

Abstract
We show here that the amino acid residues contributing to the active sites of the vanadate containing haloperoxidases are conserved within three families of acid phosphatases; this suggests that the active sites of these enzymes are very similar. This is confirmed by activity measurements showing that apochloroperoxidase exhibits phosphatase activity. These observations not only reveal interesting evolutionary relationships between these groups of enzymes but may also have important implications for the research on acid phosphatases, especially glucose-6-phosphatase—the enzyme affected in von Gierke disease—of which the predicted membrane topology may have to be reconsidered.
Keywords: vanadate, haloperoxidase, glucose-6-phosphatase, acid phosphatases
Haloperoxidases are enzymes catalyzing the two electron oxidation of a halide (X−) to the corresponding hypohalous acid according to Eq. 1.
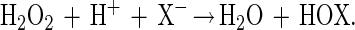 |
1 |
HOX may further react with a broad range of nucleophilic acceptors to form a diversity of halogenated compounds. These haloperoxidases are named after the most electronegative halide they are able to oxidize, and thus a chloroperoxidase (CPO) is able to oxidize chloride, bromide, and iodide.
Three classes of haloperoxidases have been identified. One of these consists of enzymes without a prosthetic group and as such have been detected in a number of bacteria (1, 2). The remaining two classes are the so-called heme-containing haloperoxidases—exemplified by the CPO from the fungus Caldariomyces fumago (3) or myeloperoxidase which is present in white blood cells (4)—and the vanadium-containing haloperoxidases that bind a vanadate ion (VO43−) as a prosthetic group. Enzymes representing these two classes not only differ in the nature of their prosthetic group but also in at least two other aspects: catalytic mechanism and stability. Heme-containing peroxidases catalyze the formation of the hypohalous acid by a redox mechanism, whereas in vanadate-containing peroxidases the transition metal does not change its redox state (5, 6) but may function as a Lewis acid. Vanadate-containing haloperoxidases not only posses a very high stability (7, 8) but they also resist a high concentration of their substrate (H2O2) (9) and their product (HOX) (7) that would readily inactivate the heme-containing peroxidases.
Vanadate-containing peroxidases were first discovered in seaweeds about a decade ago (10, 11); at present they are being discovered not only in an increasing number of seaweeds (12, 13) but also in fungi (8) and in a lichen (14).
The fungus Curvularia inaequalis has been shown to express a vanadium-containing CPO (V-CPO) (15), and recently the gene encoding this enzyme was cloned and sequenced (16). The V-CPO gene codes for a protein of 609 amino acids with a calculated molecular mass of 67,488 kDa. Database searches using the entire V-CPO sequence showed very little sequence similarity to other known proteins (16). The recent determination of the crystal structure of this enzyme at 2.1-Å resolution (17) revealed that the protein has an overall cylindrical shape and measures approximately 50 × 80 Å. The secondary structure is mainly α-helical with two four-helix bundles as main structural motifs of the tertiary structure. The vanadium binding center is located on top of the second four-helix bundle and the residues binding the prosthetic group span a length of approximately 150 residues in the primary structure. The metal is coordinated to the Nɛ2 of His-496 while five residues (Lys-353, Arg-360, Ser-402, Gly-403, and Arg-490) donate hydrogen bonds to the non-protein oxygens of vanadate. His-404 is proposed to be an acid-base group in catalysis (17). Alignment of this V-CPO with the partial amino acid sequence of the vanadium-containing bromoperoxidase (V-BPO) from the seaweed Ascophyllum nodosum (10), an enzyme of which the catalytic mechanism is considered to be analogous to that of the V-CPO (7), revealed three stretches of high similarity in the regions providing the metal anion binding site, whereas the similarity turned out to be very low in the intervening regions.
Here we report on a more extensive database search showing that similar stretches are also present in three families of acid phosphatases which were previously considered unrelated. Furthermore, we show that apo-CPO can also function as an acid-phosphatase, indicating that the active site of V-CPO is very similar to that of the acid phosphatases. The implications of these findings are discussed.
MATERIALS AND METHODS
Strains, Medium, and Growth Conditions.
Because it is difficult to obtain pure apo-enzyme in sufficient amounts from C. inaequalis as needed for our experiments, we decided to use recombinant enzyme produced from the C. inaequalis V-CPO gene in a newly developed Saccharomyces cerevisiae expression system (18). This enabled us to produce large quantities of recombinant enzyme (r-CPO), which after activation with vanadate behaves kinetically very similar to the enzyme as isolated from C. inaequalis.
r-CPO protein was isolated from yeast strain BJ1991 (Matα, leu2, trp1, ura3-251, prb1-1122, pep4-3) transformed with plasmid TNT4 (18) carrying the V-CPO gene under transcriptional control of the yeast Gal1 promotor.
To isolate the r-CPO, yeast transformants carrying TNT4 were grown until stationary phase in starter cultures containing 0.67% yeast nitrogen base without amino acids (YNB-WO from Difco), 2% (wt/vol) glucose, and 20 μg/ml (wt/vol) uracil. Starter cultures were diluted 1:10 in 1% (wt/vol) yeast extract (Difco), 1% casein hydrolysate (Difco), and 1% (wt/vol) glucose until the end of the log phase after which the cultures were induced by the addition of galactose to a final concentration of 4% (wt/vol) and allowed to grow for another 2 days.
The isolation of the recombinant protein to purity was performed as described (18) with the addition of a HPLC MonoQ step (Pharmacia) as described in ref. 16.
Enzymatic Assays.
The standard assay for determining CPO activity of V-CPO is the chlorination of 50 μM monochlorodimedon, which is monitored spectrometrically at 290 nm under conditions of 1 mM H2O2, 5 mM Cl−, and 100 mM citrate (pH 5.0). The change in extinction coefficient is 20.0 mM−1·cm−1.
A standard assay for determining phosphatase activity is measuring the hydrolysis of para-nitrophenyl phosphate (p-NPP). After incubation with this substrate reaction mixtures (see legends) were quenched with NaOH and production of para-nitrophenol (p-NP) was measured at 410 nm using an extinction coefficient of 18.3 mM−1·cm−1 at pH 12. Data were corrected for nonenzymatic hydrolysis of p-NPP at low pH.
Activation of apo-CPO.
Apo-CPO was activated by addition of 100 μM vanadate in 100 mM Tris·SO4 (pH 8.0) as described for the native protein (8). Excess vanadate was removed by dialysis against the desired buffer.
RESULTS
The results of our database search are presented in the alignment of Fig. 1A. Included in this alignment are the acid phosphatases found in our database search, the partial amino acid sequence from the V-BPO of A. nodosum, and the deduced partial amino acid sequence of the V-CPO from the fungus D. biseptata (P.B. unpublished data). It is clear that in this alignment nearly all residues coordinating vanadate in V-CPO are conserved. The only exceptions are residues Gly-76 and Gln-83 of the bcrC gene product aligning to Lys-353 and Arg-360 of CPO of C. inaequalis, respectively, and residue Trp-25 of A. nodosum, also aligning to Lys-353. The alignment strongly suggests that the binding pocket for vanadate in the peroxidases is similar to the phosphate-binding site in the aligned acid phosphatases. This is in agreement with the structural resemblance of vanadate and phosphate, with the observation that the vanadium-containing haloperoxidases rapidly loose their activity in phosphate-containing buffers and that reconstitution of the apo-BPO by vanadate is inhibited in the presence of phosphate (28). Furthermore, vanadate is recognized as a potent inhibitor of many different phosphatases (29). Fig. 1A also shows the high variability in sequence and length of the regions (which primarily form loop structures) connecting the active site residues. It is noteworthy that according to the dendrogram based on the alignment (Fig. 1B), the V-BPO of A. nodosum is not more similar to the CPOs as it is to the acid phosphatases.
Figure 1.

(A) Alignment of V-CPO of C. inaequalis with the enzymes proposed to contain a structurally similar active site. Residues identical in 50% or more of the sequences are boxed. The residues contributing to the active site of V-CPO are given underneath the alignment together with the secondary structure elements in which they are present. ▪, Residue hydrogen-bonded to vanadate in V-CPO; ♦, histidine proposed to be the acid-base group of V-CPO. •, histidine covalently linked to vanadate in V-CPO; ✫, residue shown to be essential for glucose-6-phosphatase (G-6-Pase) activity; ((), α-helix; ➱, β-sheet; , loop. (B) A dendrogram based on the alignment. The group of membrane-bound phosphatases is marked with an M, and the group of soluble phosphatases is marked with an S. pgpB, phosphatidyl glycerophosphate phosphatase B from Escherichia coli (19) and Haemophillus influenzae (20); bcrC, gene product from Bacillus licheniformis (21); G-6-Pase, glucose-6-phosphatase from mouse (22), rat (23) and human (24); phoC, phoN, class A bacterial acid phosphatase from Morganella morganii (25), Providencia stuartii (GenBank accession no. X64820X64820), Salmonella typhimurium (26), and Zymomonas mobilis (27); Apyrase ATP-diphosphohydrolase from Shigella flexneri (GenBank accession no. U04539U04539); V-BPO partial deduced amino acid sequence from A. nodosum (13); V-CPO partial deduced amino acid sequence from Drechslera biseptata (GenBank accession no. Y11123Y11123) and C. inaequalis (16) (of which the complete sequence is known). The alignment was created after a database search (Swiss-Prot) with small stretches of amino acid residues from the active site of V-CPO of C. inaequalis using the EMBL blitz server. The alignment was created with the seqapp and seqvu programs, and the dendrogram was created using the treeview program (from R. D. M. Page, 1996).
The conservation of the active sites among the presented enzymes, both in structure and residues binding the anions, prompted us to determine whether the apo-CPO from C. inaequalis could also act as a phosphatase.
The putative phosphatase activity of apo-r-CPO was assayed by measuring the release of p-NP from p-NPP, a commonly used phosphatase substrate. p-NPP hydrolysis was found to correlate linearly with the amount of enzyme added (data not shown), demonstrating that the apo-r-CPO has phosphatase activity.
The phosphatase activity of apo-r-CPO suggests that vanadate and p-NPP compete for the same binding site in the apo-enzyme, implying that phosphatase and peroxidase activity are mutually exclusive. The results presented in Fig. 2 show that this is indeed the case. Fully activated r-CPO was incubated in the presence of 0.5 mM p-NPP at pH 5.0. CPO activity showed a rapid decrease in time, whereas this was not observed in the absence of p-NPP. This indicates that at this pH vanadate is easily exchanged for p-NPP. The decrease of CPO activity could also be prevented by the addition of extra (100 μM) vanadate, which is in line with the Kd of V-CPO for vanadate at this pH (7). Incubation of fully activated r-CPO with p-NPP at pH 7.0 did not result in p-NPP hydrolysis while incubation at low pH did (results not shown). This is in agreement with results obtained earlier by our group (30), showing that that the Kd of V-CPO for vanadate increases strongly at low pH, thus enabling p-NPP to compete effectively for the binding site.
Figure 2.

Inactivation of V-CPO upon incubation with the phosphatase substrate p-NPP. V-CPO was incubated in 0.5 mM p-NPP at 25°C in 100 mM citrate (pH 5.0). At different time points samples were taken and CPO activity was measured by following the chlorination of monochlorodimedon (MCD, ɛ290 nm = 20.2 mM−1·cm−1) into dichlorodimedon (ɛ290 nm = 0.2 mM−1·cm−1). The assay contained 100 mM citrate (pH 5.0), 50 μM MCD, 1 mM H2O2, and 5 mM Cl−.
Fig. 3 depicts the activity versus the substrate concentration as determined at pH 5.0, showing Michaelis–Menten behavior. By nonlinear regression we calculated a KM of 51 μM and a maximal turnover of 1.7 min−1. It is obvious that the active site of CPO is not optimized for phosphatase activity. For example, the maximal turnover of p-NPP for various acid phosphatases is in the order of 102–103 s−1 (31–33). However, the KM compares well to the values (100–200 μM) reported for these acid phosphatases. We also determined the kinetic parameters of p-NPP hydrolysis catalyzed by apo-r-CPO as a function of pH. The Vmax was only affected by a factor of 2 in the pH range 3.7–8.0, having an optimum at pH 5.0 (results not shown). The KM for p-NPP was unaffected in the pH range 4.5–8.0, but increased strongly in the pH range 3.7–4.5 (KM 1.9 mM at pH 3.9; results not shown), indicating that protonation of either a group on the free enzyme or the substrate itself is the cause of the reduced affinity for p-NPP.
Figure 3.

Phosphatase activity of apo-r-CPO as a function of p-NPP concentration. apo-r-CPO (100 nM) was incubated with different concentrations of p-NPP at 25°C in 100 mM citrate (pH 5.0). After 6 h reaction mixtures were quenched with NaOH, and production of p-NP was measured at 410 nm using ɛ410 nm = 18.3 mM−1·cm−1 at pH 12.
The finding that the apo-CPO of C. inaequalis has phosphatase activity raises the interesting question whether the homologous phosphatases show peroxidase activity. We are currently designing experiments to answer this.
DISCUSSION
We have presented evidence for the existence of structurally similar active sites in vanadium-containing peroxidases and three classes of acid phosphatases. The similarity was deduced from the alignment of the presented enzymes and confirmed by the presence of phosphatase activity in apo-CPO.
The common architecture of the active sites of the presented enzymes may have important implications for research in the acid phosphatase field. The dendrogram of Fig. 1B shows that the aligned phosphatases can be divided into two groups. The first group (marked with an M in the dendrogram) is formed by membrane-bound enzymes. The most interesting protein family contained in this group—from an anthropocentric point of view—is that of mammalian G-6-Pase, a key enzyme in gluconeogenesis and glucose homeostasis, also known to be efficiently and competitively inhibited by vanadate (34). G-6-Pase deficiency is the cause of glycogen storage disease type 1a (von Gierke disease). Four missense mutations in the gene encoding human G-6-Pase were recently identified (24). A subsequent near-saturation mutagenesis study (35) revealed that Arg-83 (marked with an asterisk in the alignment of Fig. 1A) was absolutely required for enzyme activity. In our alignment this residue corresponds to Arg-360 of V-CPO, a residue donating hydrogen bonds to vanadate. Because it is known that a histidine is the phosphate acceptor during catalysis of G-6-Pase, the authors (35) also mutated four conserved histidines predicted to reside on the same (luminal) side of the endoplasmic receptor membrane as Arg-83. His-119 (also marked with an asterisk in Fig. 1A) turned out to be the only histidine absolutely required for phosphatase activity. This is in agreement with our alignment since His-119 of G-6-Pase corresponds to His-404 of V-CPO, a residue which may function as an acid-base group in catalysis (17). His-176 of G-6-Pase was not mutated but we predict that mutation of this residue will also abolish enzyme activity. His-176 of G-6-Pase corresponds to His-496 of V-CPO, the residue covalently linking the metal. It is likely therefore, that in G-6-Pase His-176, and not His-119 as suggested (35), is the phosphate acceptor. Consequently, Arg-83, His-119, and His-176 of G-6-Pase are situated at the same side of the endoplasmic receptor membrane, which calls for a reevaluation of the current topology model of this enzyme (35).
To one protein in the group of the membrane-bound phosphatases, the bcrC gene product, no function has been assigned yet (21). bcrC is one of the three B. licheniformis genes found in an operon conveying resistance toward the antibiotic bacitracin. One of the genes of this operon, bcrA, was identified as coding for a soluble ATP-binding protein with similarity to the so-called ABC transporters. Podlesek et al. (21) established that all three gene products of the bcr locus are necessary for bacitracin resistance and proposed that they are components of an ATP-binding transport system but the precise function of the hydrophobic bcrB and bcrC gene products was not established. Based on our alignment, we predict that bcrC codes for a membrane-bound phosphatase.
The second group of phosphatases (marked with an S in the dendrogram of Fig. 1B) is formed by the secreted class A bacterial acid phosphatases (25), a class of enzymes showing considerable sequence similarity, including a perfectly conserved sequence GSYPSGHT. This is also an important cluster in the alignment of Fig. 1A because it contains a histidine aligning to the proposed acid-base group His-404 of V-CPO. Based on the sequence similarity between the apyrase from Shigella flexneri and the class A acid phosphatases, we propose that the apyrase also belongs to this class.
The above-presented data indicate that the vanadium containing haloperoxidases and the presented phosphatases have divergently evolved from a common ancestor. Strikingly, the opposite—convergent evolution—also seems to have occurred since the active site of V-CPO resembles that of another class of acid phosphatases—the high molecular weight acid phosphatases—both in geometry and in residues contributing to the active site. In this context, however, it is important to stress that no sequence similarity is found with this class of enzymes. The resemblance is most evident from the crystal structure of the active site of rat prostatic acid phosphatase complexed with vanadate (36) which is bound in the same trigonal bipyramidal geometry as in V-CPO. This coordination is analogous to the transition state in the reaction mechanism of several phosphatases (37). Furthermore, in rat prostatic acid phosphatase the vanadate is also covalently linked to a histidine (His-12) while Arg-11, Arg-15, Arg-79, and His-257 are within hydrogen-bonding distance. The binding of vanadate to His-12 gives crystallographic evidence for the proposed role of a histidine as a nucleophile in the formation of the phosphoryl adduct (37). Our data suggest that such a role can also be fulfilled by His-496 in apo-CPO and thus most probably also by the corresponding histidines of the phosphatases contained in the alignment.
Using a S. cerevisiae expression system, we are now producing site-directed mutants for the CPO active-site residues. The resulting mutants will be analyzed with respect to both peroxidase and phosphatase activity. We anticipate that the results of these studies will give important clues to research in the peroxidase field as well as to the research in the acid phosphatase field.
Acknowledgments
This work was supported by the Netherlands Foundation for Chemical research and was made possible by financial support from the Netherlands Organization for Scientific Research, the Netherlands Technology Foundation, and the Netherlands Association of Biotechnology Centres.
ABBREVIATIONS
- CPO
chloroperoxidase
- V-CPO
vanadium-containing CPO
- r-CPO
recombinant CPO
- V-BPO
vanadium-containing bromoperoxidase
- p-NPP
para-nitrophenyl phosphate
- G-6-Pase
glucose-6-phosphatase
- p-NP
para-nitrophenol
References
- 1.Wiesner W, Van Pée K H, Lingens F. J Biol Chem. 1988;263:13725–13732. [PubMed] [Google Scholar]
- 2.Bantleon R, Altenbuchner J, Van Pée K H. J Bacteriol. 1994;176:2339–2347. doi: 10.1128/jb.176.8.2339-2347.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hager L P, Morris D R, Brown F S, Eberwein H. J Biol Chem. 1966;241:1769–1777. [PubMed] [Google Scholar]
- 4.Zeng J, Fenna R E. J Mol Biol. 1992;226:185–207. doi: 10.1016/0022-2836(92)90133-5. [DOI] [PubMed] [Google Scholar]
- 5.De Boer E, Boon K, Wever R. Biochemistry. 1988;27:1629–1635. [Google Scholar]
- 6.Arber J M, De Boer E, Garner C D, Hasnain S S, Wever R. Biochemistry. 1989;28:7968–7973. doi: 10.1021/bi00445a062. [DOI] [PubMed] [Google Scholar]
- 7.Van Schijndel J W P M, Barnett P, Roelse J, Vollenbroek E G M, Wever R. Eur J Biochem. 1994;225:151–157. doi: 10.1111/j.1432-1033.1994.00151.x. [DOI] [PubMed] [Google Scholar]
- 8.Vollenbroek E G M, Simons L H, Van Schijndel J W P M, Barnett P, Balzar M, Dekker H L, Van der Linden C, Wever R. Biochem Soc Trans. 1995;23:267–271. doi: 10.1042/bst0230267. [DOI] [PubMed] [Google Scholar]
- 9.Liu T-N, M’Timkulu T, Geigert J, Wolf B, Neidleman S L, Silva D, Hunter-Cevera J C. Biochem Biophys Res Commun. 1987;142:329–333. doi: 10.1016/0006-291x(87)90277-4. [DOI] [PubMed] [Google Scholar]
- 10.Vilter H. Phytochemistry. 1984;23:1387–1390. [Google Scholar]
- 11.De Boer E, Van Kooyk Y, Tromp M G M, Plat H, Wever R. Biochim Biophys Acta. 1986;869:48–53. [Google Scholar]
- 12.Wever R. In: Biochemistry of Global Change. Oremland R S, editor. New York: Chapman & Hall; 1991. pp. 811–824. [Google Scholar]
- 13.Vilter H. In: Metal Ions in Biological Systems. Sigel H, Sigel A, editors. Vol. 31. New York: Dekker; 1995. pp. 326–362. [PubMed] [Google Scholar]
- 14.Plat H, Krenn B E, Wever R. Biochem J. 1987;248:277–279. doi: 10.1042/bj2480277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Schijndel J W P M, Vollenbroek E G M, Wever R. Biochim Biophys Acta. 1993;1161:249–256. doi: 10.1016/0167-4838(93)90221-c. [DOI] [PubMed] [Google Scholar]
- 16.Simons L H, Barnett P, Vollenbroek E G M, Dekker H L, Muijsers A O, Messerschmidt A, Wever R. Eur J Biochem. 1995;229:566–574. doi: 10.1111/j.1432-1033.1995.tb20499.x. [DOI] [PubMed] [Google Scholar]
- 17.Messerschmidt A, Wever R. Proc Natl Acad Sci USA. 1996;93:392–396. doi: 10.1073/pnas.93.1.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barnett, P., Hondmann, D. H., Simons, L. H., Ter Steeg, P. F. & Wever, R. (1995) International Patent Application (PCT) WO 95/27046.
- 19.Icho T. J Bacteriol. 1988;171:5117–5124. doi: 10.1128/jb.170.11.5117-5124.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fleischmann R D, Adams M D, White O, Clayton R A, Kirkness A R, et al. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 21.Podlesek Z, Comino A, Herzog-Vilikonja B, Zgur-Bertok D, Komel R, Grabnar M. Mol Microbiol. 1995;16:969–976. doi: 10.1111/j.1365-2958.1995.tb02322.x. [DOI] [PubMed] [Google Scholar]
- 22.Shelly L L, Lei K J, Pan C J, Sakata S F, Ruppert S, Schutz G, Yang Chan J. J Biol Chem. 1993;268:21482–21485. [PubMed] [Google Scholar]
- 23.Lange A J. Biochem Biophys Res Commun. 1994;201:302–309. doi: 10.1006/bbrc.1994.1702. [DOI] [PubMed] [Google Scholar]
- 24.Lei K J, Shelly L L, Pan C J, Sidbury J B, Yang Chou J. Science. 1993;262:580–583. doi: 10.1126/science.8211187. [DOI] [PubMed] [Google Scholar]
- 25.Thaller M C, Berlutti F, Schippa S, Lombardi G, Rossolini G M. Microbiology. 1994;140:1341–1350. doi: 10.1099/00221287-140-6-1341. [DOI] [PubMed] [Google Scholar]
- 26.Kasahara M, Nakata A, Shinigawa H. J Bacteriol. 1991;173:6760–6765. doi: 10.1128/jb.173.21.6760-6765.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pond J L, Eddy C K, Mackenzie K F, Conway T, Borecky D J, Ingram L O. J Bacteriol. 1989;171:767–774. doi: 10.1128/jb.171.2.767-774.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tromp M G M, Van T T, Wever R. Biochim Biophys Acta. 1991;1079:53–56. doi: 10.1016/0167-4838(91)90023-s. [DOI] [PubMed] [Google Scholar]
- 29.Stankiewicz P J, Tracey A S, Crans D C. In: Metal Ions in Biological Systems. Sigel H, Sigel A, editors. Vol. 31. New York: Dekker; 1995. pp. 660–662. [Google Scholar]
- 30.Van Schijndel J W P M, Simons L H, Vollenbroek E G M, Wever R. FEBS Lett. 1993;336:239–242. doi: 10.1016/0014-5793(93)80811-8. [DOI] [PubMed] [Google Scholar]
- 31.Ostanin K, Saeed A, Van Etten R L. J Biol Chem. 1994;269:8971–8978. [PubMed] [Google Scholar]
- 32.Van Etten R L, Waymack P P. Arch Biochem Biophys. 1991;288:634–645. doi: 10.1016/0003-9861(91)90246-f. [DOI] [PubMed] [Google Scholar]
- 33.Porvari K S, Herrala A M, Kurkela R M, Taavitsainen P A, Lindqvist Y, Schneider G, Vihko P T. J Biol Chem. 1994;269:22642–22646. [PubMed] [Google Scholar]
- 34.Singh J, Nordlie R A, Jorgenson R A. Biochim Biophys Acta. 1981;678:477–482. doi: 10.1016/0304-4165(81)90129-x. [DOI] [PubMed] [Google Scholar]
- 35.Lei K J, Pan C J, Liu J L, Shelly L L, Yang Chou J. J Biol Chem. 1995;270:11882–11886. doi: 10.1074/jbc.270.20.11882. [DOI] [PubMed] [Google Scholar]
- 36.Lindqvist Y, Schneider G, Vihko P. Eur J Biochem. 1994;221:139–142. doi: 10.1111/j.1432-1033.1994.tb18722.x. [DOI] [PubMed] [Google Scholar]
- 37.Van Etten R L, Hickey M E. Arch Biochem Biophys. 1977;183:250–259. doi: 10.1016/0003-9861(77)90438-6. [DOI] [PubMed] [Google Scholar]