

Summary
Following infection in humans, herpes simplex virus (HSV) establishes latency in the sensory nerve ganglia from where it periodically reactivates, whereas the virus efficiently establishes latency but rarely reactivates in murine models. HSV inhibits MHC class I antigen presentation to CD8 T cells efficiently in humans but poorly in mice, but whether this is a crucial determinant of its ability to reactivate is not known. Here we show that antigen-specific CD8 T cells prevent the in vivo reactivation of wildtype HSV in mice. By contrast, antigen-specific CD8 T cells do not prevent the reactivation of recombinant HSVs that inhibit murine MHC class I in the mouse in a manner similar to wildtype HSV in humans, despite the presence of these cells in the ganglia of latently-infected mice. These findings indicate that efficient inhibition of MHC class I by HSV is a key factor in its ability to reactivate in humans.
Introduction
Herpes simplex virus (HSV) is one of the most common human pathogens, infecting 50-70% of the world population (Smith and Robinson, 2002). While HSV infection can cause serious or fatal disease in neonates and the immunocompromised, and ocular infection may lead to corneal scaring and blindness in normal hosts (Liesegang, 2001; Looker and Garnett, 2005; Whitley and Roizman, 2001), it typically manifests as lesions at mucosal surfaces. These recurrent lesions occurring periodically over the lifetime of an infected individual reflect reactivation of latent HSV infection, which is established in the sensory nerve ganglia at the time of primary infection. This life-long infection results in limited morbidity but facilitates transmission to new hosts and thereby helps to sustain the high prevalence of HSV infection in the human population.
The traditional model of HSV latency posits that viral protein production is halted and the virus is maintained in a quiescent state in which only an untranslated mRNA, LAT, is produced. However careful analysis of latently-infected murine ganglia has revealed infrequent, but readily detectable viral protein production, indicating that latency is leakier than previously thought (Feldman et al., 2002). These proteins may be processed and presented by MHC class I to virus-specific CD8 T cells, allowing detection and repression of overt viral reactivation (Khanna et al., 2004a). Consistent with this notion, CD8 T cells have been found to co-localize with latently-infected cells in the trigeminal ganglia of both humans and mice (Hufner et al., 2006; Khanna et al., 2003; Pereira and Simmons, 1999; Theil et al., 2003). In humans, a substantial number of these cells are HSV-specific and bear markers associated with an effector or effector memory phenotype (Verjans et al., 2007); many of these cells express the activation markers CD69 and granzyme B and are in close proximity to neurons expressing HSV LAT, suggesting ongoing exposure to viral antigens. In mice, these T cells are predominantly HSV-specific, often have TCRs clustered at points of contact with adjacent neurons, and express CD69 and granzyme B, indicating recent antigen exposure (Khanna et al., 2003; van Lint et al., 2005). These HSV-specific CD8 T cells prevent reactivation in vitro in a partially IFN-γ-dependent manner (Khanna et al., 2003; Liu et al., 2001; Liu et al., 2000). In vivo reactivation after hyperthermic stress or UV-exposure is increased in IFN-γ- and IFN-γR-deficient mice (Cantin et al., 1999; Minami et al., 2002). These findings indicate that antigen presentation to CD8 T cells is ongoing during latency, and that these HSV-specific CD8 T cells may limit reactivation. Accordingly, eliciting a robust CD8 T cell response is the objective of several potential therapeutic HSV vaccines (Koelle, 2006).
To counter the CD8 T cell response, HSV encodes multiple immune evasion strategies, including inhibition of antigen presentation by MHC class I (Tortorella et al., 2000). HSV ICP47 binds to the transporter associated with antigen presentation (TAP) 1/2 complex preventing peptides from being loaded onto nascent MHC class I heavy chains (Hill et al., 1995). An ICP47-deletion virus was shown to be less neurovirulent, and this decrease was due to CD8 T cells as depletion of these cells restored neurovirulence (Goldsmith et al., 1998). While ICP47 effectively blocks MHC class I antigen presentation on infected human cells, inhibition of presentation by murine cells is only marginally reduced, due to the low affinity of ICP47 for murine TAP1/2 (Tomazin JV 1998; Jugovic JV 1998; (Ahn et al., 1996; Tomazin et al., 1996). These findings suggest that the effects of ICP47 in vivo are likely to be much greater in humans than in mice.
To mimic in mice the effects of ICP47 in humans, we previously generated recombinant HSVs (rHSVs) that also express either human cytomegalovirus (HCMV) US11 (designated 27US11) or murine CMV m152 (designated 27m152) in addition to gfp, and a control virus (designated 27gfp) that expresses gfp but not an MHC class I inhibitor (Orr et al., 2005). Both 27US11 and 27m152 reduced surface expression of murine MHC class I, rendering infected cells less susceptible to recognition and killing by CD8 T cells, leading to a loss of CD8 T cell-mediated control and increased virulence during acute infection (Orr et al., 2005). We now show that HSV-mediated inhibition of MHC class I antigen presentation during latency abrogated CD8 T cell control of viral reactivation using both in vitro and in vivo models of HSV reactivation. Further, in the case of m152-mediated inhibition, this immune evasion strategy also reduced the frequency and activation of HSV-specific CD8 T cells in the latently-infected trigeminal ganglia. These data demonstrate that latent HSV is under immune surveillance, and that the virus is able to counteract this surveillance and to reactivate in the presence of HSV-specific CD8 T cells.
Results
Clearance of Primary Infection and Establishment of Latency
We previously demonstrated that viral inhibition of MHC class I resulted in increased viral titers during primary infection of mice inoculated in the footpad (Orr et al., 2005). To determine whether viral inhibition of MHC class I affected the course of primary ocular infection, we analyzed viral shedding by sequentially swabbing both eyes of infected mice over a 10 day period, and measured viral titers in homogenates of eyes and trigeminal ganglia from additional mice sacrificed at intervals over the first 14 days of infection. The amounts of virus shed from the eyes were similar for 27gfp, 27US11 and 27m152 through day 7, and shedding ceased by day 10 post-infection (Fig. 1a). Viral abundance in eye homogenates followed a similar pattern with the greatest amounts at day one and clearance by day 14 (Fig. 1b). Whereas peak titers in the trigeminal ganglia were observed on day 3, virus was also cleared from this site by day 14 (Fig. 1c). 27m152 demonstrated a replication advantage over 27gfp in both tissues by day 3 post-infection, while 27US11 demonstrated a similar advantage in the trigeminal ganglia but not in eye homogenates. Nonetheless, by 14 days post-infection these viruses were uniformly cleared, irrespective of their ability to inhibit MHC class I.
Figure 1. Viral inhibition of MHC class I does not prolong or prevent the clearance of primary infection.
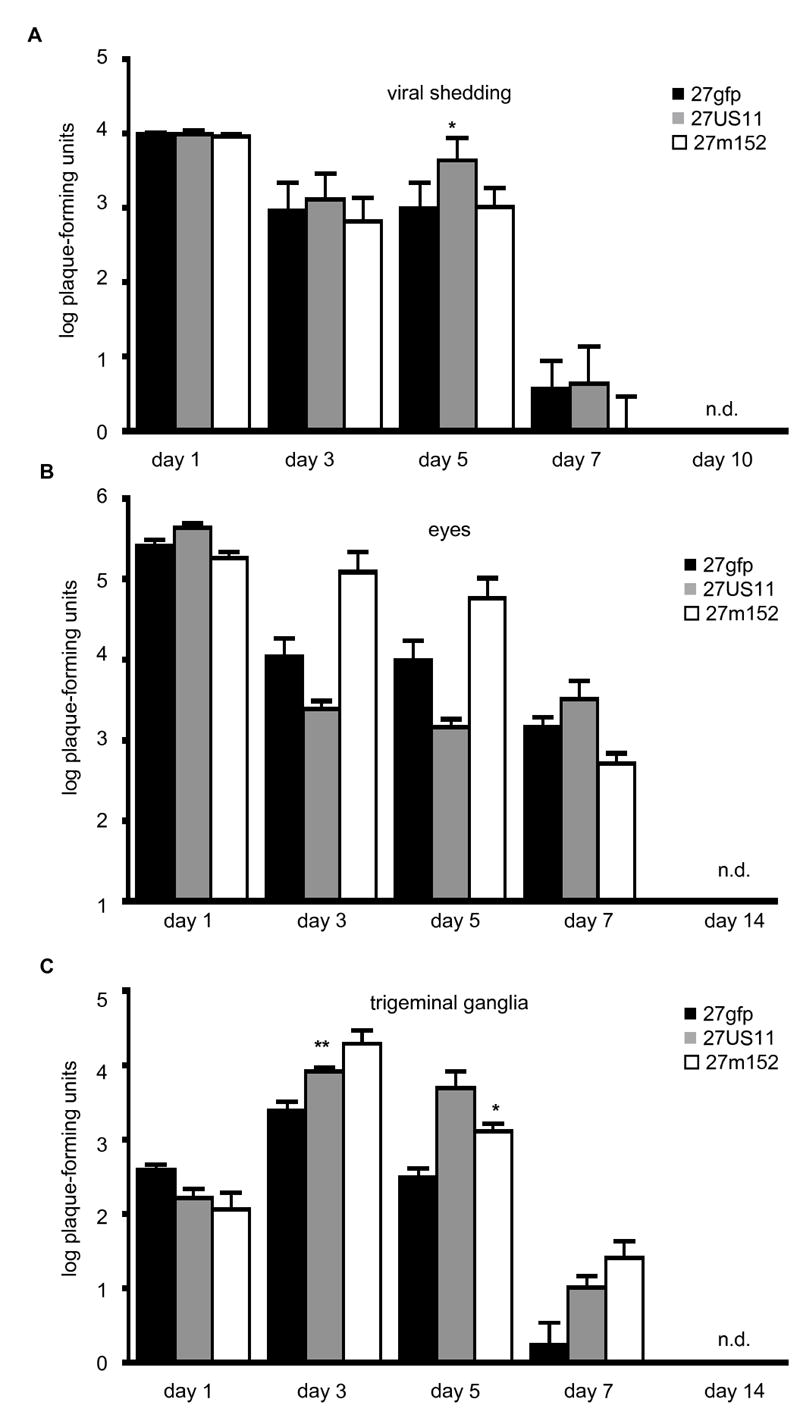
Female C57Bl/6 mice were infected with 2×105 pfu/eye of the indicated viruses. (A) 8 mice/virus were serially swabbed for viral shedding from both eyes on the indicated days. Alternatively, on the indicated days 4 mice/virus were euthanized and the (B) eyes and (C) trigeminal ganglia were harvested. Viral titers were determined by plaque formation on Vero cells. Statistically significant increases in viral titer compared to 27gfp on the same day are indicated (* p<0.05 and ** p<0.01). Error bars = one SEM. n.d. = not detected.
Previous reports have demonstrated that the amount of latent virus correlates with the amount of virus in the initial inoculum. Since 27US11 and 27m152 both replicated to higher titers than 27gfp in the trigeminal ganglia by day 3 and maintained this advantage until all viruses were cleared, we hypothesized that viral inhibition of MHC class I would result in an increased amount of latent virus. Surprisingly, at 2 weeks post-infection the numbers of latent viral copies in the trigeminal ganglia were slightly reduced in 27US11 or 27m152-infected mice, as compared to 27gfp-infected animals, however these differences were not statistically significant (Fig. 2). Thus, viral inhibition of MHC class I favored replication during acute infection, but did not prevent viral clearance or increase the amount of latent virus.
Figure 2. The amount of latent viral DNA is unaltered by viral inhibition of MHC class I.
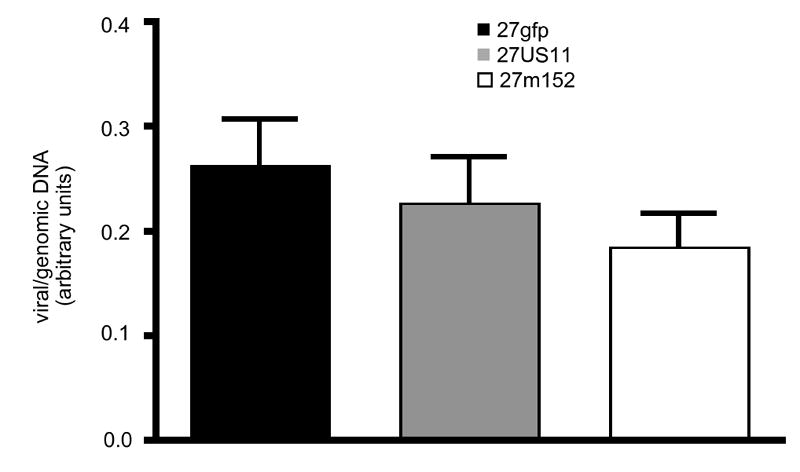
The relative quantity of viral DNA in the trigeminal ganglia of latently-infected mice 2 weeks post-infection was determined by realtime PCR for viral genomic DNA and normalized to the number of copies of the murine adipsin gene in each sample. 16-24 ganglia/virus were analyzed. Error bars = one SEM. Data are pooled from 2 experiments with similar results. There were no statistically significant differences among the groups.
CD8 T Cell Response During Latency
As previously reported with wildtype HSV (Khanna et al., 2003), two weeks after primary infection with 27gfp HSV-specific CD8 T cells were readily found within latently-infected trigeminal ganglia as detected by staining with H-2Kb dimer loaded with the immunodominant HSV peptide gB498-505 (Wallace et al., 1999) (Fig. 3 A). Whereas mice latently-infected with 27US11 and 27gfp had a similar frequency of HSV-specific CD8 T cells in the trigeminal ganglia at this time point, there were significantly fewer HSV-specific cells in ganglia latently-infected with 27m152 (Fig. 3 A). The frequencies of HSV-specific CD8 T cells did not change from two (Fig. 3 A) to five (Fig. 3 B) weeks post-infection, indicating that this number is set by two weeks post-infection. A similar decrease in the frequency of HSV-specific CD8 T cells in 27m152-infected mice was observed by intracellular staining for IFN-γ after re-stimulation with the gB peptide; this finding corroborates the results obtained by H-2Kb-gB dimer staining and indicates that the fraction of HSV gB-specific CD8 T cells capable of producing IFN-γ directly ex vivo was similar for the three viruses (Fig. 3 C). By five weeks post-infection, the majority of the gB-specific CD8 T cells in the trigeminal ganglia of 27gfp-infected mice expressed granzyme B. In 27m152-infected mice, we observed a subtle though reproducible and statistically significant (p<0.001) reduction in the fraction of gB-specific CD8 T cells expressing granzyme B ex vivo, as compared to 27gfp-infected mice, suggesting that fewer of these antigen-specific CD8 T cells had recently encountered cognate peptide-MHC (van Lint et al., 2005) (Fig. 3 D). Together these data indicate that inhibition of MHC class I by 27m152, but not by 27US11, reduced the number and degree of activation of gB-specific CD8 T cells within the latently-infected ganglia. The greater impact of m152 compared to US11 on HSV-specific CD8 T cells may be due to the more efficient blockade by m152 of antigen presentation by cells expressing H-2b (Orr et al., 2005).
Figure 3. Viral inhibition of MHC class I reduces the number and activation state of HSV-specific CD8 T cells in latently-infected ganglia.
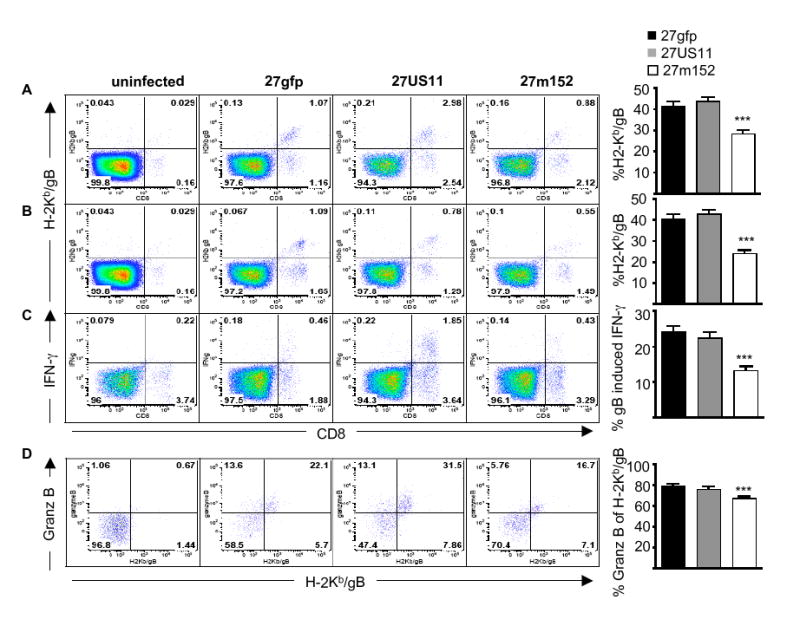
HSV gB-specific CD8 T cells in latently-infected trigeminal ganglia at (A) 14-16 or (B and C) 32-46 days post-infection were identified (A and B) by binding of H-2Kb DimerX loaded with HSV gB498-505 peptide or (C) by gB peptide-induced IFN-γ production. (D) The activation status of HSV gB-specific CD8 T cells was determined as the percentage of H-2Kb-gB DimerX-positive CD8 T cells that contained intracellular granzyme B directly ex vivo. The cells shown in the plots are gated on total cells (A, B and C) or (D) CD8 T cells in the trigeminal ganglia. The bar graphs indicate the mean percentage ± SEM (A, B and C) of CD8 T cells that are specific for HSV gB or (D) of HSV gB-specific CD8 T cells that express granzyme B ex vivo. Data are pooled from (A) three, (B) five, (C) four and (D) two experiments with similar results; in each experiment 8-10 ganglia/virus were analyzed. The difference between mice infected with 27m152 compared to 27gfp in each panel was statistically significant (P<0.0001). Error bars = one SEM.
In Vitro Reactivation
HSV-specific CD8 T cells can prevent in vitro reactivation of wildtype HSV from explanted latently-infected ganglia (Khanna et al., 2003; Liu et al., 2000). In good agreement with these reports, we found that HSV-specific CD8 T cells efficiently prevented reactivation of 27gfp from cultures of dissociated, latently-infected ganglia that were explanted 14 days post-infection (Fig. 4 A). Reactivation was prevented by CD8 T cells, as addition of blocking CD8 antibody resulted in 100% reactivation by all three viruses (Fig. 4 B). However, 27m152 and 27US11 reactivated much more readily from latently-infected ganglia in the presence of HSV-specific CD8 T cells (Fig. 4 A). Thus, by limiting antigen presentation by MHC class I, 27m152 and, to a lesser extent, 27US11 were able to reactivate more efficiently in the presence of virus-specific CD8 T cells.
Figure 4. 27m152 and 27US11 reactivate in the presence of HSV-specific CD8 T cells.
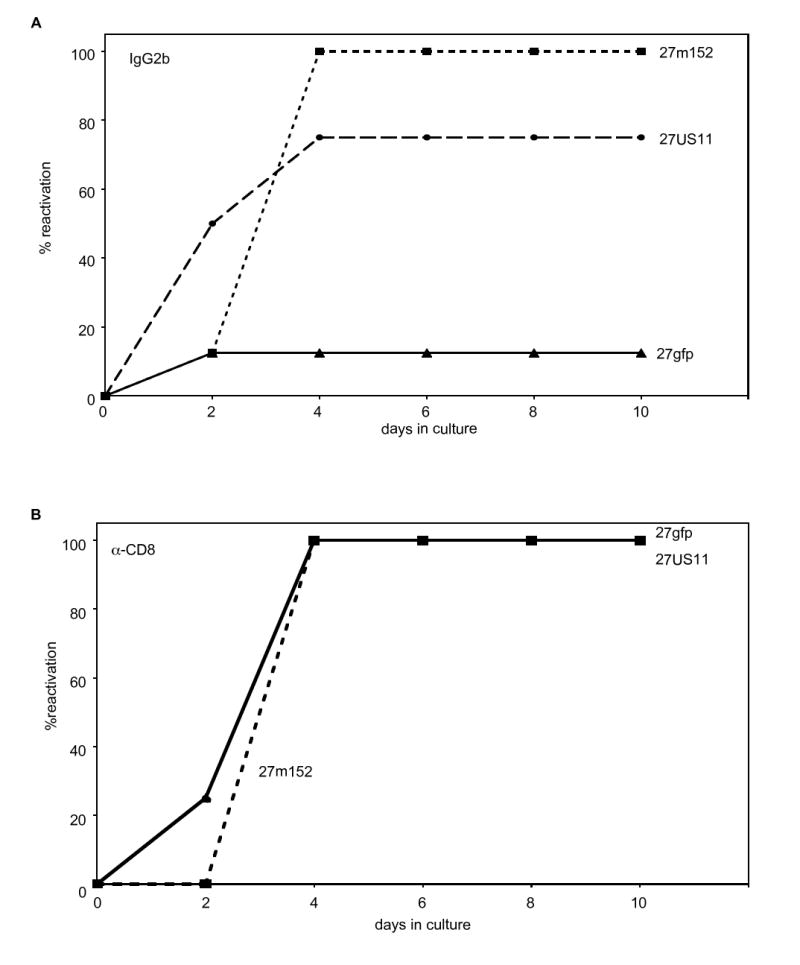
Reactivation of rHSVs in vitro from explanted, latently-infected trigeminal ganglia in the presence of effector HSV-specific CD8 T cells plus IL-2 and (A) IgG2b isotype control or (B) α-CD8 monoclonal antibody (n = 8 trigeminal ganglia/group). (A) The increased frequencies of reactivation for 27US11 (P = 0.01) and 27m152 (P = 0.0017) compared to 27gfp were statistically significant. (B) The results for 27gfp and 27US11 data were identical and thus the symbols for 27US11 are not apparent in the figure.
In Vivo Reactivation
Although spontaneous reactivation is rare in mice, reactivation can be induced by hyperthermia or exposure to UV light (Laycock et al., 1991; Sawtell and Thompson, 1992). Since the latter is a known cause of HSV reactivation in humans (Rooney et al., 1991), we examined the impact of MHC class I inhibition on reactivation in vivo as determined by the recovery of lytic virus from the eyes and trigeminal ganglia following exposure of latently-infected mice to UV irradiation. These rHSVs were made in the KOS strain of HSV-1 that reactivates less frequently than many other HSV strains (Sawtell et al., 1998), allowing us to determine more readily whether viral inhibition of MHC class I increases the reactivation frequency in vivo. The frequency of reactivation five weeks after infection was significantly higher for mice infected with 27US11 or 27m152 compared to mice infected with 27gfp (Fig. 5B). In a portion of the mice we quantified the amount of virus in the homogenates from the trigeminal ganglia and eyes separately (Fig. 5A). Although the absolute numbers of plaque forming units recovered were low, in the few mice in which 27gfp reactivated, virus was recovered only from the trigeminal ganglia. Conversely, a subset of the animals infected with 27US11 and 27m152 had virus recovered from their eyes in addition to their trigeminal ganglia. In no case, was virus recovered from eye but not trigeminal ganglia homogenates. Consistent with the low levels of virus detected in these tissue homogenates, we rarely detected viral shedding in the swabbed eyes with any of these viruses (data not shown). The increased rate of reactivation in mice infected with 27US11 and 27m152 was not simply due to decreased numbers of HSV-specific CD8 T cells, as 27US11 reactivated more frequently than 27gfp even though the number and activation status of these cells in the trigeminal ganglia of 27US11-infected mice were similar to 27gfp-infected mice (Fig. 3).
Figure 5. Viral inhibition of MHC class I promotes reactivation in vivo.
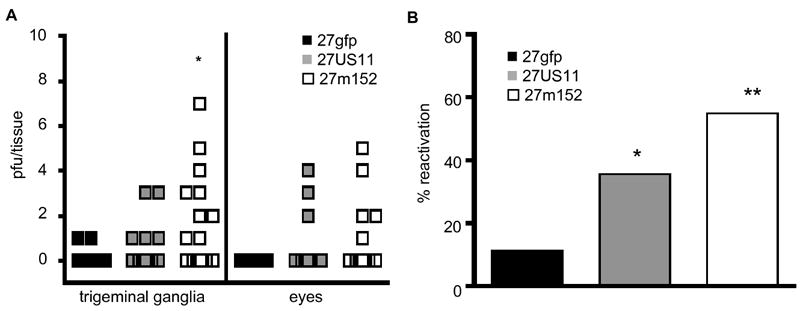
In vivo reactivation from the trigeminal ganglia and eyes of latently-infected mice following UV-exposure 5 weeks post-infection. (A) The numbers of recovered pfu’s from the trigeminal ganglia and eyes are shown. In some mice virus was recovered from both sites, in others only in the trigeminal ganglia. Virus was never recovered from the eyes but not the trigeminal ganglia. Data represent 18-19 mice/virus and were pooled from 2 experiments with similar results. (B) The overall frequency of reactivation in the trigeminal ganglia for each virus and treatment group is shown. Data are from 27-31 mice/virus pooled from 3 experiments, including the two experiments shown in A and a third experiment in which reactivation was determined on a combined homogenate of the ganglia and eyes. Statistically significant increases versus 27gfp are indicated (* p<0.05 and ** p<0.01).
To test directly whether the increased rate of reactivation of 27US11 and 27m152 was due to the ability of these viruses, but not 27gfp, to efficiently evade control by CD8 T cells, we depleted these cells in latently-infected mice prior to UV exposure. CD8 depletion, but not treatment with an IgG2b isotype control antibody, led to a markedly increased frequency of 27gfp reactivation that was similar to the frequency of reactivation of 27US11 and 27m152 (Fig. 6). Conversely, depletion of CD8 T cells had little impact on the already high reactivation frequency of 27US11 and 27m152. These results indicate that inhibition of MHC class I antigen presentation by 27US11 and 27m152 increased the probability of reactivation by limiting the efficacy of CD8 T cells in preventing reactivation.
Figure 6. Inhibition of MHC class I abrogates CD8 T cell prevention of HSV reactivation.
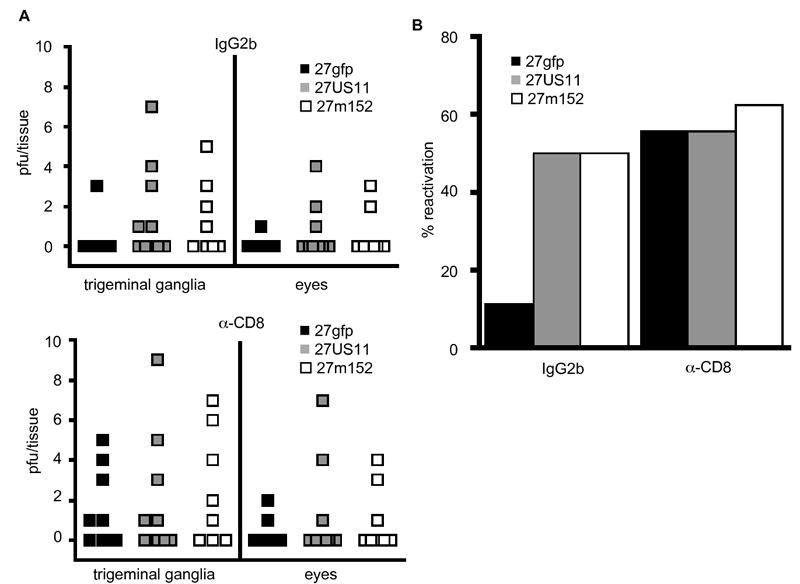
In vivo reactivation of rHSV following UV-exposure in 8-10 latently-infected mice treated with α-CD8 or IgG2b isotype control Ab. (A) The numbers of recovered pfu’s from both the trigeminal ganglia and eyes are shown. In some mice virus was recovered from both sites, in others only from the trigeminal ganglia. Virus was never recovered from the eyes but not the trigeminal ganglia. (B) The overall frequency of reactivation in the trigeminal ganglia for each virus and treatment group is indicated. The results shown are from one of two experiments performed with similar results in both.
Discussion
Previous studies have shown that HSV-specific CD8 T cells are in close association with latently-infected murine ganglia, and that these cells prevent HSV reactivation in vitro (Khanna et al., 2003; Liu et al., 2000). However, a caveat regarding the applicability of these studies to HSV infection in humans is the ability of HSV to efficiently inhibit human but not murine MHC class I antigen presentation to CD8 T cells (Ahn et al., 1996; Jugovic et al., 1998; Tomazin et al., 1996; Tomazin et al., 1998). Using rHSVs that express either HCMV US11 or MCMV m152, both of which inhibit murine MHC class I antigen presentation (Orr et al., 2005), we found that CD8 T cell control of reactivation is muted by this immune evasion strategy. Although viral inhibition of MHC class I increased the viral burden during primary infection, it neither prevented the clearance of primary infection nor increased the number of viral genomes in the latently-infected trigeminal ganglia. Although the establishment of latency was not altered, the numbers of HSV-specific CD8 T cells residing in the latently-infected ganglia were diminished in mice infected with 27m152. This decrease in cell numbers coincided with a decreased activation state of these cells, suggesting that ongoing antigen presentation is required to maintain HSV-specific CD8 T cells in latently-infected ganglia. Independent of the alteration of CD8 T cell numbers, both 27US11 and 27m152 reactivated efficiently in the presence of HSV-specific CD8 T cells in vitro, whereas the control virus, 27gfp, did not. Using an UV-induced model of reactivation we found that viral inhibition of MHC class I enhanced the rate of viral reactivation in vivo. This enhancement reflected the ability of 27US11 and 27m152 to evade CD8 T cell-mediated control, as depletion of CD8 T cells resulted in efficient reactivation by 27gfp and comparable rates of reactivation by all three viruses. These results demonstrate that HSV reactivation is under immunological surveillance by CD8 T cells, but that CD8 T cell-mediated control is subverted by efficient viral inhibition of MHC class I.
Although 27US11 and 27m152 both demonstrated an increased frequency of reactivation in vitro and in vivo, the effects of m152 were consistently stronger than US11. Additionally, expression of 27m152, but not 27US11, reduced the frequency and activation status of HSV-specific CD8 T cells within the latently-infected trigeminal ganglia. This difference may be due to a greater inhibition of MHC class I in cells infected with 27m152 than in cells infected with 27US11, as we previously reported (Orr et al., 2005). However, we cannot exclude the possibility that additional functions of m152, including inhibition of NKG2D ligand expression on the surface of infected cells, may contribute to the greater effects observed with this virus (Ehrlich et al., 2005; Krmpotic et al., 2002; Lodoen et al., 2003). During primary infection, both 27US11 and 27m152 replicated to higher titers in the trigeminal ganglia, but only 27m152 replicated to higher titers in the periphery. This difference likely results, at least in part, from the ability of m152 to inhibit recognition by CD8 T cells even in the periphery, where MHC class I expression is higher, whereas US11 was only able to do so in the ganglia, where MHC class I expression is low compared to the periphery.
Several studies have demonstrated that the rate of reactivation correlates with the numbers of latently-infected neurons in the ganglia, suggesting that these numbers may be the prime determinant of reactivation (Lekstrom-Himes et al., 1998; Maggioncalda et al., 1996; Sawtell, 1998). We found that the amount of latent virus was not affected by viral inhibition of MHC class I, yet the rate of reactivation was significantly higher for both 27US11 and 27m152. These findings suggest that the degree to which the virus evades local CD8 T cell-mediated control is a second determinant of the frequency of reactivation, independent of the amount of latent virus.
The canonical view of latency holds that production of viral proteins in the ganglia is abrogated, removing the source of antigenic peptides that are presented by MHC class I to CD8 T cells. However careful analysis of latently-infected murine trigeminal ganglia reveals low but readily detectable amounts of viral proteins, indicating that latency is less silent than previously thought (Feldman et al., 2002). These data suggest a model in which a subset of viral proteins is transcribed during latency at relatively low levels. These proteins are in turn processed and presented by MHC class I to CD8 T cells residing in the ganglia (Khanna et al., 2004b). If these cells mount a vigorous, partially IFN-γ-mediated response, viral reactivation is aborted (Decman et al., 2005). Conversely if this response is muted or absent, such as in the absence of IFN-γ or its receptor, then viral reactivation proceeds (Cantin et al., 1999).
It was previously thought that uninfected neurons do not express MHC class I, and that MHC class I expression was induced only transiently in response to acute infection with HSV, during which time CD8 T cells helped to control the infection (Pereira and Simmons, 1999; Pereira et al., 1994). However, it has recently been appreciated using more sensitive methods that neurons do express MHC class I at low levels, and that expression is regulated in part by neuronal activity (Goddard et al., 2007). These findings, along with the observations that viral antigens distinct from LAT are expressed at low levels during latency (Feldman et al., 2002), that CD8 T cells co-localize with latently-infected cells in the trigeminal ganglia of both humans and mice (Hufner et al., 2006; Khanna et al., 2003; Theil et al., 2003), and that a substantial number of these cells are HSV-specific and bear markers indicating recent antigen exposure (Khanna et al., 2003; van Lint et al., 2005; Verjans et al., 2007), suggest that latently-infected ganglia are under active immunological surveillance by CD8 T cells. Despite the presence of HSV-specific CD8 T cells, subclinical reactivation is a common occurrence in humans, suggesting that these cells are inefficient in controlling HSV reactivation from latently-infected human sensory neurons where MHC class I expression is low and thus the impact of ICP47 is much more pronounced (Crespi et al., 2007). Our data support this conclusion, as inhibition of murine MHC class I by 27US11 or 27m152 dramatically increased the rate of reactivation within the trigeminal ganglia, whereas the recovery of reactivated virus from the eye was less frequent. Our findings are also consistent with the recent finding that the primary site of immunological control of HSV-2 reactivation in humans is in the periphery, where HSV-specific CD8 T cells localize to recurrent HSV skin lesions and where infiltration by these cells correlates with viral clearance, rather than in the ganglia (Zhu et al., 2007).
Our data support this "actively testing the waters" model of HSV latency and reactivation (Cantin et al., 1995; Hufner et al., 2006; Khanna et al., 2004a). CD8 T cells can act as sentinels to terminate nascent reactivation in mice, but efficient inhibition of MHC class I allows HSV to evade CD8 T cell control and shift the balance in favor of reactivation. These findings suggest that efficient inhibition of MHC class I by HSV is an important determinant of this virus’ ability to reactivate and thereby to be transmitted to others over the lifetime of an infected individual. These findings also have implications for the development of a therapeutic vaccine for HSV, as induction of virus-specific CD8 T cells may not alone be sufficient to prevent reactivation or clear latent infection in humans, although they may limit the frequency and/or duration of recurrent lesions.
Experimental Procedures
Viruses
Generation of the recombinant HSVs 27gfp, 27US11, and 27m152 was described previously (Orr et al., 2005). Briefly, targeting constructs containing HCMV US11 or MCMV m152 under the HCMV IE promoter, eGFP under the EF promoter, and GPT under the PGK promoter were flanked with DNA homologous to HSV UL26 and UL27. Targeting constructs were linearized and electroporated into Vero cells. Cells were then infected with KOS strain HSV-1. Recombinant viruses were isolated based on expression of gfp and plaque-purified. A control virus designated 27gfp containing only gfp and gpt was generated in a similar manner.
Mice and Infection
6-8 week old female C57Bl/6 were purchased from Jackson Labs (Bar Harbor, Maine) and maintained in the University of Washington SPF facility. Prior to infection mice were anesthesized with a mixture of ketamine and xylazine. Corneas were abraded with a 30g needle passed 6 times across the cornea. Mice were inoculated in both eyes with 2 × 105 pfu of the indicated virus in 3 μL. Mice were monitored daily for development of ataxia. Affected mice were euthanized. All studies were approved by the University of Washington Institutional Animal Care and Use Committee.
Quantification of Lytic Virus
Mice were sacrificed by administration of Beuthanasia-D solution (Schering-Plough Union, NJ). Trigeminal ganglia and eyes were removed and snap-frozen on dry ice. Frozen tissue was homogenized and plated on Vero cells. To quantify viral shedding mice were swabbed in both eyes. Swabs were soaked in 0.5 mL media and the media transferred directly to Vero cells. Plaques were visualized 72-96 hours later by addition of crystal violet dye.
Detection of Latent Virus
Total DNA was isolated from latently-infected trigeminal ganglia using a DNEasy Tissue kit (Qiagen, Valencia, CA) following the Total DNA from Animal Tissues protocol. Relative abundances of viral and murine DNA were determined by real-time PCR for HSV ICP47 or murine adipsin, respectively, using SYBR Green (Stratagene, Cedar Creek, TX). The following primers were used: ICP47 forward CACGTTTGCAGCGCACATGC, ICP47 reverse CTTGCGCACCTGGTGATGTT, murine adipsin forward AAGTGATCGCTACAAGTTGT, murine adipsin reverse ACTTGCACGGAAGCCATGTA.
Quantification of CD8 T Cell Response
On the indicated day mice were sacrificed as above and exsanguinated. Trigeminal ganglia were harvested and dissociated by treatment with 3 mg/mL collagenase type I (Sigma-Aldrich, St Louis, MO) for 90 minutes at 37°C. Ganglia were then triturated into single-cell suspensions. Cells were stimulated with 5 μM of the immunodominant HSV peptide glycoprotein B498-505 (gB498-505) (United Biochemical Research, Seattle, Washington) for 5 hours in the presence of monensin, followed by surface staining with α-CD8 and α-CD45.2, permeabilized using the Intracellular Staining Kit from BD Biosciences, and stained for intracellular IFN-γ. Unstimulated lymphocytes were used as a negative control. Antigen specific IFN-γ production = percentage CD8+ IFN-γ+ stimulated – unstimulated. Alternatively, freshly isolated cells were stained with α-CD8, α-CD45.2, and H-2Kb dimerX loaded with either HSV gB peptide or an irrelevant peptide according to manufacturer’s specifications and stained intracellularly for granzyme B. DimerX and antibodies and were purchased from BD Biosciences Pharmingen.
In Vitro Reactivation
Latently-infected trigeminal ganglia were harvested 14 days post-infection and treated as for CD8 T cell quantification. Single cell suspensions were cultured in the presences of 100 U/mL IL-2, 2.5 × 106 total activated lymphocytes isolated from the draining lymph nodes of mice infected 6 days prior, and 150 μg of either α-CD8 (clone 2.43) or IgG2b isotype control. Cultures were assayed for reactivation daily by transferring 150 μL to Vero cells, which were monitored for plaque formation as described above. Fresh media containing IL-2 was added to keep culture volumes and content constant.
In Vivo Reactivation
Five weeks after acute infection, latently-infected mice were exposed to 945 mJ/cm2 UVB light using a handheld UV transilluminator (Ultra-Violet Products model UVM-57, Upland, CA) with a peak emission at 302 nm. Mice were covered such that only the eyes received UV-exposure. Three days post-exposure mice were sacrificed and assayed for lytic virus in the eyes and trigeminal ganglia as described for quantification of lytic virus. Animals were scored as positive for reactivation if lytic virus was recovered from the trigeminal ganglia. CD8 T cells were depleted from some mice by i.p. injection of 200 μg of α–CD8 (clone 2.43) or IgG2b isoytpe control on days −3 and −2 relative to UV treatment.
Statistical analysis
Statistical differences in viral titers and frequency of HSV-specific CD8 T cells were determined by two-tailed unpaired Student t test. Statistical significance for in vitro and in vivo reactivation were determined by Chi square test. Statistical differences in the titers of reactivated virus were determined by unpaired Mann-Whitney test. Statistics were determined with Prism software (GraphPad Software, Inc).
Acknowledgments
This work was supported by grant HD18184 from the NICHD, NIH. MTO was supported by a Howard Hughes Medical Institute Pre-doctoral Training Fellowship and JAS was supported by training grant CA09537 from the NCI, NIH and a Predoctoral Emphasis Pathway in Tumor Immunology training grant from the Cancer Research Institute.
Abbreviations
- rHSV
recombinant herpes simplex virus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn K, Meyer TH, Uebel S, Sempe P, Djaballah H, Yang Y, Peterson PA, Fruh K, Tampe R. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. Embo J. 1996;15:3247–3255. [PMC free article] [PubMed] [Google Scholar]
- Cantin E, Tanamachi B, Openshaw H. Role for gamma interferon in control of herpes simplex virus type 1 reactivation. J Virol. 1999;73:3418–3423. doi: 10.1128/jvi.73.4.3418-3423.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantin EM, Hinton DR, Chen J, Openshaw H. Gamma interferon expression during acute and latent nervous system infection by herpes simplex virus type 1. J Virol. 1995;69:4898–4905. doi: 10.1128/jvi.69.8.4898-4905.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi CM, Cumberland WG, Wald A, Corey L, Blower S. Longitudinal Study of Herpes Simplex Virus Type 2 Infection Using Viral Dynamic Modeling. Sex Transm Infect. 2007 doi: 10.1136/sti.2006.022020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decman V, Kinchington PR, Harvey SA, Hendricks RL. Gamma interferon can block herpes simplex virus type 1 reactivation from latency, even in the presence of late gene expression. J Virol. 2005;79:10339–10347. doi: 10.1128/JVI.79.16.10339-10347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich LI, Ogasawara K, Hamerman JA, Takaki R, Zingoni A, Allison JP, Lanier LL. Engagement of NKG2D by cognate ligand or antibody alone is insufficient to mediate costimulation of human and mouse CD8+ T cells. J Immunol. 2005;174:1922–1931. doi: 10.4049/jimmunol.174.4.1922. [DOI] [PubMed] [Google Scholar]
- Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, Margolis TP. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc Natl Acad Sci U S A. 2002;99:978–983. doi: 10.1073/pnas.022301899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard CA, Butts DA, Shatz CJ. Regulation of CNS synapses by neuronal MHC class I. Proc Natl Acad Sci U S A. 2007;104:6828–6833. doi: 10.1073/pnas.0702023104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith K, Chen W, Johnson DC, Hendricks RL. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J Exp Med. 1998;187:341–348. doi: 10.1084/jem.187.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill A, Jugovic P, York I, Russ G, Bennink J, Yewdell J, Ploegh H, Johnson D. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1995;375:411–415. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- Hufner K, Derfuss T, Herberger S, Sunami K, Russell S, Sinicina I, Arbusow V, Strupp M, Brandt T, Theil D. Latency of alpha-herpes viruses is accompanied by a chronic inflammation in human trigeminal ganglia but not in dorsal root ganglia. J Neuropathol Exp Neurol. 2006;65:1022–1030. doi: 10.1097/01.jnen.0000235852.92963.bf. [DOI] [PubMed] [Google Scholar]
- Jugovic P, Hill AM, Tomazin R, Ploegh H, Johnson DC. Inhibition of major histocompatibility complex class I antigen presentation in pig and primate cells by herpes simplex virus type 1 and 2 ICP47. J Virol. 1998;72:5076–5084. doi: 10.1128/jvi.72.6.5076-5084.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna KM, Bonneau RH, Kinchington PR, Hendricks RL. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity. 2003;18:593–603. doi: 10.1016/s1074-7613(03)00112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna KM, Lepisto AJ, Decman V, Hendricks RL. Immune control of herpes simplex virus during latency. Curr Opin Immunol. 2004a;16:463–469. doi: 10.1016/j.coi.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Khanna KM, Lepisto AJ, Hendricks RL. Immunity to latent viral infection: many skirmishes but few fatalities. Trends Immunol. 2004b;25:230–234. doi: 10.1016/j.it.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Koelle DM. Vaccines for herpes simplex virus infections. Curr Opin Investig Drugs. 2006;7:136–141. [PubMed] [Google Scholar]
- Krmpotic A, Busch DH, Bubic I, Gebhardt F, Hengel H, Hasan M, Scalzo AA, Koszinowski UH, Jonjic S. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat Immunol. 2002;3:529–535. doi: 10.1038/ni799. [DOI] [PubMed] [Google Scholar]
- Laycock KA, Lee SF, Brady RH, Pepose JS. Characterization of a murine model of recurrent herpes simplex viral keratitis induced by ultraviolet B radiation. Invest Ophthalmol Vis Sci. 1991;32:2741–2746. [PubMed] [Google Scholar]
- Lekstrom-Himes JA, Pesnicak L, Straus SE. The quantity of latent viral DNA correlates with the relative rates at which herpes simplex virus types 1 and 2 cause recurrent genital herpes outbreaks. J Virol. 1998;72:2760–2764. doi: 10.1128/jvi.72.4.2760-2764.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesegang TJ. Herpes simplex virus epidemiology and ocular importance. Cornea. 2001;20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- Liu T, Khanna KM, Carriere BN, Hendricks RL. Gamma interferon can prevent herpes simplex virus type 1 reactivation from latency in sensory neurons. J Virol. 2001;75:11178–11184. doi: 10.1128/JVI.75.22.11178-11184.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Khanna KM, Chen X, Fink DJ, Hendricks RL. CD8(+) T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J Exp Med. 2000;191:1459–1466. doi: 10.1084/jem.191.9.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodoen M, Ogasawara K, Hamerman JA, Arase H, Houchins JP, Mocarski ES, Lanier LL. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J Exp Med. 2003;197:1245–1253. doi: 10.1084/jem.20021973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looker KJ, Garnett GP. A systematic review of the epidemiology and interaction of herpes simplex virus types 1 and 2. Sex Transm Infect. 2005;81:103–107. doi: 10.1136/sti.2004.012039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggioncalda J, Mehta A, Su YH, Fraser NW, Block TM. Correlation between herpes simplex virus type 1 rate of reactivation from latent infection and the number of infected neurons in trigeminal ganglia. Virology. 1996;225:72–81. doi: 10.1006/viro.1996.0576. [DOI] [PubMed] [Google Scholar]
- Minami M, Kita M, Yan XQ, Yamamoto T, Iida T, Sekikawa K, Iwakura Y, Imanishi J. Role of IFN-gamma and tumor necrosis factor-alpha in herpes simplex virus type 1 infection. J Interferon Cytokine Res. 2002;22:671–676. doi: 10.1089/10799900260100150. [DOI] [PubMed] [Google Scholar]
- Orr MT, Edelmann KH, Vieira J, Corey L, Raulet DH, Wilson CB. Inhibition of MHC class I is a virulence factor in herpes simplex virus infection of mice. PLoS Pathog. 2005;1:e7. doi: 10.1371/journal.ppat.0010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira RA, Simmons A. Cell surface expression of H2 antigens on primary sensory neurons in response to acute but not latent herpes simplex virus infection in vivo. J Virol. 1999;73:6484–6489. doi: 10.1128/jvi.73.8.6484-6489.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira RA, Tscharke DC, Simmons A. Upregulation of class I major histocompatibility complex gene expression in primary sensory neurons, satellite cells, and Schwann cells of mice in response to acute but not latent herpes simplex virus infection in vivo. J Exp Med. 1994;180:841–850. doi: 10.1084/jem.180.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JF, Bryson Y, Mannix ML, Dillon M, Wohlenberg CR, Banks S, Wallington CJ, Notkins AL, Straus SE. Prevention of ultraviolet-light-induced herpes labialis by sunscreen. Lancet. 1991;338:1419–1422. doi: 10.1016/0140-6736(91)92723-f. [DOI] [PubMed] [Google Scholar]
- Sawtell NM. The probability of in vivo reactivation of herpes simplex virus type 1 increases with the number of latently infected neurons in the ganglia. J Virol. 1998;72:6888–6892. doi: 10.1128/jvi.72.8.6888-6892.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Poon DK, Tansky CS, Thompson RL. The latent herpes simplex virus type 1 genome copy number in individual neurons is virus strain specific and correlates with reactivation. J Virol. 1998;72:5343–5350. doi: 10.1128/jvi.72.7.5343-5350.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Thompson RL. Rapid in vivo reactivation of herpes simplex virus in latently infected murine ganglionic neurons after transient hyperthermia. J Virol. 1992;66:2150–2156. doi: 10.1128/jvi.66.4.2150-2156.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Robinson NJ. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J Infect Dis. 2002;186(Suppl 1):S3–28. doi: 10.1086/343739. [DOI] [PubMed] [Google Scholar]
- Theil D, Derfuss T, Paripovic I, Herberger S, Meinl E, Schueler O, Strupp M, Arbusow V, Brandt T. Latent herpesvirus infection in human trigeminal ganglia causes chronic immune response. Am J Pathol. 2003;163:2179–2184. doi: 10.1016/S0002-9440(10)63575-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomazin R, Hill AB, Jugovic P, York I, van Endert P, Ploegh HL, Andrews DW, Johnson DC. Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. Embo J. 1996;15:3256–3266. [PMC free article] [PubMed] [Google Scholar]
- Tomazin R, van Schoot NE, Goldsmith K, Jugovic P, Sempe P, Fruh K, Johnson DC. Herpes simplex virus type 2 ICP47 inhibits human TAP but not mouse TAP. J Virol. 1998;72:2560–2563. doi: 10.1128/jvi.72.3.2560-2563.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorella D, Gewurz BE, Furman MH, Schust DJ, Ploegh HL. Viral subversion of the immune system. Annu Rev Immunol. 2000;18:861–926. doi: 10.1146/annurev.immunol.18.1.861. [DOI] [PubMed] [Google Scholar]
- van Lint AL, Kleinert L, Clarke SR, Stock A, Heath WR, Carbone FR. Latent infection with herpes simplex virus is associated with ongoing CD8+ T-cell stimulation by parenchymal cells within sensory ganglia. J Virol. 2005;79:14843–14851. doi: 10.1128/JVI.79.23.14843-14851.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verjans GM, Hintzen RQ, van Dun JM, Poot A, Milikan JC, Laman JD, Langerak AW, Kinchington PR, Osterhaus AD. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc Natl Acad Sci U S A. 2007;104:3496–3501. doi: 10.1073/pnas.0610847104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace ME, Keating R, Heath WR, Carbone FR. The cytotoxic T-cell response to herpes simplex virus type 1 infection of C57BL/6 mice is almost entirely directed against a single immunodominant determinant. J Virol. 1999;73:7619–7626. doi: 10.1128/jvi.73.9.7619-7626.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley RJ, Roizman B. Herpes simplex virus infections. Lancet. 2001;357:1513–1518. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- Zhu J, Koelle DM, Cao J, Vazquez J, Huang ML, Hladik F, Wald A, Corey L. Virus-specific CD8+ T cells accumulate near sensory nerve endings in genital skin during subclinical HSV-2 reactivation. J Exp Med. 2007;204:595–603. doi: 10.1084/jem.20061792. [DOI] [PMC free article] [PubMed] [Google Scholar]