

Abstract
The validity of the thermodynamic hypothesis of protein folding was explored by simulating the evolution of protein sequences. Simple models of lattice proteins were allowed to evolve by random point mutations subject to the constraint that they fold into a predetermined native structure with a Monte Carlo folding algorithm. We employed a simple analytical approach to compute the probability of violation of the thermodynamic hypothesis as a function of the size of the protein, the fraction of the total number of possible conformations which are kinetically accessible, and the roughness of the free-energy landscape. It was found that even if the folding is under kinetic control, the sequence will evolve so that the native state is most often the state of minimum free energy.
Keywords: protein evolution, lattice models, folding simulation, folding kinetics
Understanding how proteins fold not only is one of the most interesting theoretical problems in molecular biophysics but also has far-reaching medical and biotechnological consequences. Levinthal pointed out that it is impossible for an unfolded protein to find the native state by randomly searching through the entire space of possible conformations (1). This led him to postulate that a protein must follow a specific path that guides it to the native state, and therefore folding must be under kinetic control. According to him, “If the final folded state turned out to be the one of lowest configurational energy, it would be a consequence of biological evolution and not of physical chemistry” (2). In contrast, Anfinsen concluded from the results of his numerous denaturation–renaturation experiments that the native state of the protein is indeed the global minimum of free energy, a conjecture that he called the thermodynamic hypothesis of protein folding (3).
The debate between these two viewpoints has continued, with numerous experimentalists and theoreticians investigating whether proteins reach their global energy minimum in a pathway-independent manner under thermodynamic control, or whether they follow a specific pathway to a possibly local minimum under kinetic control. Experiments have suggested that some small monodomain proteins obey the thermodynamic hypothesis (4, 5). There are also examples of proteins where the active state is not the thermodynamically most stable state. For instance, the plasminogen activator inhibitor (PAI-1) active conformation is metastable, and the protein takes a more stable inactive conformation within hours (6). Similar observations have been made with other members of the serpin family. Protein misfolding, as is the case in many diseases such as Alzheimer’s disease, Creutzfeldt–Jakob disease, and bovine spongiform encephalopathy, have been attributed to kinetic traps or folding to an alternate state of lower energy (7). Similarly, it has been possible to modify proteins so that they are no longer able to fold, although the stability of the native state is unaltered (8, 9).
On the theoretical side, Thirumalai used molecular dynamics simulations to support his hypothesis that proteins fold into a metastable state (10). Shakhnovich and co-workers questioned whether a protein could consistently find the same native state by using this mechanism (11), and they proposed that a sufficiently large “energy gap” separating the native state from others was a necessary and sufficient condition for rapid folding (12), consistent with the results of simple models based on spin-glass theory (13, 14). Such an energy gap would necessarily imply the thermodynamic hypothesis. In other work, Shakhnovich showed that lattice proteins under strong evolutionary pressure to fold as quickly as possible evolve so that the native state is a deep and global minimum (15). On the other hand, proteins have not evolved to fold at the maximum possible rate. In addition, in lattice models almost the entire conformation space is kinetically accessible, so the absence of other deeper minima is not surprising. Onuchic, Wolynes, and Dill and their co-workers postulated that the distinguishing characteristic of foldable proteins is the existence of a “folding funnel” that directs the folding protein into the native state without the need for a definite pathway (16–18). This approach leaves open the possibility of kinetically inaccessible lower-energy states outside of the folding funnel. More elaborate theoretical models have been developed that have further explored the relationship between the free-energy landscape and the folding kinetics, including the role of traps and intermediates in the folding process (19–21).
In this paper we attempt to address the challenge posed by Levinthal, and investigate whether the thermodynamic hypothesis could result through the process of protein evolution. During evolution, protein sequences undergo random mutations. If the mutation does not interfere with the folding and function of the protein, it is possible for this mutation to be accepted and fixed in the population. Mutations that destabilize the native state are likely to interfere with successful folding, and thus will have a low acceptance rate. Conversely, mutations that destabilize alternative deep minima are more likely to be accepted. With evolutionary time, energy minima representing nonnative states are likely to become higher in energy than the native state, so that the thermodynamic hypothesis becomes fulfilled. This can occur even if the folding is under kinetic control.
To test this hypothesis, we performed simulations of protein evolution by using simple lattice model proteins. We first designed protein sequences that fold under kinetic control to a native state different from the ground state of lowest energy. We then allowed the protein to evolve by random mutations. Mutations were accepted only if the protein still folded into the original native conformation. At each generation we calculated the energies of the native state, the initially designed ground state, and the current ground state of the sequence. As expected, the native state generally became the ground state, although there was some fraction of the time where this was not the case. We then developed a simple analytical model to estimate the probability that a protein under kinetic control would obey the thermodynamic hypothesis.
METHODS
Lattice Model.
We used a simple model of 16-residue proteins confined to a two-dimensional lattice, as shown in Fig. 1. Each residue occupies one lattice site in a square lattice with an excluded volume and lattice parameter of unit length. All 802,075 compact and noncompact conformations were enumerated, enabling us to identify the global energy minimum by evaluating the energies of all possible conformations.
Figure 1.

Examples of the two-dimensional lattice model used in this study, representing the target native structure (a) and target ground state structure (b) chosen for the simulations. The dotted line in conformation b indicates the interaction between residue 13 and 16 that is designed into the initial sequence as an unfavorable contact.
We used a simple energy function of the form
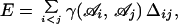 |
1 |
where γ(𝒜i𝒜j) is the contact potential between residue type 𝒜i at position i and residue type 𝒜j at position j, and Δij is equal to one if residues i and j are not adjacent in sequence but are on adjacent lattice sites, and zero otherwise. We used statistical potentials of interaction between two residues as derived by Miyazawa and Jernigan, which implicitly includes the effect of interaction of the residues with the solvent (table VI in ref. 22). The global energy minimum state for any sequence is the conformation of minimum energy among the 802,075 conformations. We used the index q to quantify the similarity between any two structures, equal to the fraction of the total number of contacts that are common between the structures (0 ≤ q ≤ 1), with pairs of identical structures having q = 1.
Folding Kinetics.
For a given sequence of amino acids, folding simulations were carried out to determine whether the protein could successfully find the target native state. At each Monte Carlo time step a local conformational change was made [tail-wag, corner move, or crankshaft rotation (23)] and the resulting new conformation was accepted with a probability P based on the Metropolis algorithm:
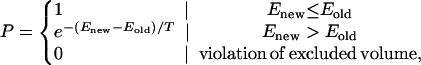 |
2 |
where Enew is the energy of resulting conformation after the Monte Carlo move and Eold is the energy of the existing conformation, and the Boltzmann constant has been set equal to one. We chose a working temperature of T = 0.085 where the folding kinetics were relatively rapid yet the native state was sufficiently stable so that the folded state population would weakly dominate that of the unfolded states during the later part of the simulations. Each simulation was carried out for 10 million time steps.
The protein was considered to successfully fold to the native state if the sequence adopted the target structure or a structure with sufficient similarity to it [q ≥ 0.88 (=8/9)], for more than 50% of the time during the final 2 million time steps of the simulation in at least five of ten simulation runs, each run starting from a random initial conformation. We also performed the simulations with a stricter folding criterion, where the protein had to be in the target state or in a state with q ≥ 0.88 for 70% of the time in at least seven of ten different runs. As results from both folding measures yielded similar qualitative results, we focus on the runs made under the first criterion.
Sequence Evolution.
We created an initial starting sequence that folded into a metastable native state that differed from a target ground state, violating the thermodynamic hypothesis. The target native state and ground state chosen in our study are shown in Fig. 1. The ground state was designed to be kinetically inaccessible by making the interaction between residues 13 and 16 unfavorable, preventing formation of the central nucleus. Starting from a random sequence, the initial sequence was designed by using a simple hill-climbing algorithm by changing the amino acid residues at random and accepting the change if it lowered the harmonic mean of the energy of the target native state (ENS) and the energy of the target ground state (EGS) while keeping EGS at a lower value than ENS. The residues at position 13 and 16 were fixed during this search. This optimization was performed until the target ground state became the global energy minimum. We generated three different sequences that successfully folded into the target native state in at least seven of ten runs, spending more than 85% of the time during those runs in the native state. We then made random mutations in the sequence and performed kinetic simulations of the resulting sequence to see whether the protein could still successfully fold to the same native state, before deciding whether or not to accept the mutation. Simulations of evolution based on the kinetic considerations on the folding of the protein were carried out for two cases, one where the restrictions on the residues at position 13 and 16 were maintained throughout the evolutionary runs and the other where it was relaxed after the initial generation.
RESULTS AND DISCUSSION
Five different evolutionary runs were carried out for each of the three initial sequences where all residues were allowed to mutate, each run consisting of 150 generations proceeded by a 100-generation pre-equilibrium. Fig. 2 shows the energies of the native state, the nonnative state with the lowest energy, and the initial ground state conformation in three of the evolutionary runs. The generations where the nonnative state with the lowest energy is below the energy of the native state corresponds to the violation of the thermodynamic hypothesis. After the pre-equilibration, the average percentage of time when the hypothesis holds was 93.4% for sequence 1, 92.1% for sequence 2, and 87.0% for sequence 3. Fig. 2a represents a typical result, where the percentage of time the hypothesis holds is about 90%. Fig. 2 b and c represents extreme cases, the former showing no violations of the thermodynamic hypothesis after pre-equilibration, and the latter violating this hypothesis for approximately one-third of the simulation. When the thermodynamic hypothesis failed, the nonnative ground state was in general not similar to the native state, with a q value between these states of 0.43, comparable to the average q value of 0.38 between any two random semicompact structures having at least 7 of a possible 9 interresidue contacts.
Figure 2.

The energies of the native state (solid line), nonnative state of lowest energy (dotted line), and initial ground state(dashed line) for various generations for three different runs. (a) Representative plot for a typical simulation. (b and c) The extreme cases for which the hypothesis is least (b) and most (c) violated.
Our criterion for successful folding was quite generous. As we would expect, a more stringent criterion decreased the fraction of the time when the thermodynamic hypothesis was violated. Defining successful folding as reaching a native-like state 70% of the time for 7/10 of the folding simulations increased the percentage of time when the hypothesis was fulfilled to 94%.
Another set of five runs, three with sequence 1 and one each with sequences 2 and 3, were carried out where the designed unfavorable contact between residues 13 and 16 in the initial sequences was maintained throughout the run. In this manner, not all of the conformations would be kinetically accessible. The thermodynamic hypothesis held for these runs 87% of the time.
As indicated by these simulations, there is a finite probability for any random structure to have a lower energy than the native structure. This structure will not interfere with the folding process as long as this state is kinetically inaccessible on the time-scale of folding. Because of the astronomically large number of conformations available to a protein, it is possible that a significant fraction of the conformation space is not kinetically accessible. There is therefore some probability that a conformation in this fraction will be lower in energy than the native state, and the thermodynamic hypothesis will be violated. To explore this issue in more detail we used the Random Energy Model (24) and estimated the probability that the thermodynamic hypothesis is violated by considering the probability of finding a state with a lower energy than that of the native state among the kinetically inaccessible conformations. In this model we assume the simplest possible criterion for foldability: that the native state is sufficiently stable with respect to all of the other accessible conformations. While this criterion can be justified on the basis of ideas borrowed from the physics of spin glasses (13, 14, 25) and has been supported by lattice simulations (11, 12), it is significantly simpler than the more sophisticated models that have been developed by other researchers (19–21, 26, 27).
The total number of conformational states for a protein of length N is given by esN, where s is the effective entropy per residue. All of these available states cannot be explored during the folding time-scale, and some may be inaccessible because of the presence of kinetic barriers. Let us assume that a fraction ρ of all conformations are kinetically accessible. The number of states kinetically accessible to the protein chain is then ρesN. Following the random energy model, the probability density of unfolded states with energy E, n(E), was represented as a Gaussian centered at E = 0
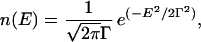 |
3 |
where Γ is the width of the distribution of energies of the various conformational states, which is related to the degree of ruggedness in the protein energy landscape (13). This model explicitly neglects any correlations between the energy levels of different conformations, including conformations that share structural similarities, an issue that has been explored by other investigators (21).
Assuming that the accessible states are at equilibrium, the probability that the protein is in the ground state at any time, PNS, is given by the Boltzmann expression
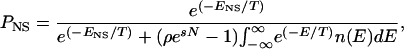 |
4 |
in which we have neglected the possibility of nonnativelike outliers in n(E). As such outliers would greatly reduce PNS, they can reasonably be expected to be absent among the kinetically accessible conformations in natural proteins. If we have an estimate of PNS and Γ, we can use this equation to solve for ENS as a function of ρ for any given N.
Because the native state is the state of lowest energy among accessible conformations, we can estimate the probability of having an inaccessible state with energy less than ENS, meaning that the thermodynamic hypothesis is violated. Let us assume that the distribution of energies of the inaccessible states depends mostly on the overall composition of the protein, and thus is not overly different from the distribution of energies of the nonnative accessible states represented in Eq. 3. P(∃E < ENS), the probability of having a state with energy less than ENS among the (1 − ρ)esN inaccessible states, is then equal to one minus the probability that all of the inaccessible states have energy higher than ENS
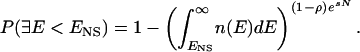 |
5 |
On the basis of hydrogen-exchange experiments, the ΔG for the global unfolding process for cytochrome c has been estimated to be −13.0 kcal/mol at 30°C (28). This provides us an estimate of PNS as a function of ρ. Wolynes and co-workers have estimated the effective conformational entropy s to be approximately 0.6 per monomer unit (29). They also made an estimate of Γ2/Tf2 as ranging between 22 and 36, consistent with values obtained from other experiments (29). Using these numbers, we calculated the probability that the thermodynamic hypothesis is violated for a 120-residue protein as a function of ρ, for different values of Γ2/Tf2, as shown in Fig. 3a. Fig. 3b shows the probability of violation as a function of ρ for various protein lengths, for Γ2/Tf2 equal to 36.
Figure 3.

Logarithm of the probability of violation of the hypothesis P(∃E < ENS) as a function of ρ. (a) For various Γ2/Tf2, the ruggedness of the protein energy landscape, for a protein 120 residues long. (b) For various N, the length of the protein chain for Γ2/Tf2 = 36.
As can be seen in these plots, the probability that the thermodynamic hypothesis is satisfied is quite large as long as more than a minuscule fraction of the conformational landscape is accessible. It might be expected that the longer the chain of the protein, the greater is the possibility of finding an alternate state lower in energy than that of the native state. Contrary to this expectation, increasing the length of the protein for a constant ρ increases the number of accessible states, requiring a further decrease in ENS to maintain the population of the native state, decreasing the possibility of violation of the thermodynamic hypothesis. This effect might be decreased or reversed if ρ increases with increasing N.
CONCLUSION
The thermodynamic hypothesis is a statement concerning the nature of the native state, that this native state represents the ground state of lowest free energy. This hypothesis does not necessarily imply thermodynamic control of the folding process, where folding occurs to the native state because it is the conformation of lowest free energy. In contrast, the assumption of kinetic control is a statement about how the native state is determined by the folding process. If the thermodynamic hypothesis is violated, then the folding must be under kinetic control. The inverse is not necessarily true—it is possible, as illustrated here, for the folding to be under kinetic control and for the thermodynamic hypothesis to be satisfied. As a consequence, there is no conflict between kinetic control and the thermodynamic hypothesis, and demonstrations of kinetic control do not necessarily demonstrate that the thermodynamic hypothesis is wrong.
There is a tendency to expect that the results of evolution necessarily represent adaptation. In actuality, evolution can result in many modifications that themselves do not give comparative advantage, as is the situation in our model. Even if folding is under kinetic control and there is no evolutionary advantage to a protein satisfying the thermodynamic hypothesis, the process of random mutation may still result in the native state becoming the state of lowest energy. Levinthal may have suggested a way in which Anfinsen’s conclusions can be justified.
The current model focuses on the selective pressure acting of the protein to form a stable structure, which is only one of the factors necessary for the protein to fulfill its specific functional role. Other investigators have investigated the complementary problem of modeling protein evolution, given strong constraints on preserving functionality (30). In particular, there may be cases where there is an evolutionary advantage to not fulfilling the thermodynamic hypothesis, or where metastability is a consequence of some functional need. The instability of the active conformation of the plasminogen activator 1 (PAI-1) is believed, for instance, to have a selective advantage (6). In this case, our model would not be applicable. Similarly, this model would not hold for proteins that have been modified in the laboratory and are thus not the product of natural evolution (8, 9).
The thermodynamic hypothesis has been the basis behind many approaches to predict protein structure, by looking for the conformation of lowest free energy. Some methods, such as genetic and threading algorithms and landscape smoothing, employ search strategies not available to the protein in its own search (31–35). Our results suggest that these methods may be appropriate, even if the native state of the protein is determined by kinetic considerations.
Acknowledgments
We thank Kurt Hillig for computational assistance and Nicolas Buchler for helpful discussions. Financial support was provided by the College of Literature, Science, and the Arts, the Horace H. Rackham School of Graduate Studies, National Institutes of Health Grant LM05770, and National Science Foundation Equipment Grant BIR9512955.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Levinthal C. In: Mossbauer Spectroscopy in Biological Systems. Debrunner P, Tsibris J C M, Munck E, editors. Urbana: Univ. of Illinois Press; 1969. pp. 22–24. [Google Scholar]
- 2.Levinthal C. J Chim Phys. 1968;65:44–45. [Google Scholar]
- 3.Anfinsen C. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 4.Kim P S, Baldwin R L. Annu Rev Biochem. 1990;59:631–660. doi: 10.1146/annurev.bi.59.070190.003215. [DOI] [PubMed] [Google Scholar]
- 5.Dill K A. Biochem. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 6.Berkenpas M B, Lawrence D A, Ginsburg D. EMBO J. 1995;14:2969–2977. doi: 10.1002/j.1460-2075.1995.tb07299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas P J, Qu B, Pederson P L. Trends Biochem Sci. 1995;20:456–459. doi: 10.1016/s0968-0004(00)89100-8. [DOI] [PubMed] [Google Scholar]
- 8.Mitraki A, Fane B, Haase-Pettingell C, Sturtevant J, King J. Science. 1991;253:54–58. doi: 10.1126/science.1648264. [DOI] [PubMed] [Google Scholar]
- 9.Baker D, Sohl J L, Agard D A. Science. 1992;356:263–265. doi: 10.1038/356263a0. [DOI] [PubMed] [Google Scholar]
- 10.Honeycutt J D, Thirumalai D. Proc Natl Acad Sci USA. 1990;87:3526–3529. doi: 10.1073/pnas.87.9.3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Šali A, Shakhnovich E I, Karplus M J. J Mol Biol. 1994;235:1614–1636. doi: 10.1006/jmbi.1994.1110. [DOI] [PubMed] [Google Scholar]
- 12.Šali A, Shakhnovich E I, Karplus M J. Nature (London) 1994;369:248–251. doi: 10.1038/369248a0. [DOI] [PubMed] [Google Scholar]
- 13.Bryngelson J D, Wolynes P G. Biopolymers. 1990;30:171–188. [Google Scholar]
- 14.Goldstein R A, Luthey-Schulten Z A, Wolynes P G. Proc Natl Acad Sci USA. 1992;89:4918–4922. doi: 10.1073/pnas.89.11.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gutin A M, Abkevich V I, Shakhnovich E I. Proc Natl Acad Sci USA. 1995;92:1282–1286. doi: 10.1073/pnas.92.5.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leopold P E, Montal M, Onuchic J N. Proc Natl Acad Sci USA. 1992;89:8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryngelson J D, Onuchic J N, Socci N D, Wolynes P G. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 18.Dill K A, Chan H S. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 19.Socci N D, Onuchic J N, Wolynes P G. J Chem Phys. 1996;104:5860–5868. [Google Scholar]
- 20.Onuchic J N, Luthey-Schulten Z, Wolynes P G. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 21.Plotkin S S, Wang J, Wolynes P G. J Chem Phys. 1997;106:2932–2948. [Google Scholar]
- 22.Miyazawa S, Jernigan R L. Macromolecules. 1985;18:534–552. [Google Scholar]
- 23.Socci N D, Onuchic J N. J Chem Phys. 1994;101:1519–1528. [Google Scholar]
- 24.Derrida B. Phys Rev Lett. 1980;45:79–82. [Google Scholar]
- 25.Bryngelson J D, Wolynes P G. Proc Natl Acad Sci USA. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pande V S, Grosberg A Y, Tanaka T. J Chem Phys. 1994;101:8246–8257. [Google Scholar]
- 27.Pande V S, Grosberg A Y, Tanaka T. Proc Natl Acad Sci USA. 1994;91:12972–12975. doi: 10.1073/pnas.91.26.12972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai Y, Englander S W. Proteins. 1996;24:145–151. doi: 10.1002/(SICI)1097-0134(199602)24:2<145::AID-PROT1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 29.Onuchic J N, Wolynes P G, Luthey-Schulten Z, Socci N D. Proc Natl Acad Sci USA. 1995;92:3626–3630. doi: 10.1073/pnas.92.8.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saito S, Sasai M, Yomo T. Proc Natl Acad Sci USA. 1997;94:11324–11328. doi: 10.1073/pnas.94.21.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowie J U, Luthy R, Eisenberg D. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 32.Jones D T, Taylor W R, Thornton J M. Nature (London) 1992;358:86–89. doi: 10.1038/358086a0. [DOI] [PubMed] [Google Scholar]
- 33.Dandekar T, Argos P. Protein Eng. 1992;5:637–645. doi: 10.1093/protein/5.7.637. [DOI] [PubMed] [Google Scholar]
- 34.Sun S. Protein Sci. 1993;2:762–785. doi: 10.1002/pro.5560020508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheraga H A. Biophys Chem. 1996;59:329–339. doi: 10.1016/0301-4622(95)00126-3. [DOI] [PubMed] [Google Scholar]