] [PubMed] [Google Scholar]
- 33.Cosman M, Fiala R, Hingerty B E, Amin S, Geacintov N E, Broyde S, Patel D J. Biochemistry. 1994;33:11518–11527. doi: 10.1021/bi00204a014. [DOI] [PubMed] [Google Scholar]
- 34.Cosman M, Hingerty B E, Luneva N, Amin S, Geacintov N E, Broyde S, Patel D J. Biochemistry. 1996;35:9850–9863. doi: 10.1021/bi9605346. [DOI] [PubMed] [Google Scholar]
- 35.Cosman M, de los Santos C, Fiala R, Hingerty B E, Singh S B, Ibanez V, Margulis L A, Live D, Geacintov N E, Broyde S, Patel D J. Proc Natl Acad Sci USA. 1992;89:1914–1918. doi: 10.1073/pnas.89.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de los Santos C, Cosman M, Hingerty B E, Ibanez V, Margulis L A, Geacintov N E, Broyde S, Patel D J. Biochemistry. 1992;31:5245–5252. doi: 10.1021/bi00138a002. [DOI] [PubMed] [Google Scholar]
- 37.Cotton C U, Reuss L. J Gen Physiol. 1991;97:667–686. doi: 10.1085/jgp.97.4.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takemura T, Sato F, Saga K, Suzuki Y, Sato K. J Membr Biol. 1991;119:211–219. doi: 10.1007/BF01868726. [DOI] [PubMed] [Google Scholar]
- 39.Foskett J K. Am J Physiol. 1990;259:C998–C1004. doi: 10.1152/ajpcell.1990.259.6.C998. [DOI] [PubMed] [Google Scholar]