

Abstract
Dioxo-MoVI complexes of general formula Tp*MoO2(p-SC6H4Dn) (6a-6c) (where Tp* = hydrotris(3,5-dimethyl-pyrazol-1-yl)borate and Dn= dendritic unit) have been synthesized and characterized by spectroscopy and mass spectrometry. 1H NMR spectra of the metal complexes indicate that the Cs local symmetry about the metal core does not change by the incorporation of dendritic functionality at the thiophenolato ring. Electrochemical data show ∼20 mV change in the redox potential in the complexes with dendritic ligands suggesting a very small perturbation in the redox orbital, which is also supported by small changes in the electronic spectra. The peak-to peak separation (ΔEp) increases from 125 mV in 6(a) to 240 mV in 6(c), suggesting sluggish electron transfer in molecules with larger dendritic ligands.
Introduction
In mononuclear molybdenum enzymes, encapsulation of the Mo-center is universal and probably is a necessary protective feature of the catalytic site from forming unwanted dinuclear centers. In the crystal structure of oxidized plant sulfite oxidase (pSO), the [MoO2]2+ unit is embedded inside the large protein matrix.1 Similar features were observed in the crystal structure of nitrate reductases and DMSO reductases.2,3,4,5 The enzymes mentioned above are thought to function via oxygen atom transfer (OAT) process and are often referred to as oxotransferases.6
A number of discrete dioxo-MoVI complexes react with oxygen atom abstractors like phosphines, and a common occurrence in the process is the formation of dimeric complexes (equation 1, where Tp* = hydrotris(3,5-dimethyl-pyrazol-1-yl)borate).7,8,9,10,11,12,13,14,15 Unless the dinuclear compounds are in rapid equilibrium with the mononuclear compounds, formation of dinuclear compounds terminates the catalytic process. Two general approaches have been used to prevent the dinucleation. The first of the two, the electrostatic control is an approach where the nucleating unit carries a charge which inhibits two such units to come close to form a dinuclear species. This approach uses mononuclear centers that remain charged before and after the atom transfer. An example of this approach is the use of dianionic maleonitrile (C4N2S22-) as a ligand.16 This approach has been used to probe reaction of [MoVIO2]2+ centers with tertiary phosphines as well as other substrates. The second approach involves steric control where sterically bulky ligands have been used or by grafting reacting units onto polymeric support which physically inhibit the formation a dinuclear species. The second approach involves the use of a sterically crowded ligand architecture that prevents the formation of dinuclear species. Both approaches have had mixed success in preventing dinuclear species. For example, the sterically encumbered pyridine thiolate ligand system or hydro(trispyrazolyl)borate ligand systems stabilize the dinuclear compounds under certain conditions. The formation of the dinuclear compounds in these systems underscores the pervasive nature of the dinucleation reaction especially with smaller ligand architectures.
| (1) |
In this paper we offer a different approach where we have used larger dendritic ligand system to prevent the dinucleation. In general, the dendritic ligand architectures provide an opportunity to accomplish encapsulation on the molecular scale.17 Indeed, dendritic ligand architecture has been employed to protect catalytically competent reactive Zn-centers of an enantioselective catalyst from forming thermodynamically favorable Zn-O-Zn units.18,19,20 We have built the steric bulk asymmetrically keeping three coordination sites of a dioxo-Mo(VI) core invariant, through coordination by the hydrotris(3,5-dimethyl-pyrazol-1-yl)borate ligand (Tp*), and the open site is coordinated by a dendritic thiolato ligand (Figure 1). Dendritic thiolato ligand systems have been used in the past to understand the redox chemistry of metal sulfur cores such as [Fe4S4]21 and [MoOS4]22. Here we disclose the syntheses and characterization of dioxo-Mo(VI) complexes containing dendritic thiolato ligands.
Figure 1.
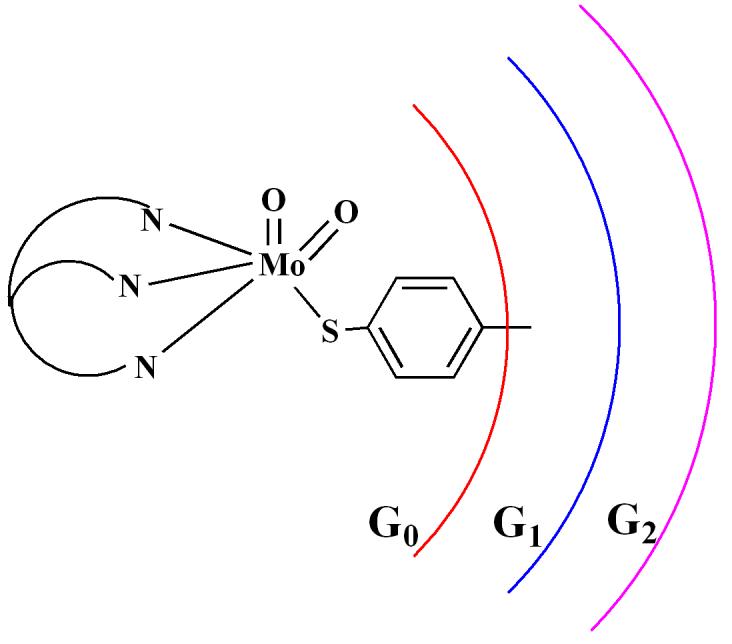
Schematic representation of the dioxo-Mo(VI) complexes used in this study.
Experimental
Unless specified, all the reactions were conducted in oven-dried glassware under dry argon atmosphere using standard Schlenk techniques. Tp*MoO2Cl and Tp*MoO2(SPh) were prepared using published procedures.23 The G1-ester dendron (3-[2-amino-3-(2-ethoxycarbonyl-ethoxy)-2-(2-ethoxycarbonyl-ethoxymethyl)-propoxy]-propionic acid ethyl ester), compounds 1 and 2 were prepared by following the procedures described earlier.24 All work up and chromatographic purifications of complexes were conducted in air using distilled solvents. The solvents were purified as follows: methylene chloride (CH2Cl2) from CaH2; tetrahydrofuran (THF) from Nabenzophenone; acetonitrile from CaH2 followed by Li2CO3-KMnO4 and finally from P2O5; methanol (MeOH) and ethanol was purified from Mg. Triethylamine and pyridine were dried by distilling over KOH pallets. For adsorption chromatography, silica gel (60 Å, 63-200 μm) from Sorbent Technologies was used.
Room temperature 1H and 13C NMR spectra were recorded using a Bruker ACP-300 spectrometer at 300.133 MHz and 75.469 MHz frequencies, respectively. Deuterated solvents for NMR were obtained from Cambridge Isotope Laboratories and were used without purification. Infrared spectra were recorded using a Perkin-Elmer FT-IR 1760X spectrometer on NaCl plates or in KBr pellets. Electrospray ionization Mass spectra (ESI MS) were collected on a Micromass ZMD mass spectrometer using both negative and positive ionization modes. Acetonitrile solutions of samples were injected via a syringe pump with a flow rate of 0.1 – 0.2 ml/min. Electronic spectra of complexes were recorded in a Cary 3 spectrophotometer equipped with temperature controlled cell holders. Cyclic voltammograms (CVs) of complexes were recorded in a Bioanalytical systems (BAS) model CV-50W using a standard three electrode system consisting of Pt-disk working electrode, Pt-wire auxiliary and reference electrodes. Measurements were performed in dry and degassed acetonitrile using 0.1M Bu4NClO4 as the supporting electrolyte and using Fc+/Fc as an internal standard. All potentials are reported with respect to the Fc+/Fc couple and without correction for the junction contribution. The OAT reactivity of dioxo-Mo(VI) complexes with tertiary phosphines were probed by ESI-MS. In the ESI-MS method, solutions of both metal complex and phosphine were prepared in MeCN separately and mixed at room temperature prior to the injection into the mass spectrometer using a syringe pump at a flow rate of 200 μl/min.
Synthesis of 3. To a solution of 1 (6.4 g, 5.75 mmol) in ethanol (95%, 120 mL) an aqueous solution of NaOH (1.84 g, 46 mmol in 40 mL water) was added and the mixture was stirred for 2 days. The solvent was removed to dryness under reduced pressure. The resulted solid was redissolved in water and acidified to pH∼3 with 0.5N HCl. The resulting white precipitate was extracted with ethyl acetate, dried over anhydrous MgSO4, and evaporated to dryness to yield a white gummy solid. Yield = 82% (4.44 g). 1H NMR (dmso-d6): δ = 2.5 (t, J=6Hz, 12H), 3.7 (t, J=6Hz, 12H ), 3.79 (s, 12H), 7.61 (d, J=7Hz, 4H), 7.82 (d, J=6Hz, 4H). 13C NMR (dmso-d6): δ = 34.52, 60.22, 66.61, 67.94, 126.08, 129.63, 130.24, 140.69, 166.52, 172.50. IR (NaCl): 3185, 2818, 2649, 1719, 1593, 1532, 1487, 1188, 843, 758. ESI−-MS (CH3OH): m/z: 943 [M-H]−. (M = C40H51O20N2S2, 943).
Synthesis of 4. To a solution of 3 (2.87 g, 3.04 mmol) in THF (40 mL) and DMF (0.2 mL) oxalyl chloride (11.6 mL, 91.2 mmol) at 0°C was slowly added and the resulting light yellow solution was gradually warmed to room temperature and stirred for 6 h. Evaporation of the solvent under reduced pressure yielded a yellow solid, which was redissolved in CH2Cl2. To this solution a mixture of G1-ester dendron (9.2 g, 21.9 mmol) and triethylamine (4.6 mL, 32.85 mmol) in CH2Cl2 (25 mL) was added at 0°C and stirred for overnight at room temperature. The reaction was quenched by water and the organic layer was separated, dried over anhydrous MgSO4 and evaporated to yield a light brown gummy liquid. The target compound was collected in pure form as the first yellow band from a silica gel column using a mixture of 30% MeCN in toluene as the eluent. Yield = 72% (7.24 g). 1H NMR (CDCl3): δ = 1.20-1.34 (m, 54H), 2.40-2.61 (m, 48H), 3.60-3.85 (m, 96H), 4.00-4.20 (m, 36H), 6.20 (s, 4H), 6.57 (s, 2H, NH), 7.40 (d, 2H, J = 8.0 Hz), 7.72 (d, 4H, J = 8.0 Hz). 13C NMR (CDCl3): δ = 13.08, 33.81, 50.28, 59.12, 65.23, 65.80, 67.55, 68.0, 124.98, 127.08, 129.21, 136.24, 166.48, 169.99. IR (NaCl): 3385; 2978, 2938, 1729, 1721, 1712, 1651. ESI+-MS (CH3CN): m/z: 1695 [M+Na+H]2+ 1137 [M+K+2H]3+ (M = C154H250O68N8S2, 3366).
Synthesis of 5. At 0°C to a solution of 4(1.17 g, 0.48 mmol) in EtOH and THF (10mL/ 10mL) NaBH4 (75 mg, 1.92 mmol) was added and the reaction mixture was stirred for additional 4h. The solvent was removed under reduced pressure and water (10 mL) was added to the resulting light yellow solid. The pH of the aqueous solution was adjusted to ∼ 4.0 with 0.5N HCl. The resulting off white precipitate was extracted with ethyl acetate (2 × 25 mL). The organic layer was dried over anhydrous MgSO4 and evaporated under reduced pressure to afford a yellow gummy compound with 50% (0.6 g) yield. 1H NMR (CDCl3): δ = 1.06-1.15 (m, 27H), 2.35-2.55 (m, 24H), 3.50-3.87 (m, 48H), 4.00-4.18 (m, 18H), 5.24 (s, 1H), 6.53 (s, 2H), 6.75 (s, 1H), 7.25 (m, 2H), 7.70 (m, 2H). 13C NMR (CDCl3): δ = 14.01, 34.82, 60.30, 66.64, 69.03, 69.54, 127.65, 128.17, 167.44, 171.37. IR (NaCl): 3389, 2978, 2931, 2872, 2554, 1731, 1654. ESI−-MS (CH3CN): m/z: 1682 [M-H]− (M = C77H126O34N4S1, 1683).
Synthesis of 6(a). 4-Mercapto-N-methyl-benzamide24 (210 mg, 1.26 mmol) was dissolved in CH2Cl2 (12 mL) and Et3N (2 mL, 2.52 mmol) was added to this solution. The solution was transferred to a suspension of Tp*MoO2Cl (690 mg, 1.5 mmol) in CH2Cl2 (10mL). The reaction mixture was stirred for 8 h and monitored using TLC. The solvent was evaporated under reduced pressure and the remaining solid was chromatographed on silica gel using 30% MeCN in toluene. Yield = 35% (260 mg). 1H NMR (300 MHz, CDCl3): δ = 2.36 (s, 3H); 2.40 (s, 6H); 2.62 (s, 3H); 2.67 (s, 6H); 3.00 (d, 3H, J = 4.5 Hz); 5.84 (s, 1H); 5.87 (s, 2H); 7.63 (d, 2H, J = 8 Hz); 7.71 (d, 2H, J = 7.0 Hz). 13C NMR (75.5 MHz, CDCl3): δ = 12.49, 12.77, 14.59, 15.19, 26.75, 107.45, 126.71, 129.23, 130.96, 132.55, 144.65, 147.08, 148.61, 153.26, 153.75, 154.30, 167.96. IR (KBr, cm−1): 3413 (νNH); 2929 (νCH); 2548 (νBH); 1654 (w,νCO), 1543; 1448; 1417; 1365; 1208; 1072; 1045; 931, 900 (νMoO2); 860; 810; 758; 680. ESI+-MS (CH3CN): m/z: 616 [M+Na]+. (M = C23H30O3N7SBMo, 593).
Synthesis of 6(b). It was synthesized by following the procedure similar to that described for 6(a) using 2and Tp*MoO2Cl. Yield = 45%. 1H NMR (300 MHz, CDCl3): δ = 1.23 (t, 9H); 2.35 (s, 3H); 2.39 (s, 6H); 2.55 (t, 6H); 2.60 (s, 3H); 2.66 (s, 6H); 3.73 (t, 6H); 3.84 (s, 6H); 4.15 (q, 6H); 5.86 (s, 3H); 6.55 (s, 1H); 7.51 (d, 2H, J = 7.4 Hz); 7.80 (d, 2H, J = 7.4 Hz). 13C NMR (75.5 MHz, CDCl3): δ = 12.35, 12.77, 14.18, 15.20, 35.05, 59.60, 60.42, 66.81, 69.27, 106.86, 107.42, 127.81, 129.11, 132.36, 144.60, 145.72, 152.18, 153.74, 154.29, 166.57, 171.43. IR (KBr, cm−1): 3423 (νNH); 2980, 2930, 2876 (νCH); 2549 (νBH); 1731 (νCO), 1665 (νCO), 1594; 1543; 1528; 1484; 1447; 1370; 1265; 1187; 1113; 1078; 1034; 928, 898 (νMoO2); 860; 810; 760. ESI+-MS (CH3CN): m/z: 984 [M+H]+, 1006 [M+Na]+ (M = C41H60O12N7SBMo, 983).
Synthesis of 6(c). It was synthesized by following the procedure similar to that described for 6(a) using 5 and Tp*MoO2Cl. Yield = 15%. 1H NMR (300 MHz, CDCl3): 1.20-1.29 (m, 27H); 2.20-2.80 (m, 42H); 3.50-3.90 (m, 48H); 4.00-4.20 (m, 18H); 5.82 (s, 2H); 5.85 (s, 1H); 6.54 (m, 4H); 7.32 (d, 2H, J = 7.4 Hz); 7.69 (d, 2H, J = 7.4 Hz). 13C NMR (75.5 MHz, CDCl3): δ = 12.19, 12.68, 14.11, 29.57, 30.21, 34.95, 60.32, 66.71, 69.16, 106.66, 107.21, 127.63, 127.98, 144.06, 151.87, 166.60, 171.42. IR (KBr, cm−1): 3378 (νNH); 2916 (m, νCH); 2547 (νBH); 1735 (br,νCO); 1594; 926, 897 (νMoO2); 860; 759; 694. ESI+-MS (CH3CN): m/z: 1078 [M+2Na]+2 (M = C92H147O36N10SBMo, 2109).
Result and Discussion
Synthesis
Dendritic disulfide ligands were synthesized by following the divergent approach,25 where dendrization of thiophenols was achieved by linking the amine and the acid terminals at its para-position. Synthesis of zero and first generation of disulfide molecules have been published elsewhere.24 The second generation of disulfide ligand 4, was synthesized by hydrolyzing the ester functionality of the first generation disulfide 1 with ethanolic solution of sodium hydroxide. The completion of the hydrolysis reaction and formation of 3 was confirmed by ensuring the complete absence of the ester signals in 1H NMR spectra. The terminal acid groups were then coupled with the amine terminals of G1-ester dendron. The disulfide linkage was cleaved by NaBH4 in 1:1 mixture of EtOH and THF. Compounds 2 and 5, obtained as yellow liquids, are moderately sensitive to air, and decompose in DMSO presumably due to oxidation.
The molybdenum precursor complex, Tp*MoO2Cl, and the first member of dendritic dioxo-complexes, Tp*MoO2(SPh), were synthesized by following the published procedure.23 Dendritic dioxo molybdenum complexes were synthesized from the nucleophilic displacement of the chloride group in Tp*MoO2Cl with dendritic thiolato ligands in the presence of Et3N in CH2Cl2 (Scheme 2). The progress of reactions was monitored by TLC. Due to the bulky nature of the dendritic ligands the reaction required longer time (∼ 6h for 6(b) and 10h for 6(c)) for completion than the smaller analogs (e.g., <1h Tp*MoO2(SPh)). Complexes 6(a-c) were purified by adsorption chromatography on silica gel using a mixture of 30% MeCN in toluene. Complex 6(c) was further purified by size exclusion chromatography on Biobead SX3 using toluene. All dioxo-MoVI complexes were obtained as brown materials, where 6(a) was a solid while both 6(b) and 6(c) were gummy materials. Solutions of all complexes were stable in air for several hours.
Scheme 2.

Characterization
Dendritic thiol and disulfide ligands were characterized by infrared, solution 1H and 13C NMR spectroscopy, and mass spectrometry. Unlike compound 1, no ethyl (ester) groups could be observed in the 1H NMR spectrum of the disulfide, 3, which was further analyzed by ESI-MS confirming a clean hydrolysis reaction. The disulfide of G2-Ester (compound 4) exhibits broad 1H NMR signals, and the integration of the aromatic vs aliphatic protons is consistent with the molecular formulation. ESI-MS exhibited a multiply charged molecular ion for 4. The 1H NMR spectrum of the thiol G2-Ester (compound 5) is similar to 4 and the infrared spectra exhibits a characteristic signal due to the S-H group around 2554 cm−1. In the 1H NMR spectra, aromatic signals move towards higher field in disulfides compared to respective thiols, presumably due to the higher electron withdrawing nature of two sulfur atoms in the disulfide compounds.
All dioxo-MoVI complexes were characterized by 1H and 13C NMR, Infra-red, and electronic spectroscopies and mass spectrometry. The 1H and 13C NMR spectra of complexes supported the local Cs symmetry of the dioxo-molybdenum complexes similar to the smaller member of the series, Tp*MoO2(SPh). The methyl protons on the pyrazole rings follow the 6:3:6:3 ratio and methyne protons follow the 2:1 ratio indicating that the dendrimer bulk did not affect the Cs local symmetry at the metal centers. The para-substituted thiophenols exhibit two sets of doublets for the thiophenolato ring similar to the aromatic signals of corresponding disulfide compounds. The 1H NMR spectrum of 6(c) contained broad signals due to the presence of dendritic bulk and no spin-spin splitting was resolved. However, the integration agreed well with the expected number of protons. Infrared spectra of the dioxo complexes exhibited two strong bands for a cis-MoO2 unit ca. 930 cm−1 and 900 cm−1 assigned as the symmetric and asymmetric vibrational modes, respectively. Other characteristic bands of the ligand architecture e.g., a sharp B-H stretch at ∼ 2545 cm−1, a broad stretch for N-H function in the range 3378-3420 cm−1, as well as the C=O, and C-H stretches were also observed. For complex 6(a) only one aromatic C=O amide stretch was observed at 1654 cm−1, but in 6(b) aromatic amide C=O and ester C=O stretches were observed at 1665 and 1731 cm−1, respectively. In the case of 6(c) a broad C=O signal indicating several unresolved C=O stretches was observed, which is consistent with the presence of several amide and ester groups.
The UV/visible spectra of dioxo complexes were recorded in acetonitrile at room temperature. A broad band near 415 nm (ε > 1000 M−1cm−1) due to the S → Mo charge transfer band was observed in all complexes. There was no significant change in the band positions from 6(a) to 6(c) indicating similar electronic environment around the metal center in these molecules. It is also consistent with the similar 13C NMR chemical shifts of para-carbon of the para-substituted thiophenol ligands. Earlier we have demonstrated that 13C NMR spectra can serve as a sensitive reporter of the electronic environment about the thiophenol ring.24 A strong transition around 275 nm can be assigned to intraligand charge transfer transition, which increases in intensity going from 6(a) to 6(c), but is absent in Tp*MoO2(SPh).
Positive mode electrospray ionization mass spectra (ESIMS) of complexes 6(a-c) were recorded in acetonitrile solution containing 0.001% methanolic solution of trifluroacetic acid solution. Generally, peak clusters assignable to [M+H]+ and/or [M+Na]+ were observed. Thus, molecular ions for 6(a) and 6(b) were observed and their isotopic distributions agreed well with the theoretical values. For 6(c), the molecular ion peak was not observed, however, a peak cluster for doubly charged ion (associated with Na+ ions) could be detected. The isotope distribution pattern of the observed peaks is consistent with the theoretically predicted isotope distribution pattern for the molecular peaks.
Redox Reactivity
Room temperature redox chemistry of the dendritic dioxo-Mo(VI) complexes was investigated using cyclic voltammetry in MeCN. Figure 2 shows cyclic voltammograms recorded in MeCN, and all potentials are listed in Table 1. All the complexes showed a well-behaved redox wave due the Mo(VI/V) couple similar to those reported earlier.26 No other reductive response was observed within the solvent window. In all cases the anodic and cathodic currents follow a liner relationship with the square root of the scan rate indicating a diffusion control process. Redox potentials of dendritic complexes are less negative than Tp*MoO2(SPh) showing the electron withdrawing nature of the substituent on the thiophenolato ligands (i.e., amide functionality) that makes the metal center electron deficient, and therefore lowers the redox potential. A similar behavior was observed in the case of other oxomolybdenum complexes, where the redox potential changes with the substituent constant (σp).22 The dendritic-substituents on the thiophenolato ligand impart no significant change observed in the redox potential (E1/2) of the complexes suggesting the electronic perturbation from the ligand environment was nearly the same. However, the peak-to-peak separation (ΔE) progressively increased with the increase in the bulk around the Mo center. The ΔE values changed from 125 mV for 6(a) to 215 mV for 6(b) to 240 mV for 6(c) suggesting the rate of electron transfer becomes sluggish with increasing steric bulk. Such a behavior is well documented for different encapsulated redox centers, where the irreversibility of electron transfer increases with the increase in the generation of dendrimer around the redox active site.22,27,28,29
Figure 2.
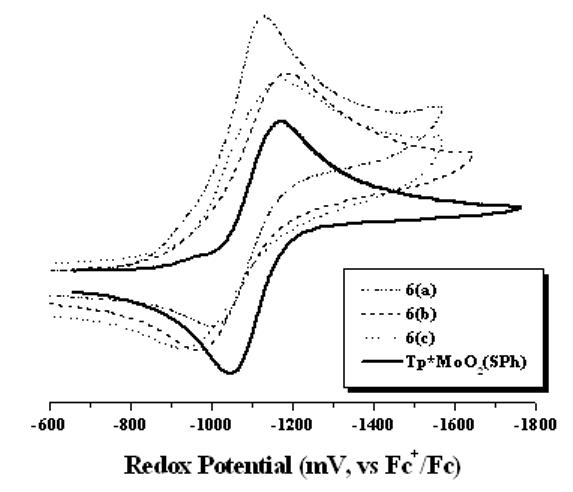
Room temperature cyclic voltammograms of dioxo-Mo(VI) complexes.
Table 1.
Electrochemical data recorded in MeCN (potentials are referenced with respect to the Fc+/Fc redox couple).
| Complex | E1/2 (ΔE), mV |
|---|---|
| Tp*MoO2(SPh) | −1182 (102) |
| 6(a) | −1066 (125) |
| 6(b) | −1077 (215) |
| 6(c) | −1052 (240) |
Oxygen Atom Transfer
The oxygen atom transfer (OAT) reactions can be viewed as a two-electron redox process, and in the case where the metal center serves as an oxo donor is reduced by two electrons. It has now been established that the overall OAT reactions from dioxo-MoVI complexes with phosphine proceed through at least in two steps.30 The metal center is reduced in the first step to form a phosphine oxide bound intermediate complex. In the presence of a coordinating solvent, phosphine oxide is replaced by a solvent molecule in the second step. In recent years we have isolated and structurally characterized several phosphoryl intermediate complexes, and established the solid state molecular structures.31,32,33,34 In parallel, we have established that in situ mass spectrometry provides a useful tool in detecting the intermediate molecules. To this end, both fast atom bombardment (FAB)33 and electrospray ionization (ESI)35 mass spectrometry have been successfully employed. In the present case, we have probed the OAT reactivity of the dioxo-molecules with tertiary phosphines using ESI-MS.
ESI MS is a powerful technique for detecting the molecular ion peaks without significant fragmentations.36 It has been successfully used for detecting the unstable species generated during the OAT reaction from a discrete molybdenum complex with phosphines. In the present case, complexes 6(a) to 6(c) were reacted with triphenyl phosphine in acetonitrile solution and were analyzed by mass spectrometry. Room temperature reactions of the dioxo-complexes with triphenyl phosphine were probed in acetonitrile using mass spectrometry. The positive mode ESI MS showed a peak cluster whose mass-to-charge and isotope distribution pattern agree well with a PPh3 bound molybdenum species. Figure 3 shows the changes in the ESI MS of complex 6(b) after the addition of triphenyl phosphine solution. A peak cluster at 1246 (m/z) formed with 100% intensity indicating the formation of a OPPh3 coordinated complex (Figure 3). Figure3 also shows two less intense peaks ∼ 1232 (m/z) and ∼1274 (m/z). Identification of these peaks is difficult ascertain due to their low intensity, but presumably they are due to the loss of a CH2 group (the peak ∼1232 m/z), a defect in the dendrimer and its acetonitrile associated species (the peak ∼1274 m/z). In addition to the major peak ∼1246 (m/z), a weak peak cluster observed at ∼967 (m/z) agrees well with the formation of the monooxo-product suggesting that OPPh3 has been dissociated. Similar results were also obtained from the reaction between less crowded 6(a) and PPh3 in acetonitrile. Interestingly, when complex 6(c) was reacted with PPh3 under similar conditions, no peak cluster were detected that could be assigned as the OPPh3 bound species. However, under similar conditions, compound 6(c) reacts with PEt3, and a peak cluster around m/z 731 (100%) could be observed. Based on the isotope distribution pattern and mass-to charge ratio this peak cluster could be assigned as a [M-O]3+ ion associated with Na+ and K+. Another peak cluster at 1037 can be assigned as [M-O]2+ ion associated with Na+ and K+ ions. The mass spectral data indicate the formation of a monooxo molybdenum complex in the reaction.
Figure 3.
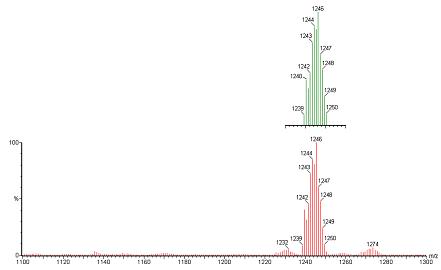
Positive mode ESIMS of a reaction mixture between 6(b) and PPh3 in MeCN. [complex] ∼ 1 mM and [PPh3] ∼0.1 M the peak cluster around m/z of 1246 is consistent with the formulation C59H75O12N7SBPMo(M) [M+H+PPh3]3. The top green pattern is the calculated isotope distribution pattern for the molecular formula 6(b)+PPh3; and bottom red is the observed spectrum which also shows small impurities
Our previous studies have shown that there is little difference in electronic nature of the ligands p-HSC6H4CONHCH3 and 2.24 We anticipate that the electronic nature of the 2nd generation ligand is similar to that of the 1st generation, and also the electronic structures of the metal centers in these molecules do not change appreciably and the electrophilicity of the terminal oxo-groups remain invariant. Therefore, the difference in the reactivity of between PPh3 and PEt3 with 6(c) may be attributed to two major factors – nucleophilic control arising from a difference in the basicity of the phosphines and steric control arising the size of the phosphines as well the ligands architecture surrounding the molybdenum center. Furthermore, our in depth kinetic investigation on TpiprMoO2(OPh) demonstrates that introduction of phenyl group into the tertiary phosphines enhances the overall rate of the reaction.30 We surmise that the differential reactivity of 6(c) with PPh3 and PEt3 is due to the steric restriction at the metal center imposed by the ligand architecture, such that the dendritic ligand architecture imposes size selectivity at the metal center. Interestingly, shape selectivity imposed by dendritic ligands has been demonstrated in manganese porphyrin chemistry.19
Summary
The embedded nature of the molybdenum center appears to be a common feature of mononuclear molybdenum enzymes. The effect of the encapsulation on the metal center has been investigated by using synthetic molecules where encapsulated [MoO2]2+ centers have been stabilized by substituted thiophenolato ligands. In this case we have used one anchoring point for the dendrimer growth, and therefore caution must be exercised in viewing these molecules as completely encapsulated metal centers. All complexes have been thoroughly characterized by 1H NMR, 13C NMR, infrared, and electronic spectroscopies. The electronic spectra show no significant change in the position and intensity for the band designated as S → Mo change donation. Electrochemical analysis of complexes shows one electron process for Mo(VI)/Mo(V) redox process, and expectedly irreversibility of the voltammograms increase with increasing steric bulk. All complexes react with tertiary phosphines generating intermediate complexes, as evidenced by ESI mass spectrometry with no evidence for the formation of μ-oxo-dimeric species.
Scheme 1.

Acknowledgments
We thank Ms Archana Nigam for experimental assistance, and the National Institutes of Health for partial financial support of this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schrader N, Fischer K, Theis K, Mendel R, Schwarz G, Kisker C. Structure. 2003;11:1251. doi: 10.1016/j.str.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Moura JJG, Brondino CD, Trincao J, Romao MJ. J. Biol. Inorg. Chem. 2004;9:791. doi: 10.1007/s00775-004-0573-9. [DOI] [PubMed] [Google Scholar]
- 3.Arnoux P, Sabaty M, Alric J, Frangioni B, Guigliarelli J-M, Adriano D, Pignol D. Nature Struc. Biol. 2003;10:928. doi: 10.1038/nsb994. B. [DOI] [PubMed] [Google Scholar]
- 4.Jormakka M, Richardson D, Byrne B, Iwata S. Structure. 2004;12:95. doi: 10.1016/j.str.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 5.Bertero MG, Rothery RA, Palak M, Hou C, Lim D, Blasco F, Weiner JH, Strynadka NCJ. Nature Struc. Biol. 2003;10:681. doi: 10.1038/nsb969. [DOI] [PubMed] [Google Scholar]
- 6.Enemark JH, Cooney JJA, Wang J-J, Holm RH. Chem. Rev. 2004;104:1175. doi: 10.1021/cr020609d. [DOI] [PubMed] [Google Scholar]
- 7.Bhattacharjee S, Bhattacharjee R. J. Chem. Soc., Dalton Trans. 1992:1357. [Google Scholar]
- 8.Hinshaw CJ, Peng G, Singh R, Spence JT, Enemark JH, Bruck MA, Kristofzski J, Merbs SL, Ortega RB, Wexler PA. Inorg. Chem. 1989;28:4483. [Google Scholar]
- 9.Roberts SA, Young CG, Cleland WE, Jr., Ortega RB, Enemark JH. Inorg. Chem. 1988;27:3044. [Google Scholar]
- 10.Yoshinaga N, Ueyama N, Okamura T, Nakamura A. Chem. Lett. 1990:1655. [Google Scholar]
- 11.Das SK, Choudhury PK, Biswas D, Sarkar S. J. Am. Chem. Soc. 1994;116:9061. [Google Scholar]
- 12.Purohit S, Koley AP, Prasad LS, Monoharan PT, Ghosh S. Inorg. Chem. 1989;28:3735. [Google Scholar]
- 13.Berg JM, Holm RH. J. Am. Chem. Soc. 1985;107:917. [Google Scholar]
- 14.Schultz BE, Gheller SF, Muetterties MC, Scott MJ, Holm RH. J. Am. Chem. Soc. 1993;115:2714. [Google Scholar]
- 15.Cervilla A, Corma A, Fornes V, Llopis E, Perez F, Rey F, Ribera A. J. Am. Chem. Soc. 1995;117:6781. [Google Scholar]
- 16.Lorber C, Plutino MR, Elding LI, Nordlander E. Ebbe. J. Chem. Soc., Dalton Trans. 1997:3997. [Google Scholar]
- 17.Hecht S, Frechet JMJ. Angew. Chem. Int. Ed. 2001;40:74. doi: 10.1002/1521-3773(20010105)40:1<74::aid-anie74>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 18.Hu Q-S, Pugh V, Sabat M, Pu L. J. Org. Chem. 1999;64:7528. [Google Scholar]
- 19.Bhyrappa P, Young JK, Moore JS, Suslick KS. J. Am. Chem. Soc. 1996;118:5708. [Google Scholar]
- 20.Suslick KS. In: In The Porphyrin Hand Book. Kadish K, Smith K, Guillard R, editors. Academic Press; NY: 1999. [Google Scholar]
- 21.Representative examples include: Chasse TL, Sachdeva R, Li Q, Li Z, Randall J, Gorman CB. J. Am. Chem. Soc. 2003;125:8250. doi: 10.1021/ja035515f. Chasse TL, Yohannan JC, Kim N, Li Q, Li Z, Gorman CB. Tetrahedron. 2003;59:3853. Gorman CB, Smith JC. J. Am. Chem. Soc. 2000;122:9342. Gorman CB, Smith JC, Hager MW, Parkhurst BL, Sierzputowska-Gracz H, Haney C. J. Am. Chem. Soc. 1999;121:9958.
- 22.Basu P, Nemykin VN, Sengar RS. Inorg. Chem. 2003;42:7489. doi: 10.1021/ic034821r. Mondal S, Basu P. Inorg. Chem. 2001;40:192. doi: 10.1021/ic0011866. McNaughton RL, Mondal S, Nemykin VN, Basu P, Kirk ML. Inorg. Chem. 2005;44:8216. doi: 10.1021/ic0482281.
- 23.Roberts SA, Young CG, Kipke CA, Cleland WE, Jr., Yamanouchi K, Carducci MD, Enemark JH. Inorg. Chem. 1990;29:3650. [Google Scholar]
- 24.Sengar RS, Nemykin VN, Basu P. New J. Chem. 2003;27:1115. [Google Scholar]
- 25.Newkome GR, Lin X, Young JK. Synlett. 1992:53. [Google Scholar]
- 26.Xiao Z, Gable RW, Wedd AG, Young CG. J. Am. Chem. Soc. 1996;118:2912. [Google Scholar]
- 27.Weyermann P, Diederich F. Helv. Chim. Acta. 2002;85:599. [Google Scholar]
- 28.Stone DL, Smith DK, McGrail PT, Terry P. J. Am. Chem. Soc. 2002;124:856. doi: 10.1021/ja0117478. [DOI] [PubMed] [Google Scholar]
- 29.Cardona CM, Mendoza S, Kaifer AE. Chem. Soc. Rev. 2000;29:37. [Google Scholar]; Chow H-F, Chan Y-KI, Fung P-S, Mong TK-K, Nongrum MF. Tetrahedron. 2001;57:1565. [Google Scholar]
- 30.Kail BW, Pérez LM, Zarić SD, Millar AJ, Young CG, Hall MB, Basu P. Chem Eur. J. 2006;12:7501–7509. doi: 10.1002/chem.200600269. [DOI] [PubMed] [Google Scholar]
- 31.Nemykin VN, Laskin J, Basu P. J. Am. Chem. Soc. 2004;126:8604. doi: 10.1021/ja049121f. [DOI] [PubMed] [Google Scholar]
- 32.Millar AJ, Doonan CJ, Smith PD, Nemykin VN, Basu P, Young CG. Chem. Eur. J. 2005;11:3255. doi: 10.1002/chem.200401101. [DOI] [PubMed] [Google Scholar]
- 33.Smith PD, Millar AJ, Young CG, Ghosh A, Basu P. J. Am. Chem. Soc. 2000;122:9298. [Google Scholar]
- 34.Nemykin VN, Basu P. Inorg. Chem. 2005;44:7494. doi: 10.1021/ic0508324. [DOI] [PubMed] [Google Scholar]
- 35.Nemykin VN, Basu P. Dalton Trans. 2004:1928. doi: 10.1039/b403964e. [DOI] [PubMed] [Google Scholar]
- 36.Cole RB, editor. Electrospray ionization mass spectrometry: fundamentals, instrumentation, and applications. Willey; New York: 1997. p. 570. [Google Scholar]