

Abstract
Cholesterol has been proposed to play a critical role in regulating neurotransmitter release and synaptic plasticity. The neuronal porosome/fusion pore, the secretory machinery at the nerve terminal, is a 12−17 nm cup-shaped lipoprotein structure composed of cholesterol and a number of proteins, among them calcium channels, and the t-SNARE proteins syntaxin-1 and SNAP-25. During neurotransmission, synaptic vesicles dock and fuse at the porosome via interaction of their v-SNARE protein with t-SNAREs at the porosome base. Membrane-associated neuronal t-SNAREs interact in a circular array with liposome-associated neuronal v-SNARE, to form the t-/v-SNARE ring complex. The SNARE complex along with calcium is required for the establishment of continuity between opposing bilayers. Here we show that although cholesterol is an integral component of the neuronal porosome and is required for maintaining its physical integrity and function, it has no influence on the conformation of the SNARE ring complex.
Keywords: cell secretion, porosome/fusion pore, cholesterol, neurotransmission
1. Introduction
The organization of biological membranes is critical to normal cellular function. The role of cholesterol in the organization of biological membranes and in disease is well documented (Maxfield and Tabas, 2005). The fine tuned composition of lipid and cholesterol at the cellular membrane, have been suggested to play crucial role in compartmentalizing various membrane proteins for specific cellular activities (Mukherjee and Maxfield, 2004). Cholesterol has been proposed to play a critical role in regulating neurotransmitter release (Zamir and Charlton, 2006) and synaptic plasticity (Koudinov and Koudinova, 2001). Therefore the participation of cholesterol in porosome structure/function and in SNARE-induced membrane fusion at the nerve terminal is hypothesized. Porosomes are supramolecular lipoprotein structures at the cell plasma membrane, where secretory vesicles dock and fuse to release intravesicular contents to the outside of the cell (Jena, 2005). The neuronal porosome, the secretory machinery at the nerve terminal, is a 12−17 nm cup-shaped lipoprotein structure composed of cholesterol (Jeremic et al. 2006), and a number of proteins, among them calcium channels, and the t-SNARE proteins syntaxin-1 and SNAP-25 (Cho and Jeremic et al., 2004). During neurotransmission, synaptic vesicles dock and fuse at the base of porosomes via interaction of their v-SNARE protein wlth t-SNAREs at the Porosome base. It has been demonstrated that membrane-associated neuronal t-SNAREs interact in a circular array with liposome-associated neuronal v-SNARE, to form the t-/v-SNARE ring complex (Cho and Kelly et al., 2002; Cho et al., 2005). The SNARE complex (Weber et al., 1998) along with calcium (Cho and Kelly et al., 2002; Jeremic et al., 2004a; Jeremic et al., 2004b), is required for the establishment of continuity between opposing bilayers. Furthermore, porosome-associated t-SNAREs (Jena et al., 2003) and calcium channels (Cho and Jeremic et al., 2005) are known to interact. Similarly, the important role of cholesterol on the distribution of t-SNAREs (Lang et al., 2001) and calcium channels (Jeremic et al., 2006; Taverna et al., 2004) at the nerve terminal, has previously been reported. However the precise role of membrane cholesterol on neurotransmission remains unclear.
The role of cholesterol on the integrity of the neuronal porosome, and its influence on the assembly of neuronal t-/v-SNARE complex, was therefore the focus of this study. Our results demonstrate a significant inhibition in interactions between porosome-associated t-SNAREs and calcium channels following depletion of membrane cholesterol. Since calcium is critical to SNARE-induced membrane fusion (Cho and Kelly et al., 2002; Jeremic et al., 2004a; Jeremic et al., 2004b), the loss of interaction between SNAP-25, Syntaxin-1, and calcium channel at the neuronal porosome complex, would seriously compromise or even abrogate neurotransmission at the nerve terminal.
2. Materials and methods
2.1 Synaptosome and synaptosome membrane preparation
Synaptosomes and synaptosomal membrane were prepared from rat brains according to the method of Cho and Jeremic et al. (2004). Whole rat brain from Sprague-Dawley rats weighing 100−150 g, were isolated and placed in ice-cold buffered sucrose solution (5 mM Hepes, pH 7.4, 0.32 M sucrose), supplemented with protease inhibitor cocktail (Sigma, St. Louis, MO). The brain tissue was homogenized using 8−10 strokes in a Teflon-glass homogenizer. The total homogenate was centrifuged for 3 min at 2,500 x g, and the supernatant fraction was further centrifuged for 15 min at 14,500 x g, to obtain a pellet. The resultant pellet was resuspended in buffered sucrose solution, and loaded onto a 3−10−23% Percoll gradient. After centrifugation at 28,000 x g for 6 min, the enriched synaptosomal fraction was collected at the 10−23% Percoll gradient interface. To isolate synaptosomal membrane, isolated synaptosomes were diluted with 9 vol of ice-cold water (hypotonic lyses of synaptosomes to release synaptic vesicles) and immediately homogenized using 3 strokes in a Teflon-glass homogenizer, followed by 30 min incubation on ice. The homogenate was then centrifuged for 20 min at 25,500 x g, and the resultant pellet enriched in synaptosomal membrane was collected.
2.2 Immunoisolation and immunoanalysis of neuronal porosome
To isolate the neuronal porosome complex, SNAP-25 specific antibody conjugated to protein A-sepharose (Santa Cruz, CA) was used. Solubilized synaptosomal membrane preparations in ice-cold Triton-Lubrol buffer (0.5% Lubrol; 1 mM benzamidine; 5 mM Mg-ATP; 5 mM EDTA; 0.5% Triton X-100, in phosphate buffered saline or PBS), in the absence or presence of saponin (0.5% w/v) and supplemented with protease inhibitors (Sigma, St. Louis, MO), was used. SNAP-25 antibody conjugated to protein A-sepharose, was incubated with the solubilized homogenate overnight in the cold room, followed by washing (3X) with 40 vol. of wash buffer (500 mM NaCl, 10 mM Tris, 2 mM EDTA, pH 7.5). The immunoisolated sample attached to the immuno-sepharose beads was eluted using low pH buffer to obtain the porosome complex, which was immediately brought to neutral pH. Immunoanalysis was then performed on the SNAP-25 immunoisolates. Protein concentrations were estimated by Bradford method (Bradford 1976), and resolved using 10% SDS-PAGE. The resolved proteins were electrotransferred to 0.2 mm thick nitrocellulose membrane for immunoblot analysis using specific antibodies. The nitrocellulose membranes were incubated for 1 h at room temperature in blocking buffer (5% non-fat milk in PBS containing 0.1% Triton X-100 and 0.02% NaN3), and immunoblotted for 1 h at room temperature using specific primary antibody. The immunoblotted nitrocellulose sheets were washed (3X) in PBS containing 0.1% Triton X-100, and incubated for 1 h at room temperature in horseradish peroxidase-conjugated secondary antibody at a dilution of 1:3000 in blocking buffer. The immunoblots were washed (3X) in PBS, processed for enhanced chemiluminescence, and photographed using a Kodak Image Station 414.
2.3 Resolution of cholesterol and proteins in solubilized synaptosomal membrane using gel filtration chromatography
Freshly isolated synaptosomal membrane preparations from rat brain, were solubilized in ice-cold Triton/Lubrol (1% w/v) PBS, and 0.5 mg of total solubilized protein in a volume of 300 μl, were loaded onto a calibrated 70 ml Sephadex G-200 gel filtration column. After void volume, 20 fractions each of 1.5 ml were collected and subsequently assayed for cholesterol content using Sigma Infinity cholesterol reagent kit according to manufacturer instruction. Similarly, each fraction was assayed for the presence of various porosome proteins, using immunoblot analysis.
2.4 Resolving the SNARE complex using continuous sucrose density gradient centrifugation
The size and partitioning of SNARE complexes in Triton/Lubrol-solubilized recombinant SNARE-reconstituted PC:PS and PC:PS:Cholesterol vesicles, was analyzed using small-scale sucrose density sedimentation method (Tanese, 1997). The solubilized vesicles were immediately loaded onto a linear 10−50% sucrose density gradient, and centrifuged at 150,000 x g at 4° C for 3 h in a Beckman Ti 100.3 bench-top ultracentrifuge to resolve complexes of various size. A total of 7 fractions were collected from the top, and subsequently analyzed for the presence of the t-SNARE syntaxin-1 (synt 1), using immunoblot analysis.
2.5 Preparation of PC:PS-associated neuronal porosome complexes and neuronal t-SNARE- and v-SNARE-associated PC:PS and PC:PS:Cholesterol vesicles
All lipids were obtained from Avanti Polar Lipids (Alabaster, AL). Preparation of lipid vesicles and their reconstitution with proteins were performed using previously published procedures (Cho and Kelly et al., 2002; Cho et al., 2005). A 10 mM lipid stock solution was prepared by mixing lipid solution in chloroform-DOPC (1,2-dioleoyl-phosphatidylcholine): DOPS (1,2-dioleoyl-phosphatidylserine) in 70:30 mol/mol ratios, or DOPC:DOPS:Cholesterol in 56:24:20 mol/mol/mol ratios in glass test tubes. The lipid mixture was dried under gentle stream of nitrogen and resuspended in decane. The lipids were then suspended in buffer containing 10 mM Hepes-NaOH [pH =7.5] and 140 mM NaCl by vortexing for 5 min. at room temperature. Vesicles were prepared following sonication for 2 min, and extrusion using an extruder with membranes of different pore size, to obtain different size vesicles. Large unilamellar vesicles were formed following sonication for 2 min. Liposomes ranging in size from 0.2−2 μm in diameter were obtained, as assessed by zeta sizer and atomic force microscopy (AFM). Protein expression and purification was performed according to published procedure (Cho and Kelly et al., 2002; Weber et al., 1998). Proteoliposomes were prepared by gently mixing either purified recombinant or native t-SNAREs or v-SNARE, or the immunoisolated neuronal porosome complex obtained from solubilized synaptosome membrane, followed by 3 freeze/thaw cycles to enhance reconstitution at the vesicles membrane. Specifically, recombinant t-/ or v-SNARE proteoliposomes were prepared by gently mixing either t-SNARE complex (Syntaxin-1/SNAP-25; final concentration 5 μg/ml) or VAMP2 (final concentration 2.5 μg/ml) with 10 mM liposomes. However, due to low copy numbers and or yield, protein concentration of immunoisolated native SNAREs was undetectable.
2.6 Photon correlation spectroscopy
(PCS) and right angle scattering, well-known techniques for the measurement of size of sub-micron particles and macromolecules, was performed using published procedure (Jeremic and Cho et al., 2005). PCS measurements were performed using a Zetasizer Nano ZS, (Malvern Instruments, UK). In a typical experiment, the size distribution of vesicles was determined using built-in software provided by Malvern Instruments. Prior to determination of vesicle hydrodynamic radius, calibration of instrument was performed using latex spheres of known size. In PCS, subtle fluctuations in the sample scattering intensity are correlated across microsecond time scales. The correlation function is calculated, from which the diffusion coefficient is determined by the instrument. Using Stokes-Einstein equation, hydrodynamics radius can be calculated from diffusion coefficient. The intensity size distribution, which is obtained as a plot of the relative intensity of light scattered by particles in various size classes, is calculated from correlation function using the built-in software. The particle scattering intensity is proportional to the molecular weight squared. Volume distribution, which assigns more realistic weights to both small and big particles, is calculated from the intensity distribution using Mie theory. The transforms of the PCS intensity distribution to volume distributions is obtained using the provided software by Malvern Instruments.
2.7 Electron microscopy
Rat brain was perfused with normal saline solution followed by phosphate buffer (pH 7.4) containing 2.5% glutaraldehyde. After perfusion, the brain was carefully removed and diced into 1-mm3 pieces. The brain slices were post-fixed in phosphate buffer containing 1.5% osmium tetroxide, dehydrated in graded ethanol and acetone, and embedded in araldite. The brain tissue blocks were appropriately trimmed and sectioned. Forty to fifty nanometer sections were obtained and mildly stained using lead citrate, followed by their examination using a JEOL JEM-100C transmission electron microscope.
Negative staining electron microscopy -
Aliquots (6 μl) porosome samples at 0.012 mg/ml concentration were adhered to carbon-coated, 400-mesh copper grids previously rendered hydrophilic by glow discharge. The grids were washed with five successive drops of deionized water and then exposed to 3 successive drops of 2% (w/v) uranyl nitrate for 1.5 min (Ted Pella, Tustin, CA). Images at 80,000 x magnification were recorded at defocus of 2.5 and 0.8 μm respectively on 4K × 4K Gatan UltraScan CCD under low electron dose conditions using a Tacnai 20 electron microscope (Philips Electron Optics/FEI, Eindhoven, The Netherlands) operating at 200 kV.
Atomic Force Microscopy-
Isolated synaptosomes, immunoisolated neuronal porosomes reconstituted in lipid membrane, and t-/v-SNARE complexes in PBS, pH 7.5, were imaged using the atomic force microscope (AFM) (BioScope III, Digital Instruments, Santa Barbara, CA). AFM imaging was performed in fluid (PBS, pH 7.5) using the “tapping” mode. All images presented in this study were obtained in the tapping mode in fluid, using silicon nitride tips with a spring constant of 0.06 N/m, and an imaging force of <200 pN. To obtain high-resolution images, imaging force of 300−500 pN was also used. Images were obtained at line frequencies of 1.98 Hz, with 512 lines per image, and constant image gains. Tip velocity 11.4 mm/s; tip spring constant 0.06 N/m; sample/line 512; integral gain 2.0; proportional gain 1.0; amplitude set point 0.12 – 0.28V; drive frequency 7.76 – 8.12 KHz; and drive amplitude 150 – 400 mV, were used. Topographical dimensions of both native and lipid-reconstituted porosomes, and the t-/v-SNARE complex were analyzed using the NanoScope IIIa version 4.43r8 software, supplied by Digital Instruments.
Briefly, atomic force microscopy was performed on lipid membrane in buffer, over a mica surface. To prepare SNARE-complexes on mica for AFM studies, freshly cleaved mica disks were placed in a fluid chamber. Two hundred microliters of imaging buffer solution containing 140 mM NaCl, 10 mM HEPES, and 1 mM CaCl2, was placed at the center of the cleaved mica disk. Reconstituted t-SNARE vesicles were deposited on mica and incubated for_1h at room temperature. v-SNARE reconstituted vesicles were then gently added to the t-SNARE membrane, and following incubation for 10 min. at room temperature, gently washed five times (two volumes/wash) using imaging buffer. SNARE complexes were measured either following dislodging the proteoliposomes, or following their solubilization using Triton X-100 (0.1% w/v), by a published procedure (Cho and Kelly et al., 2002; Cho et al., 2005). The mixture was then allowed to incubate for 10 min at room temperature, prior to imaging using the AFM.
Results and discussion
Electron micrographs of the presynaptic membrane demonstrate the presence of 12−17 nm cup-shaped porosomes, where 30−50 nm synaptic vesicles are found docked (Fig. 1a,b). A central plug-like structure is observed at the opening of the porosomes in electron micrographs. High-resolution imaging using atomic force microscopy (AFM) of isolated synaptosome preparations in buffered solution, confirms the presence of these 12−17 nm cup-shaped porosomes at the presynaptic membrane possessing a central plug (Fig. 1,2). Close examination of the porosomes by AFM reveals an array of 8 globular structures arranged at the rim of the porosome opening, with every alternate globular element tethered to the central plug of the structure (Fig. 1c,d). Using SNAP-25-specific antibody, immunoisolation of neuronal porosomes from Triton/Lubrol-solubilized synaptosome membrane is demonstrated, as it readily reconstitutes into artificial PC:PS membrane and exhibits all of the characteristic features found in the native structure (Fig. 1d). Examination of inside-out synaptosomal membrane preparations in buffer using AFM, further demonstrates, the presence of porosomes as inverted cup-shaped structures, some having synaptic vesicles still docked at their base (Fig. 2a,b). AFM measurement of the porosomes (13.05±0.91) and attached synaptic vesicles (40.15±3.14) in the cytosolic compartment of synaptosome membrane, is demonstrated in Fig. 2c (n=15). Dynamic light scattering and photon correlation spectroscopy, further confirm the size of the immunoisolated porosomes as 12−17 nm structures (Fig. 2e).
Figure 1. Neuronal fusion pore.
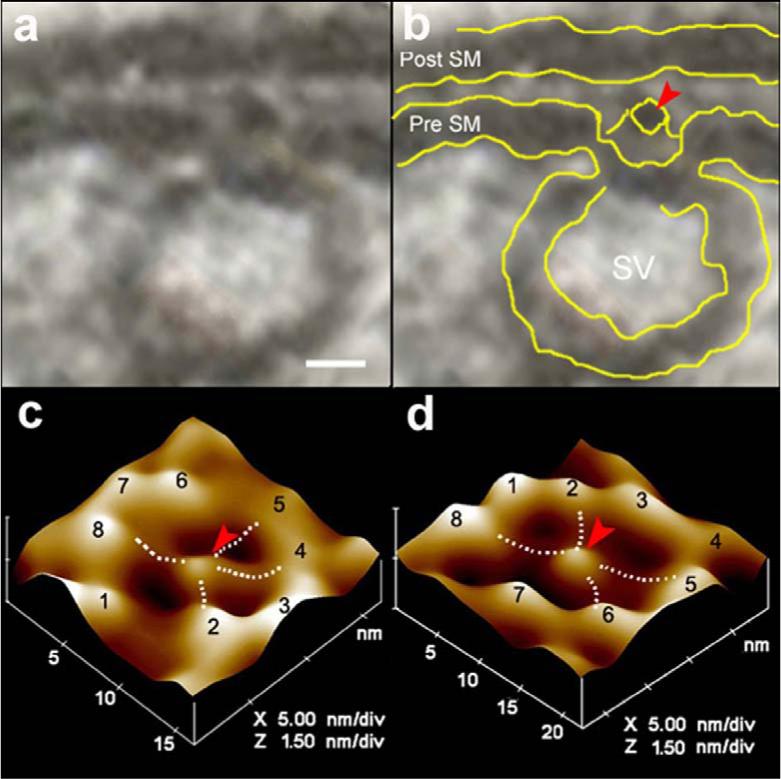
(a,b) Electron micrograph of a cup-shaped neuronal fusion pore measuring 12 nm at the presynaptic membrane, where a 30 nm synaptic vesicles is docked. Note the central plug (red arrowhead) of the fusion pore complex. Bar = 5 nm. (c) Atomic force micrograph of the surface topology of a neuronal fusion pore at the presynaptic membrane in an isolated synaptosome. (d) Atomic force micrograph of an isolated neuronal fusion pore, reconstituted in lipid membrane. Note the central plug and the 8 ridges in the atomic force micrographs of the fusion pore, both at the presynaptic membrane of the synaptosome (c) and in the isolated lipid-reconstituted complex (d). Bridges connecting the ridges with the central plug (dotted lines) are seen in the atomic force micrographs.
Figure 2. Neuronal fusion pore distribution, size and structure.

Figure 2a shows the structure and distribution of fusion pores at the cytosolic compartment of a synaptosome. Inside-out synaptosome preparations when imaged in buffer using AFM, demonstrates inverted 12−16 nm cup-shaped fusion pores, some with docked vesicles. Note one inverted cup-shaped fusion pore (green arrow heads), with a docked synaptic vesicle (red arrow heads), shown at higher magnification in Figure 2b. (b) Atomic force micrograph shows a 37 nm synaptic vesicle docked to a 14 nm fusion pore at the cytoplasmic compartment in the isolated synaptosomal membrane. (c) AFM measurement of the fusion pores (13.05±0.91) and attached synaptic vesicles (40.15±3.14) in the cytosolic compartment of synaptosome membrane, is demonstrated in Fig. 2c (n=15). (d) Schematic illustration of a neuronal fusion pore, showing the 8 vertical ridges and a central plug. (e) Photon correlation spectroscopy, further demonstrates fusion pores to measure 12−16 nm.
To test the role of cholesterol on the integrity of the neuronal porosome, Triton/Lubrol-solubilized synaptosome membrane preparation was resolved using a calibrated G-200 Sephadex gel filtration column. Immunoanalysis of the various fractions for t-SNARE proteins Syntaxin-1 (Synt 1) and SNAP-25, demonstrate their peak elution profiles to be in fractions 9 through 11, representing a complex of molecular weight >650 kD (Fig. 3a). Similarly, immunoanalysis of other porosome-associated proteins such as N-Ca2+-channel, and the calcium-binding protein synaptotagmin (Sytg 1), also reveal their co-isolation with t-SNAREs (Fig. 3a). Cholesterol assay of the same fractions, demonstrate peak elution of cholesterol in fractions 9 through 11, representing the >650 kD complex (Fig. 2b). Saponin has the ability to partition cholesterol from membrane proteins. When Triton/Lubrol/Saponin-solubilized synaptosome membrane preparation was resolved using the same G-200 Sephadex gel filtration column, no complex of molecular weight >650 kD eluted from the column, demonstrating that removal of cholesterol resulted in disassembly of the complex (data not shown). The >650 kD complex might represent the intact neuronal porosome, and to determine whether this was the case, the neuronal porosome complex was immunoisolated from Triton/Lubrol-solubilized synaptosome membrane preparations using a SNAP-25-specific antibody. When the SNAP-25 immunoisolate was resolved using the calibrated G-200 Sephadex gel filtration column, it eluted in the same fractions (Fig. 3c) as the >650 kD complex in Triton/Lubrol-solubilized synaptosome membrane preparations (Fig. 3b). Further, reconstitution of the >650 kD immunoisolated complex into artificial PC:PS membranes followed by imaging using the AFM, confirmed them to be neuronal porosomes (Fig. 3c). Additionally, negative staining electron microscopy of the >650 kD complex in absence of membrane, demonstrated the protein backbone of the porosome to be a 10−12 nm asymmetric cart-wheel structure (Fig. 3c), similar to the AFM image of the reconstituted porosome. To further test whether removal of cholesterol influenced interactions between proteins of the neuronal porosome complex, the SNAP-25 immunoisolate from Triton/Lubrol- and Triton/Lubrol/Saponin-solubilized synaptosome membrane preparations were examined. When such immunoisolates were immunoprobed using Synt 1 and N-Ca2+-channel specific antibody, the Triton/Lubrol/Saponin-solubilized immunoisolates exhibited little or no Synt 1 and N-Ca2+-channel immunoreactivity (Fig. 3d). These results demonstrate that the interaction between SNAP-25, Synt 1, and N-Ca2+-channel in the porosome complex at the presynaptic membrane is cholesterol-dependent. Hence, cholesterol is required to retain the integrity of the neuronal porosome. These results may explain why in presence of cholesterol-depleting agents, or in situations of cholesterol-imbalance at the cellular membrane in disease states, there is impaired neurotransmitter release and or loss of neuronal plasticity (Zamir and Charlton, 2006; Koudinov and Koudinova, 2001).
Figure 3. Cholesterol at the synaptosomal membrane is required to maintain the integrity of the neuronal fusion pore.

The elution profile of various fusion pore proteins (a), and cholesterol (b) in solubilized synaptosomal membrane preparation resolved on a G-200 sizing column, is demonstrated. Synaptosomal membrane were solubilized in Triton / Lubrol (1% w/v) PBS, and loaded onto Sephadex G-200 gel filtration column. Fractions 1 through 20 were collected and assayed for various fusion pore proteins following SDS-PAGE and immunoblot analysis. Note the elution Syntaxin 1 (Synt 1), SNAP-25, N-type calcium channel (N-Ca2+), and synaptotagmin (Sytg 1), primarily in fractions 9−11, a >650 kDa complex. Similarly, cholesterol elutes primarily in the same fractions (9−11), as assayed using Sigma Infinity cholesterol reagent kit. (c) When SNAP-25 -immunoisolated fusion pores from Triton-Lubrol-solubilized synaptosomal membrane was resolved using the column, the fusion pore complex eluted primarily in fractions 9 and 10, as demonstrated from immunoanalysis of the fractions for Syntaxin 1 and N-type Ca2+-channel. This is further confirmed by both atomic force microscopy (AFM) of the lipid-reconstituted fractions, and negative-staining electron microscopy (EM) of the fractions. Only fractions 9 and 10 contained the intact fusion pore as demonstrated (c). Note the 10−12 nm protein backbone of the fusion pore in the EM, demonstrating an asymmetric cart-wheel structure. (d) SNAP-25-immunoisolation of cholesterol-depleted (using saponin) synaptosomal membrane preparations, demonstrate a significant abrogation (*P<0.05; **P<0.01) of SNAP-25 binding to Syntaxin 1 and the N-type Ca2+-channel.
Since depletion of cholesterol affects the integrity of the neuronal porosome, it would be logical to speculate that cholesterol may influence the formation of appropriate t-/v-SNARE complexes, required for fusion of synaptic vesicles at the porosome complex in the nerve terminal. To test this hypothesis, the role of cholesterol in formation of the t-/v-SNARE complex, was also examined in our study. To eliminate the influence of other associated proteins at the synaptic vesicle and in the porosome complex at the presynaptic membrane, purified recombinant t-SNAREs and v-SNARE in artificial lipid membrane were used. Artificial PC:PS and PC:PS:Cholesterol membrane were reconstituted with t-SNAREs, and PC:PS and PC:PS:Cholesterol vesicles with v-SNARE. Vesicles (PC:PS) reconstituted with v-SNARE were exposed to t-SNARE-associated PC:PS membrane. Similarly, v-SNARE-associated vesicles containing cholesterol (PC:PS:Cholesterol) were exposed to t-SNARE-associated PC:PS:Cholesterol membrane. The size and arrangement of the resultant t-/v-SNARE complex formed, when t-SNARE-associated membrane interact with v-SNARE-associated vesicles, was assessed using the AFM (Fig. 4a). Surprisingly, the size and arrangement of the resultant t-/v-SNARE complexes formed either in the presence or absence of cholesterol, was found to be identical (Fig. 4b). To further determine the influence of cholesterol on the interaction between t-SNARE proteins, recombinant Synt 1 and SNAP-25 reconstituted in PC:PS and PC:PS:Cholesterol vesicles, were subjected to Triton/Lubrol solubilization, and resolved using a 10−50% linear sucrose gradient (Fig. 4c). Assay of the sucrose gradient fractions for Synt 1, demonstrated no influence of cholesterol on the resolution of t-SNARE complex in the gradient (Fig. 4c). These results demonstrate that similar to the inability of cholesterol on influencing the size and arrangement of the t-/v-SNARE complex (Fig. 3b), there is no change in the interaction between Synt 1 and SNAP-25 in the presence or absence of cholesterol (Fig. 4c). These results further suggest that cholesterol has no direct interaction with either Synt 1 or SNAP-25. If cholesterol had any direct interactions with either of the t-SNARE proteins, there would have been a shift in the distribution of Synt 1 since binding of cholesterol would have changed the buoyancy of the t-SNARE complex in sucrose gradient.
Figure 4. Cholesterol exhibits no influence on the structure and assembly of the t-/v-SNARE complex.
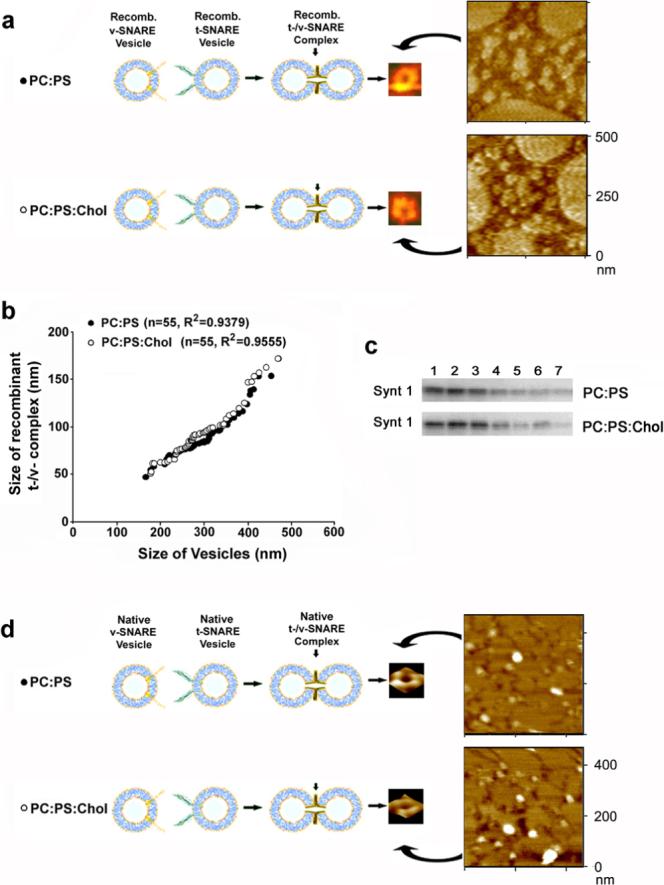
(a) Full length recombinant v-SNARE in proteoliposomes, interact with full length recombinant t-SNARE-proteoliposomes in a circular array, to form t-/v-SNARE ring complexes. (b) Size of the t-/v-SNARE ring complex is directly proportional to the size of the proteoliposomes used. Note, that presence of cholesterol has no influence on the size, structure, and assembly of the t-/v-SNARE ring complex (a, b). (c) Resolution of Triton-Lubrol-solubilized proteoliposomes (PC:PS- or PC:PS:cholesterol-t-/v-SNARE ring complexes) on a sucrose gradient, demonstrates no difference in the presence or absence of cholesterol. (d) As observed for recombinant SNAREs (a-c), no difference in the native t-/v-SNARE ring complex is detected in presence of cholesterol.
There remained however the possibility that native SNAREs may interact and behave differently with cholesterol as opposed to recombinant SNAREs, primarily due to two fundamental reasons. One, native SNAREs may behave differently due to secondary modifications, and two, native SNAREs are devoid of the extra His-tag stretch incorporated into the recombinant proteins to allow efficient purification on metal column. To test this hypothesis, native Synt 1, SNAP-25, and VAMP-1 were separately immunoisolated from Triton/Lubrol/Saponin-solubilized brain synaptosome fractions containing high salt buffer (2M PBS). The native t-SNAREs and v-SNARE were each reconstituted into their respective PC:PS and PC:PS:Cholesterol vesicles, and the size and arrangement of the resultant t-/v-SNARE complex formed was determined by high resolution AFM imaging (Fig. 4d). Results from these studies demonstrate, that similar to recombinant SNAREs, membrane-associated native t-SNAREs interact with membrane-associated native v-SNARE in a circular array to form ring complexes. Furthermore, analogous to recombinant SNAREs, cholesterol exhibited no influence on the structure and arrangement of the native t-/v-SNARE complex (Fig. 4d).
In contrast to soluble proteins, membrane-associated proteins organize differently in the absence of membrane (Cho and Kelly et al., 2002; Bowie, 2005). The hydrophobic stretch of membrane proteins that reside within the bilayer membrane, play critical roles in the 3D conformation of their extra-membranous domains. The amino acid composition and sequence of protein stretches residing within the variable and anisotropic membrane environment, dictates its own folding properties (Popot and Engelman, 1990; Engelman et al., 2003), which also influences the structure and conformation of its extra-membranous component. In agreement, it has been demonstrated that in the absence of membrane, membrane proteins t-SNAREs and v-SNARE interact without appropriate orientation and conformation, resulting in the formation of random globular structures (Cho and Kelly et al., 2002). On the contrary, when both SNAREs are residing in opposing membranes, as they do in their native state, they interact in a circular array to form t-/v-SNARE ring complexes or channels (Cho and Kelly et al. 2002). Since cholesterol is part of the neuronal porosome as are t-SNAREs, and found to regulate the integrity of the pore complex, the role of cholesterol in the formation of the t-/v-SNARE complex was explored in this study. Surprisingly however, our results show that the interaction and arrangement of membrane-associated t-SNAREs and v-SNARE is cholesterol-independent (Fig. 4). Cholesterol however, maintains the integrity of the neuronal porosome and the interactions between its component proteins (Fig. 3). In the native state, cholesterol may indirectly modulate interactions between t-SNARE proteins and other proteins of the neuronal porosome machinery. This would explain the loss of interaction between SNAP-25, Synt 1, and N-Ca2+-channel in the porosome complex following depletion of cholesterol, even though both syntaxin and calcium channels are known to interact with SNAP-25/23 proteins (Cho and Jeremic et al., 2005; Yang et al., 1999; Wiser et al., 1999). Since calcium is critical to SNARE-induced membrane fusion (Cho and Kelly et al., 2002; Jeremic et al., 2004a; Jeremic et al., 2004b), the loss of interaction between SNAP-25, Synt 1, and N-Ca2+-channel in the neuronal porosome complex as a result of loss or disbalance of cholesterol, could seriously compromise or even abrogate neurotransmission.
Acknowledgements
We thank members of the laboratory for suggestion and critical reading of the manuscript.
This work was supported by grants from the NIH (to BPJ).
Glossary
Abbreviations used:
- t-/v- SNARE
target-/vesicle-soluble N-ethylmalemide-sensitive factor attachment protein receptor
- SNAP
soluble NSF attachment protein
- VAMP
vesicle-associated membrane protein
- DOPC
1,2-dioleoyl phosphatidylcholine
- DOPS
1,2-dioleoyl phosphatidylserine
- EM
electron microscopy
- AFM
atomic force microscopy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438:612–21. doi: 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Cho SJ, Kelly M, Rognlien KT, Cho JA, Horber JK, Jena BP. SNAREs in opposing bilayers interact in a circular array to form conducting pores. Biophys J. 2002;83:2522–7. doi: 10.1016/s0006-3495(02)75263-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WJ, Jeremic A, Jena BP. Direct interaction between SNAP-23 and L-type Ca2+ channel. J Cell Mol Med. 2005;9:380–6. doi: 10.1111/j.1582-4934.2005.tb00363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WJ, Jeremic A, Jena BP. Size of supramolecular SNARE complex: membrane-directed self-assembly. J Am Chem Soc. 2005;127:10156–7. doi: 10.1021/ja052442m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WJ, Jeremic A, Rognlien KT, Zhvania MG, Lazrishvili I, Tamar B, Jena BP. Structure, isolation, composition and reconstitution of the neuronal fusion pore. Cell Biol Int. 2004;28:699–708. doi: 10.1016/j.cellbi.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Engelman DM, Chen Y, Chin CN, Curran AR, Dixon AM, Dupuy AD, Lee AS, Lehnert U, Matthews EE, Reshetnyak YK, Senes A, Popot JL. Membrane protein folding: beyond the two stage model. FEBS Lett. 2003;555:122–5. doi: 10.1016/s0014-5793(03)01106-2. [DOI] [PubMed] [Google Scholar]
- Jena BP. Molecular Machinery and Mechanism of Cell Secretion. Exp Biol Med. 2005;230:307–19. doi: 10.1177/153537020523000504. [DOI] [PubMed] [Google Scholar]
- Jena BP, Cho SJ, Jeremic A, Stromer MH, Abu-Hamdah R. Structure and Composition of the Fusion Pore. Biophys J. 2003;84:1–7. doi: 10.1016/S0006-3495(03)74949-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeremic A, Cho WJ, Jena BP. Involvement of water channels in synaptic vesicle swelling. Exp Biol Med. 2005;230:674–80. doi: 10.1177/153537020523000910. [DOI] [PubMed] [Google Scholar]
- Jeremic A, Cho WJ, Jena BP. Membrane fusion: what may transpire at the atomic level. J Biol Phys Chem. 2004a;4:139–42. [Google Scholar]
- Jeremic A, Cho WJ, Jena BP. Cholesterol is critical to the integrity of neuronal porosome/fusion pore. Ultramicroscopy. 2006;106:674–7. doi: 10.1016/j.ultramic.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Jeremic A, Kelly M, Cho JA, Cho SJ, Hörber JK, Jena BP. Calcium drives fusion of SNARE-apposed bilayers. Cell Biol Int. 2004b;28:19–31. doi: 10.1016/j.cellbi.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Koudinov AR, Koudinova NV. Essential role for cholesterol in synaptic plasticity and neuronal degeneration. FASEB J. 2001;15:1858–60. doi: 10.1096/fj.00-0815fje. [DOI] [PubMed] [Google Scholar]
- Lang T, Bruns D, Wenzel D, Riedel D, Holroyd P, Thiele L, Jahn R. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 2001;20:2202–13. doi: 10.1093/emboj/20.9.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–21. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Maxfield FR. Membrane domains. Annu Rev Cell Dev Biol. 2004;20:839–66. doi: 10.1146/annurev.cellbio.20.010403.095451. [DOI] [PubMed] [Google Scholar]
- Popot J, Engelman D. Membrane protein folding and oligomerization: the two-stage model. Biochemistry. 1990;29:4031–7. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- Tanese N. Small-scale density gradient sedimentation to separate and analyze multiprotein complexes. Methods. 1997;12:224–34. doi: 10.1006/meth.1997.0475. [DOI] [PubMed] [Google Scholar]
- Taverna E, Saba E, Rowe J, Francolini M, Clementi F, Rosa P. Role of Lipid Microdomains in P/Q-type Calcium Channel (Cav2.1) Clustering and Function in Presynaptic Membranes. J Biol Chem. 2004;279:5127–34. doi: 10.1074/jbc.M308798200. [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–72. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Wiser OM, Hernandez TA, Renstrom E, Barg S, Rorsman P, Atlas D. The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the exocytotic machinery. Proc Natl Acad Sci USA. 1999;96:248–53. doi: 10.1073/pnas.96.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SN, Larsson O, Branstrom R, Bertorello AM, Leibiger B, Leibiger IB, Moede T, Kohler M, Meister B, Berggren PO. Syntaxin 1 interacts with the L(D) sub-type of voltage-gated Ca2+ channels in pancreatic beta cells. Proc Natl Acad Sci USA. 1999;96:10164–9. doi: 10.1073/pnas.96.18.10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir O, Charlton MP. Cholesterol and synaptic transmitter release at crayfish neuromuscular junctions. J Physiol. 2006;571:83–99. doi: 10.1113/jphysiol.2005.098319. [DOI] [PMC free article] [PubMed] [Google Scholar]