

Abstract
Mannan binding lectin (MBL) is an innate immune mediator belonging to the collectin family known to bind to the surfaces of many viruses, bacteria, and fungi. However, pathogenic strains of the fungus Cryptococcus neoformans are resistant to MBL binding. To dissect the mechanism of cryptococcal resistance to MBL, we compared MBL binding to an encapsulated wild-type strain, an encapsulated ccr4Δ mutant defective in cell integrity, and an acapsular cap60Δ strain. No MBL binding was detected on wild-type C. neoformans. In contrast, the ccr4Δ mutant bound MBL to the cell wall, predominantly at the ends of enlarged buds, whereas the acapsular strain bound MBL only at the bud neck and bud scars. In addition, the ccr4Δ mutant was sensitive to the cell wall-active antifungal caspofungin and other cell wall stress inducers, and its virulence was reduced in a mouse model of cryptococcosis. Interestingly, treatment of wild-type cells with caspofungin also increased MBL binding to C. neoformans. These results suggest that both the presence of capsule and wild-type cell wall architecture preclude MBL binding to C. neoformans.
Infections by the yeast-like fungus Cryptococcus neoformans are a significant cause of complications in transplant recipients, patients with hematologic malignancies, patients on chemotherapy, and human immunodeficiency virus (HIV) patients (10). Although C. neoformans is capable of infecting patients with no obvious immune defect, infections in immunocompromised patients predominate (5). Since the majority of C. neoformans infections occur in patients with defects in cell-mediated immunity, understanding the interaction of this pathogen with components of innate immunity that are likely intact in susceptible populations may provide avenues for novel therapies. Many factors contribute to the virulence of C. neoformans, including thermotolerance, production of melanin pigments, and encapsulation. The polysaccharide capsule of C. neoformans is immunomodulatory and antiphagocytic and provides protection from innate immune mediators (2).
Mannan binding lectin (MBL) is a member of the collectin (collagen-like or C-type lectin) family of innate immune mediators (7). In humans, this family includes MBL and pulmonary surfactant proteins A to D, which have homologues in plants and nonvertebrate animals. The roles of collectins in innate immunity are manifold and include opsonization, complement fixation, cytokine induction, and direct killing of microbes (39).
MBL is composed of three to six subunits, each made up of a trimer of the MBL gene product (9, 22). The collagen-like N-terminal stalk regions align to form the trimeric subunits. The subunits are arranged in a “bouquet” with the C-terminal carbohydrate recognition domains (CRD) at one end of the molecule (20). The carbohydrate recognition domains bind high-mannose sugars in a calcium-dependent manner (7). MBL, through the catalytic activity of its associated proteases MASP1 and MASP2, activates complement promoting C3 deposition on microbial surfaces (21, 36).
Serum levels of MBL and genetic polymorphisms that affect serum levels of MBL have been demonstrated to impact susceptibility to a number of infectious diseases (11, 17, 41). Polymorphisms in MBL and its associated protease MASP2 have been shown to be risk factors for the development of invasive fungal infections in stem cell transplant recipients (18). Recent studies have demonstrated the ability of intravenous MBL to reduce mortality associated with infection by the opportunistic fungi Candida albicans and Aspergillus fumigatus in mouse models of invasive disease (23, 25). Indeed, phase I clinical trials have been performed for use of recombinant human MBL administered to patients deficient in MBL as a result of immunosuppression (30).
MBL has been shown to bind to acapsular strains of C. neoformans but not to encapsulated strains, suggesting that capsule inhibits MBL binding (33). The role of cell wall architecture in the resistance of C. neoformans to MBL binding has not been investigated. To dissect the contribution of capsule and cell wall to MBL resistance in C. neoformans, we compared the localization of MBL on the surface of serum-opsonized wild-type C. neoformans with that of an acapsular strain and a C. neoformans ccr4Δ mutant that was found to be defective in cell integrity. Our previous data suggest that the mRNA degradation machinery, of which Ccr4 is the catalytic component, regulates cell integrity in C. neoformans (6). These data, together with data from Saccharomyces cerevisiae demonstrating cell integrity defects in the ccr4Δ mutant, made the cryptococcal CCR4 homologue an attractive target to explore the role of the mRNA degradation machinery in C. neoformans cell integrity (4).
The cell integrity-deficient ccr4Δ mutant bound MBL on the cell wall, primarily in regions with increased calcofluor white intensity, whereas the acapsular strain exhibited strong MBL staining only at bud scars and the bud neck. No MBL was found to be surface associated in wild-type C. neoformans.
MATERIALS AND METHODS
Strains and media.
All strains used in these studies were derived from C. neoformans var. grubii strain H99, a clinical isolate generously provided by John Perfect, Duke University, Durham, NC. Strains were propagated on YPD (1% yeast extract, 2% peptone, 2% dextrose, 2% agar) agar at 30°C or were grown in YPD liquid cultures in an orbital shaker at 30°C unless otherwise specified.
Molecular techniques.
Construction of the CCR4 deletion cassette was performed as previously described (29). Primers used to amplify the targeting regions, and the full-length CCR4 gene for complementation are listed in Table 1. To construct the complementation cassette, the full-length CCR4 gene was amplified from genomic DNA and cloned as a BglII-XhoI fragment into pSL1180 containing the hygromycin B resistance cassette. Introduction of the CCR4 deletion cassette by biolistic transformation and introduction of the CCR4 complementation cassette by electroporation were performed as previously described. PCR screening and Southern blotting were performed by standard methods (29, 37).
TABLE 1.
Oligonucleotides
| Oligonucleotide | Sequence | Use |
|---|---|---|
| 5′CCR4KO-F | GCTTTCCCTCCGCCATCCTTCC | CCR4 knockout construct |
| 5′CCR4KO-R | GAATTCGAATTCGCTGTGGTGTGAGCGTTGCCG | CCR4 knockout construct |
| 3′CCR4KO-F | AGATCTAGATCTGTCATATCCCGGTATTCACGC | CCR4 knockout construct |
| 3′CCF4KO-R | CTCGAGCTCGAGCGATGGCTATTCTGGCGG | CCR4 knockout construct |
| 5′CCR4-C | GGAGGAAGATCTGGGCGAAAACTACCCTTGG | CCR4 complementation |
| 3′CCR4-C | CTCGAGCTCGAGCGATGGCTATTCTGGCGG | CCR4 complementation |
Spot plate assay.
To assess the sensitivity of the congenic sets to stress conditions, a spot plate assay was used. A suspension of cells of each strain was standardized to an optical density at 600 nm (OD600) of 1.0. Five microliters of each suspension and five serial 10-fold dilutions (unless otherwise indicated) were spotted on the surface of an agar plate containing the indicated stressor as described previously (16). A control plate with no addition was included for each set of experiments. The following compounds were used: 1.5 mg/ml calcofluor white (Fluorescent Brightener 28; Sigma), 0.5% Congo red (Fluka), 0.03% sodium dodecyl sulfate (SDS) (Invitrogen), and 0.5 mg/ml caffeine (Spectrum Chemical Mfg. Group). Stock solutions were prepared using the instructions provided by the Cryptococcus gene deletion database (www.genome.slu.edu/delete/screens.html).
Trypan blue uptake.
To measure the membrane permeability of cells of the congenic set, trypan blue staining was performed using a modification of a protocol described previously (16). Briefly, mid-log-phase cells of each strain were stained with a 0.4% solution of trypan blue dye before and after a shift to 37°C for 3 h. The proportion of cells stained with trypan blue was quantitated using bright-field microscopy. One hundred cells were scored positive or negative for trypan blue uptake for each strain in triplicate. Statistical significance was determined by analysis of variance of square root transformed data. Pairwise analyses were performed post hoc using Tukey's t test.
Lysing enzyme sensitivity.
Sensitivity to lysing enzymes from Trichoderma harzanium (Sigma) was determined as described previously (16). Briefly, overnight cultures of the wild type and the ccr4Δ mutant in YPD medium were washed once with phosphate-buffered saline (PBS) and once with 1 M sorbitol-20 mM sodium citrate (pH 5.8). Cells were resuspended in 1 M sorbitol-0.1 M sodium citrate-10 mM EDTA (pH 5.8), and the OD600 was normalized to 1.0. Lysing enzymes were added to a final concentration of 50 mg/ml. Cultures were incubated at 30°C with gentle agitation. The OD600 was determined in duplicate at 1-h intervals.
Assessment of caspofungin sensitivity.
The sensitivity of the wild type and the ccr4Δ strain to capsofungin was measured by a spot plate assay (see above) on YPD agar containing 32 μg/ml caspofungin. To determine the caspofungin MICs for the wild type and ccr4Δ mutant, the broth microdilution protocol adapted for C. neoformans was used (31). Briefly, 1 × 104 cells were inoculated into RPMI medium buffered to pH 7 with morpholinepropanesulfonic acid (MOPS) and supplemented with 2% dextrose. Twofold dilutions of the drug were assessed in the range from 128 to 0.125 μg/ml. Plates were incubated at 36°C for 2 days, at which time the concentration of drug in the first nonturbid well was taken as the MIC. Assays were performed in triplicate.
Assessment of capsular permeability.
To induce capsule, yeast cells were grown in 3 ml of RPMI medium in a 12-well plate incubated in a CO2-enriched environment (GasPak EZ CO2; Becton Dickinson) in a 37°C water-jacketed incubator for 4 days. Tetramethyrhodamine-dextran 2000 kDa (TMRD; Invitrogen) staining of the C. neoformans capsule on cells grown under capsule-inducing conditions was performed as described previously (15). Cells were also stained with calcofluor white and were visualized by fluorescence microscopy 15 min after staining with TMRD. Staining with AlexaFluor 488-conjugated anticapsular Fab fragments (a generous gift from Tom Kozel, University of Nevada, Reno) was performed as described previously, except that calcofluor white stain was included in the reaction mixture (15). The distance between the outside of the cell wall and the staining front was measured using Slidebook software. A statistical analysis was performed using Graph Pad software. The Student t test was used to compare mean distances.
Detection of MBL binding.
Yeast cells were grown to mid-log phase in YPD broth at 30°C and 300 rpm. Approximately 5 × 106 cells were washed in PBS and incubated for 1 h in 100% human AB serum (Sigma) at 37°C to allow opsonization. Cells were washed three times with 1 ml PBS and incubated with either anti-human MBL monoclonal antibody HYB-131-01 (Abcam) or an immunoglobulin G1 (IgG1) isotype control (anti-c-myc clone 9E10; Sigma) at a dilution of 1:250 in PBS with 0.5 mg/ml bovine serum albumin for 1 h at room temperature. After three 1-ml washes in PBS, cells were incubated with an AlexaFluor 488-conjugated anti-mouse antibody (Invitrogen) at a 1:500 dilution in PBS-bovine serum albumin for 1 h at room temperature. After three washes in PBS, cells were resuspended in 20 μl PBS with 1 μg/ml calcofluor white and embedded in 0.5% low-melting-point agarose (NuSieve GTG; FMC Bioproducts) on a standard microscope slide. Epifluorescence was detected using an Olympus IX-70 deconvolution microscope.
Virulence assay.
NIH Swiss albino mice (Jackson Labs) were inoculated in the lateral tail vein with 1 × 106 cells of the wild-type, ccr4Δ mutant, or complemented strain suspended in physiological saline. Mouse health was monitored twice daily, and moribund mice, defined as mice that were lethargic (mice which could be aroused but relapsed into slumber within 1 min or had repetitive neurological signs such as circling) or were not able to reach food or water, were sacrificed according to the University of Illinois at Chicago IACUC guidelines. Statistical significance was assessed by the Kaplan-Meier test followed by the log rank test using Graph Pad software. Necropsies were performed to isolate infected brains, which were either fixed in 4% phosphate-buffered formalin for histology or homogenized in PBS and centrifuged on a 20 to 100% discontinuous sucrose gradient at 3,000 rpm for 30 min in a swinging bucket rotor to isolate purified yeast cells. Fixed brain tissue was submitted to the University of Illinois veterinary pathology service for embedding, mounting, and staining. Sections of brain were stained with Grocott-Gomori methenamine silver stain to visualize yeast morphology. India ink staining was used to visualize capsule on yeast cells isolated from brains.
RESULTS
Construction of a C. neoformans ccr4Δ mutant.
Our previous study of a C. neoformans DEAD-box RNA helicase gene, VAD1, demonstrated a role for the mRNA degradation machinery in the regulation of cell integrity in C. neoformans (29). To probe further the role of the mRNA degradation machinery in cell integrity, we deleted CCR4 encoding the C. neoformans homologue of the S. cerevisiae gene. CCR4 encodes the poly(A)-specific 3′-to-5′ exoribonuclease responsible for mRNA deadenylation (6, 38). Based on the phenotype of the S. cerevisiae ccr4Δ mutant, we predicted that the C. neoformans mutant would be defective in cell integrity (4). To construct the C. neoformans ccr4Δ mutant, the URA5 selectable marker flanked by a sequence homologous to the genomic sequence directly upstream and downstream of the CCR4 coding sequence was transformed into an ura derivative of wild-type C. neoformans var. grubii strain H99 (H99FOA) by biolistic transformation (Fig. 1A) (37). Confirmation of PCR-positive clones by Southern blotting demonstrated that ccr4Δ-70 was a homologous recombinant (Fig. 1B). The lack of CCR4 transcript in the Δccr4-70 mutant was verified by using a Northern blot probed with a genomic fragment containing the CCR4 coding sequence (Fig. 1C). To ensure that the mutant phenotype was due to the lack of CCR4 and not to an additional, unintended mutation, the ccr4Δ-70 mutant (referred to below as ccr4Δ) was transformed with full-length CCR4 flanked by the hygromycin B resistance cassette by electroporation. Hygromycin-resistant colonies were screened by Southern blotting (data not shown) and Northern blotting (Fig. 1C) and were found to express the CCR4 transcript. One of five independently derived strains with genome-integrated complementation constructs was chosen for further study.
FIG. 1.

(A) Schematic diagram of the construct used to generate the ccr4Δ deletion mutant by homologous recombination. (B) Southern blot analysis of the ccr4Δ mutant demonstrating hybridizing bands of the predicted size for a ccr4Δ homologous recombinant compared to the wild type after digestion with BamHI. (C) Northern blot analysis demonstrating the loss of the CCR4 transcript in the ccr4Δ mutant and restoration in the complemented strain. wt, wild type.
ccr4Δ mutant exhibits cell wall defects.
To determine if Ccr4 regulates cell integrity in C. neoformans, the sensitivity of the ccr4Δ mutant to a panel of cell wall-damaging agents was compared to the sensitivities of the wild type and a complemented strain using a spot plate assay (16). As shown in Fig. 2A, the ccr4Δ mutant was hypersensitive to caffeine, SDS, and the glucan-binding dye Congo red, suggesting that the ccr4Δ mutant is defective in maintenance of cell integrity. To assess the ability of temperature stress to exacerbate the cell integrity defect, the uptake of trypan blue dye was used as a measure of cell integrity at 30 and 37°C. As demonstrated in Fig. 2B, uptake of the dye at 30°C by the ccr4Δ mutant was greater than uptake by the wild type. Whereas the shift to 37°C had no effect on trypan blue uptake by the wild type, increased uptake by the ccr4Δ mutant was exacerbated at the elevated temperature. As an additional measure of cell integrity, the sensitivity of the ccr4Δ mutant to the activity of lysing enzymes was assessed. The ccr4Δ mutant was found to be hypersensitive to lysing enzymes, as evidenced by a more rapid decrease in the OD600 over time, consistent with defects in cell wall architecture (Fig. 2C).
FIG. 2.

(A) Spot plate assay demonstrating the sensitivity of the ccr4Δ mutant to a panel of known cell wall stress inducers, including caffeine (0.5 mg/ml), SDS (0.03%), Congo red (0.5%), and calcofluor white (1.5 mg/ml). Plates were incubated for 3 days at 30°C. (B) Uptake of trypan blue dye in the wild type and the ccr4Δ mutant at mid-log growth in YPD medium at 30°C and after a shift to 37°C for 3 h. One asterisk, P < 0.05; two asterisks, P < 0.0001. (C) Sensitivity of the wild type and the ccr4Δ mutant to lysing enzymes as measured by reduction in the OD600 over time. One asterisk, P < 0.05; two asterisks, P < 0.005; three asterisks, P < 0.001. (D) Spot plate assay demonstrating sensitivity of the ccr4Δ mutant to 32 μg/ml caspofungin. Plates were incubated for 3 days at 30°C. wt, wild type; CFW, calcofluor white.
The echinocandins represent the newest class of antifungal drugs, target cell wall glucan synthesis, and are safe and effective against most ascomycetous pathogenic fungi (8). C. neoformans, however, exhibits a high level of intrinsic resistance to this class of drug (12). The cell integrity defects of the ccr4Δ mutant led us to hypothesize that loss of CCR4 sensitized C. neoformans to the antifungal echinocandin caspofungin. The sensitivity of the ccr4Δ mutant to caspofungin was compared to that of the wild type and that of the complemented strain using the spot plate assay. As demonstrated in Fig. 2D, the ccr4Δ mutant was hypersensitive to 32 μg/ml caspofungin, whereas the wild type and complemented strain were unaffected by this concentration of caspofungin. The caspofungin MICs as measured by broth microdilution were 32 μg/ml for the wild type and 16 μg/ml for the ccr4Δ strain in each of three replicate experiments. This suggests that Ccr4-dependent regulation of cell integrity is important for wild-type resistance to caspofungin.
Growth of the ccr4Δ mutant was found to be attenuated on YPD agar and on Asn salts agar at 37°C (Fig. 3A, left and center panels). The attenuated growth on Asn salts agar was suppressed by addition of sorbitol to the medium, consistent with the temperature sensitivity due to cell integrity defects (Fig. 3A, right panel).
FIG. 3.

(A) Spot plate assay showing the sorbitol-remediable 37°C growth defect of the ccr4Δ mutant on YPD agar (left panel) and on Asn salts agar containing 2% dextrose and 1 mg/ml yeast nitrogen base medium (ASN 2%DEX YNB) with (right panel) and without (center panel) 1 M sorbitol as an osmostabilant. The spots contained 5 μl of a cell suspension at an OD600 of 1.0 and five 1:5 dilutions. The plates were incubated for 3 days at 37°C. (B) Photomicrographs showing cells of the wild type (top) and the ccr4Δ mutant (bottom) grown on Asn salts agar at 37°C stained with calcofluor white. Scale bars = 5 μm. The arrowheads indicate wild-type calcofluor white accumulation at the bud necks. wt, wild type.
The effects of the ccr4Δ mutation on morphology were readily apparent during microscopic observation of cells grown on Asn salts agar at 37°C. Calcofluor white is a chitin-binding aniline dye that has been demonstrated to uniformly stain the cell wall of C. neoformans. As demonstrated in the top row of Fig. 3C, the wild type uniformly binds calcofluor white to the cell wall, with an accumulation of the dye at the bud neck. Calcofluor white staining of the ccr4Δ mutant, however, revealed a lack of uniformity in cell wall staining and heterogeneity in the accumulation of calcofluor white at the bud neck (Fig. 3C, bottom rows). Heterogeneity in calcofluor white staining has been linked to irregularity in chitin deposition (34).
Capsule production is intact in the ccr4Δ mutant.
The capsule of C. neoformans is linked to the cell wall (32). India ink staining revealed that capsule production in the ccr4Δ mutant is similar to capsule production in the wild type despite morphological abnormalities (Fig. 4A). To assess capsular porosity in the ccr4Δ mutant, diffusion of high-molecular-weight dextran and binding of an anticapsular Fab fragment (a generous gift from Tom Kozel) were compared among strains (15). Capsular permeability was found to be similar for the wild type and the ccr4Δ mutant by both TMRD diffusion (Fig. 4B) and Fab fragment 3C2 staining (Fig. 4C), suggesting that defects in cell integrity resulting from CCR4 deletion do not alter capsular porosity. For quantitation of capsular permeability, the distance between the outside of the cell wall and the staining front was measured for 10 cells using Slidebook software. There was no significant difference in the depth of Fab penetration into the capsule between the wild type and the ccr4Δ mutant. The radius of Fab exclusion was 0.9 ± 0.1 μm for the wild type and 0.9 ± 0.1 μm for the ccr4Δ mutant (P = 0.9). The radius of TMRD exclusion was 5.8 ± 0.2 μm for the wild type and 4.7 ± 0.3 μm for the ccr4Δ mutant, representing a small but significant (P < 0.01) difference in TMRD penetration between the strains. As expected, there was no exclusion of TMRD in the cap60Δ strain, and there was no staining of the cap60Δ strain by the Fab fragment, as it lacks the capsular epitope.
FIG. 4.

(A) India ink staining of the wild type and the ccr4Δ mutant grown under capsule-inducing conditions. (B and C) Analysis of capsular permeability by TMRD incorporation (B) and deposition of monoclonal antibody 3C2 Fab fragments (C) assessed 15 and 30 min after staining, respectively. Scale bars = 5 μm. wt, wild type; BF, bright field; CFW, calcofluor white.
MBL binds to the cell wall of the ccr4Δ mutant.
MBL is known to bind to mannose-rich epitopes on the surface of pathogenic fungi, including Candida albicans and Aspergillus fumigatus (28). MBL has been demonstrated to bind to acapsular mutants of C. neoformans, but binding of MBL to wild-type C. neoformans has not been shown (33). To determine if cell wall defects stemming from the loss of CCR4 increased the accessibility of MBL epitopes in the cell wall of C. neoformans, the binding of human serum MBL to the ccr4Δ mutant was compared to the binding to the wild type and the binding to an acapsular cap60Δ mutant. In cells grown to mid-log phase in YPD medium, MBL was most often found to bind to the cell wall of the ccr4Δ mutant in regions that colocalized with regions that exhibit increased calcofluor white staining relative to the wild type (Fig. 5). These regions were often found at the ends of enlarged buds, opposite the bud neck. In contrast, MBL binding was not observed on the cell wall of wild-type cells, where calcofluor white staining was found to be less intense and more uniform. Furthermore, serum-opsonized acapsular cells exhibited MBL staining at the bud neck and on circular bud scars of every cell. This staining pattern was not seen in either the wild type or the ccr4Δ mutant, suggesting that basal capsule production is able to prevent MBL binding to these vulnerable regions of the cell wall in both the wild type and the ccr4Δ mutant. Alternatively, the cap60Δ strain may also exhibit a subtle defect in cell wall architecture, as the cellular function of Cap60 is unknown. Assessment of the cap60Δ strain for sensitivity to the cell wall-damaging agents caffeine, SDS, calcofluor white, and Congo red with the spot plate assay revealed modest sensitivity to SDS (Fig. 2A). No MBL staining was seen on unopsonized controls stained in parallel. Staining of serum-opsonized cells with anti-c-myc monoclonal antibody clone 9E10 (Sigma) as an IgG1 isotype control revealed no fluorescence in addition to the background fluorescence. Enumeration of MBL-positive cells by scoring 100 cells in triplicate revealed that less than 1% of wild-type cells were MBL positive on the cell wall. Nearly 100% of cap60Δ cells were positive (99.7% ± 0.3%), with binding seen only at the bud neck and bud scars. The ccr4Δ mutant exhibited MBL staining on approximately 30% of cells (30.7% ± 0.9%), and binding was often seen as an arc of punctuate fluorescence colocalizing with increased calcofluor white fluorescence on the cell wall opposite the bud neck.
FIG. 5.

Patterns of MBL binding to the wild-type, ccr4Δ, and cap60Δ strains grown to mid-log phase at 30°C in YPD medium, followed by serum opsonization at 37°C. Cells were stained with either anti-MBL monoclonal antibody or anti-c-myc monoclonal antibody as an IgG1 isotype control, followed by an AlexaFluor 488-conjugated goat-anti-mouse IgG secondary antibody (Invitrogen). For the cap60Δ strain, the same cell is shown in two separate focal planes; the top images demonstrate MBL binding to the bud neck, and the bottom images demonstrate MBL binding at the bud scar. Scale bars = 2.5 μm. wt, wild type; BF, bright field; CFW, calcofluor white; mAb, monoclonal antibody.
To investigate the correlation of MBL binding with increased calcofluor white staining, the mean calcofluor white fluorescence intensities were compared for MBL-colocalizing regions and the regions of the cell wall negative for MBL staining in five cells (Fig. 6A). Regions of the ccr4Δ cell wall that colocalized with MBL binding exhibited significantly more intense calcofluor white fluorescence than MBL-negative regions (Fig. 6B). The increased calcofluor white staining in these regions may be indicative of defects in cell wall architecture. However, because MBL did not bind uniformly in regions with increased calcofluor white staining, the MBL epitopes were most likely not uniformly exposed in calcofluor white-accumulating regions. The binding of MBL to the cell wall of the ccr4Δ mutant did suggest that there was exposure of epitopes normally masked in C. neoformans cells with no defects in cell integrity.
FIG. 6.

(A) Colocalization of MBL staining with regions of increased calcofluor white intensity. The average intensity of calcofluor white staining in regions positive for MBL staining was compared to the intensity of calcofluor white staining in MBL-negative regions. Scale bars = 2.5 μm. (B) Graph of pooled data from the five cells shown in panel A (P < 0.0005). (C) Wild-type cells exhibit uniform calcofluor white staining. The average intensity of calcofluor white staining was measured for the half of a bud proximal to the bud scar (P) and for the half of a bud distal to the bud scar (D) for each of five budded wild-type cells. (D) Graph of pooled data from the five cells shown in panel C (P = 0.24). CFW, calcofluor white.
To assess the homogeneity of calcofluor white staining in the wild type, the intensity of calcofluor white staining on the cell wall was measured. The daughter of each of five budded wild-type cells was bisected into a region proximal to the bud neck and a region distal to the bud neck. The mean calcofluor white fluorescence intensity was calculated for each half-bud using SlideBook software (Fig. 6C). Pooled data were analyzed for statistical significance using the Student t test. No significant difference in calcofluor white fluorescence intensity was observed (Fig. 6D) (P = 0.24). This demonstrates that the heterogeneity in calcofluor white staining observed in the ccr4Δ strain is not shared by wild-type C. neoformans.
Caspofungin increases MBL binding to wild-type C. neoformans.
Cell integrity is a common target for antifungal agents. Because MBL binding to the ccr4Δ mutant correlated with caspofungin sensitivity, we investigated whether caspofungin treatment could expose MBL-binding epitopes on the surface of wild-type C. neoformans. Treatment of wild-type C. neoformans for 3 h at 30°C with 30 μg/ml caspofungin increased binding of MBL to the cell wall of wild-type C. neoformans (Fig. 7). Unlike MBL staining of the ccr4Δ mutant, caspofungin-induced staining of the wild type did not coincide with alterations in calcofluor white staining. This may indicate that the structural changes in the cell wall exhibited by the ccr4Δ mutant are different from those induced by caspofungin treatment. Diffuse intracellular green channel fluorescence that colocalized with intracellular blue channel fluorescence was most likely autofluorescence due to the cytotoxic effects of caspofungin. This diffuse fluorescence was present in the isotype control-stained cells that were treated with caspofungin but was absent in cells not treated with caspofungin but stained with either the anti-MBL antibody or the isotype control. Drug-induced binding of MBL to wild-type C. neoformans suggests that antifungals that target cell integrity may expose novel epitopes on the surface of C. neoformans, thereby modulating the host response.
FIG. 7.

Caspofungin potentiates MBL binding to wild-type C. neoformans. (A) Wild-type cells were grown to mid-log phase and incubated for 3 h in the presence of 30 μg/ml caspofungin in YPD medium at 30°C. Cells were washed in PBS, serum opsonized, and stained for MBL binding (top and middle panels) or with a monoclonal isotype control antibody (bottom panels). (B) Serum-opsonized cells of the wild type without caspofungin treatment stained for MBL binding or with an isotype control antibody. Scale bars = 2.5 μm. wt, wild type; BF, bright field; CFW, calcofluor white; mAb, monoclonal antibody.
ccr4Δ mutant has reduced virulence.
To determine the effect of CCR4 deletion on the ability of C. neoformans to cause mortality in a mouse model of cryptococcosis, 1 × 106 cells of the wild type, ccr4Δ mutant, and complemented strain were injected into the lateral tail vein of NIH Swiss albino mice. Mouse health was monitored closely, and moribund mice were sacrificed. Mice inoculated with either the wild type or the complemented strain succumbed to infection within the first week postinoculation (Fig. 8A). However, mice inoculated with the ccr4Δ mutant remained healthy, even with a large inoculum, until the fourth week postinoculation, at which time they succumbed to infection (P < 0.0001, determined using Kaplan-Meier analysis). The brains of three mice that were inoculated with the ccr4Δ mutant were harvested postmortem and plated on YPD agar containing 50 μg/ml chloramphenicol. Phenotypic analysis of the recovered strains demonstrated that the mutant phenotype was retained, suggesting that mortality was not due to spontaneous suppressor mutation (data not shown). India ink preparations of yeast cells recovered from the brains of mice inoculated with the ccr4Δ mutant showed production of capsule in vivo despite severe morphological defects (Fig. 8B). Histological sections from mice that succumbed to infection with the ccr4Δ mutant were stained with Grocott-Gomori methenamine silver stain to investigate the morphology of the ccr4Δ mutant in vivo. The ccr4Δ mutant exhibited cellular morphology similar to that seen during growth at 37°C on asparagine salts agar. The yeast cells were large and had irregular shapes, often with multiple buds (Fig. 8C).
FIG. 8.
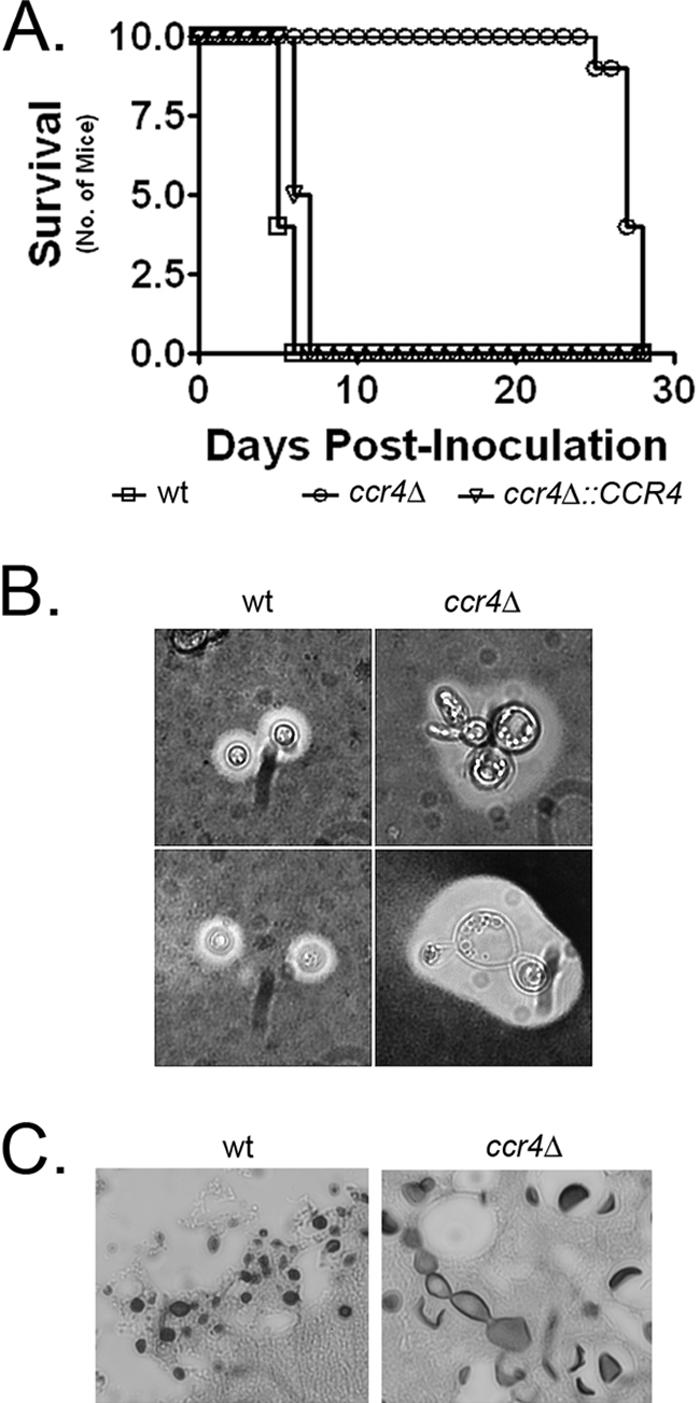
(A) Analysis of the virulence of the ccr4Δ mutant compared to the wild type and the complemented strain in a mouse model of disseminated cryptococcosis with an inoculum of 1 × 106 yeast cells (10 mice per group; P < 0.001). (B) Capsule and cell morphology of wild-type and ccr4Δ mutant cells isolated from the brains of mice at the time of death. (C) Silver-stained paraffin-embedded sections of brains from mice infected with either the wild type or the ccr4Δ mutant. wt, wild type.
DISCUSSION
The lack of MBL binding to encapsulated C. neoformans cells has been attributed mainly to protection by the large polysaccharide capsule (33). Studies have demonstrated quantitatively increased binding of MBL to acapsular strains of C. neoformans compared to the binding to encapsulated strains, which supports the contention that capsule is sufficient to protect C. neoformans from MBL binding to the cell wall. In this study we demonstrated that mature C. neoformans cell wall alone did not bind MBL; rather, MBL bound only to bud necks and bud scars of an acapsular mutant, regions whose structure differs from that of the main body of the cell wall as a result of septum formation and degradation (24). The binding of MBL to the cell wall of the ccr4Δ mutant suggests that maturation of the cell wall is a Ccr4-dependent process and that the mature cell wall serves as a protective barrier against MBL binding to mannoprotein targets of C. neoformans. Indeed, the mannoprotein of C. neoformans has been reported to be either secreted or hidden within the cell wall matrix, which may have evolved as a protective measure against lectin-like molecules produced by plants and other organisms in the environment (26, 40). Interestingly, a mannose binding lectin has recently been demonstrated to be surface localized in Acanthamoeba castellanii and has been shown to be important for virulence in this parasitic pathogen (13). C. neoformans has been shown to parasitize the phagosome of A. castellanii (35). This relationship has been hypothesized to select for maintenance of cryptococcal virulence in the environment (35).
MBL was observed to bind to the cell wall of C. neoformans when cell integrity was perturbed by mutation of CCR4 or addition of caspofungin or in the absence of capsule. These results suggest that integrity of the cell wall architecture of C. neoformans is sufficient to protect the cell from MBL binding. They also suggest that the capsule aids in protection of vulnerable regions accessible to MBL in acapsular strains. Because MBL binding was performed using serum opsonization, other serum factors may promote MBL binding to the ccr4Δ mutant. However, our results are consistent with reported binding of purified MBL to acapsular strains of C. neoformans (33). The role of MBL in the immunopathogenesis of cryptococcosis has yet to be investigated, although MBL-C. neoformans mannoprotein complexes have been shown to increase production of tumor necrosis factor alpha by monocytes (3).
MBL binding in both the ccr4Δ and cap60Δ strains correlated with accumulation of calcofluor white. In the ccr4Δ strain this colocalization occurred at the ends of enlarged buds, whereas in the cap60Δ strain the colocalization occurred at the bud neck. The correlation of MBL binding to increased calcofluor white accumulation is likely indicative of differences in cell wall architecture in the more uniform regions of the cell wall and may not be directly related to the exposure of MBL epitopes. This possibility is suggested by the potentiation of MBL binding to wild-type C. neoformans by caspofungin treatment that did not correlate with increased calcofluor white accumulation. It is unclear whether the accumulation of calcofluor white is due to increased chitin levels, decreased glucan levels, altered levels of mannoprotein, or defects in the assembly of cross-linked polymers. Future studies will investigate the structural defect in the ccr4Δ cell wall and will identify MBL binding components that could be potential immunogens.
In regions of sub-Saharan Africa, infection by C. neoformans has emerged as the most common cause of meningitis (1, 19). MBL deficiencies have been demonstrated to increase susceptibility to HIV infection, but a relationship between MBL levels and/or polymorphism and susceptibility to cryptococcal infection has not been investigated (14). Interestingly, an MBL polymorphism that results in malformation of the MBL oligomer has been demonstrated to be unique to regions of sub-Saharan Africa, correlating geographically with a high prevalence of cryptococcosis in HIV-infected populations (1, 27).
The cell integrity-defective ccr4Δ mutant was found to be sensitive to caspofungin, an echinocandin antifungal to which C. neoformans exhibits intrinsic resistance (12). Cell integrity is currently the target of the majority of antifungal agents in use. Caspofungin targets cell wall glucan synthesis, whereas the azoles and amphotericin B target cell membrane sterols. This raises the possibility that antifungal agents that target cell integrity could work in synergy with soluble innate immune mediators to eradicate infection. In a clinical scenario of antifungal failure, not only might the antifungal susceptibility of the pathogen be considered a factor, but polymorphisms in innate immune mediators such as MBL might also be considered factors.
In addition to characterization of MBL binding to C. neoformans, these results demonstrate that Ccr4 contributes to the pathogenesis of cryptococcosis by allowing C. neoformans to thrive at the host temperature. This report suggests a role for Ccr4 and the mRNA degradation pathway in the adaptation of C. neoformans to host temperature. Studies to delineate the contribution of targets of Ccr4 to cell integrity have the potential to identify structural genes involved in cell wall remodeling and synthesis, as well as potential targets for novel antifungal therapies.
Acknowledgments
We acknowledge Tom Kozel for providing the Fab fragments used in this study. We acknowledge the following institutions for access to C. neoformans genome sequence information: The Institute for Genome Research (TIGR), the C. neoformans H99 sequencing project, Duke IGSP Center for Applied Genomics and Technology (http://cgt.duke.edu/), and the Genome Sequence Centre, BC Cancer Research Centre (www.bcgsc.bc.ca).
This work was supported in part by NRSA fellowship grant AI062124 to J.C.P. and by Public Health Service grants AI045995 and AI049371 to P.R.W.
Editor: A. Casadevall
Footnotes
Published ahead of print on 23 July 2007.
REFERENCES
- 1.Bicanic, T., and T. S. Harrison. 2004. Cryptococcal meningitis. Br. Med. Bull. 72:99-118. [DOI] [PubMed] [Google Scholar]
- 2.Buchanan, K. L., and J. W. Murphy. 1998. What makes Cryptococcus neoformans a pathogen? Emerg. Infect. Dis. 4:71-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaka, W., A. F. Verheul, V. V. Vaishnav, R. Cherniak, J. Scharringa, J. Verhoef, H. Snippe, and A. I. Hoepelman. 1997. Induction of TNF-alpha in human peripheral blood mononuclear cells by the mannoprotein of Cryptococcus neoformans involves human mannose binding protein. J. Immunol. 159:2979-2985. [PubMed] [Google Scholar]
- 4.Chang, M., D. French-Cornay, H. Y. Fan, H. Klein, C. L. Denis, and J. A. Jaehning. 1999. A complex containing RNA polymerase II, Paf1p, Cdc73p, Hpr1p, and Ccr4p plays a role in protein kinase C signaling. Mol. Cell. Biol. 19:1056-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chayakulkeeree, M., and J. R. Perfect. 2006. Cryptococcosis. Infect. Dis. Clin. N. Am. 20:507-544, v-vi. [DOI] [PubMed] [Google Scholar]
- 6.Chen, J., Y. C. Chiang, and C. L. Denis. 2002. CCR4, a 3′-5′ poly(A) RNA and ssDNA exonuclease, is the catalytic component of the cytoplasmic deadenylase. EMBO J. 21:1414-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colley, K. J., M. C. Beranek, and J. U. Baenziger. 1988. Purification and characterization of the core-specific lectin from human serum and liver. Biochem. J. 256:61-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denning, D. W. 2003. Echinocandin antifungal drugs. Lancet 362:1142-1151. [DOI] [PubMed] [Google Scholar]
- 9.Dong, M., S. Xu, C. L. P. Oliveira, J. S. Pedersen, S. Thiel, F. Besenbacher, and T. Vorup-Jensen. 2007. Conformational changes in mannan-binding lectin bound to ligand surfaces. J. Immunol. 178:3016-3022. [DOI] [PubMed] [Google Scholar]
- 10.Dromer, F., S. Mathoulin-Pelissier, O. Launay, and O. Lortholary. 2007. Determinants of disease presentation and outcome during cryptococcosis: the CryptoA/D study. PLoS Med. 4:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eisen, D. P., and R. M. Minchinton. 2003. Impact of mannose-binding lectin on susceptibility to infectious diseases. Clin. Infect. Dis. 37:1496-1505. [DOI] [PubMed] [Google Scholar]
- 12.Feldmesser, M., Y. Kress, A. Mednick, and A. Casadevall. 2000. The effect of the echinocandin analogue caspofungin on cell wall glucan synthesis by Cryptococcus neoformans. J. Infect. Dis. 182:1791-1795. [DOI] [PubMed] [Google Scholar]
- 13.Garate, M., I. Cubillos, J. Marchant, and N. Panjwani. 2005. Biochemical characterization and functional studies of Acanthamoeba mannose-binding protein. Infect. Immun. 73:5775-5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garred, P., C. Richter, A. B. Andersen, H. O. Madsen, I. Mtoni, A. Svejgaard, and J. Shao. 1997. Mannan-binding lectin in the sub-Saharan HIV and tuberculosis epidemics. Scand. J. Immunol. 46:204-208. [DOI] [PubMed] [Google Scholar]
- 15.Gates, M. A., P. Thorkildson, and T. R. Kozel. 2004. Molecular architecture of the Cryptococcus neoformans capsule. Mol. Microbiol. 52:13-24. [DOI] [PubMed] [Google Scholar]
- 16.Gerik, K. J., M. J. Donlin, C. E. Soto, A. M. Banks, I. R. Banks, M. A. Maligie, C. P. Selitrennikoff, and J. K. Lodge. 2005. Cell wall integrity is dependent on the PKC1 signal transduction pathway in Cryptococcus neoformans. Mol. Microbiol. 58:393-408. [DOI] [PubMed] [Google Scholar]
- 17.Gordon, A. C., U. Waheed, T. K. Hansen, G. A. Hitman, C. S. Garrard, M. W. Turner, N. J. Klein, S. J. Brett, and C. J. Hinds. 2006. Mannose-binding lectin polymorphisms in severe sepsis: relationship to levels, incidence, and outcome. Shock 25:88-93. [DOI] [PubMed] [Google Scholar]
- 18.Granell, M., A. Urbano-Ispizua, B. Suarez, M. Rovira, F. Fernandez-Aviles, C. Martinez, M. Ortega, C. Uriburu, A. Gaya, J. M. Roncero, A. Navarro, E. Carreras, J. Mensa, J. Vives, C. Rozman, E. Montserrat, and F. Lozano. 2006. Mannan-binding lectin pathway deficiencies and invasive fungal infections following allogeneic stem cell transplantation. Exp. Hematol. 34:1435-1441. [DOI] [PubMed] [Google Scholar]
- 19.Hakim, J. G., I. T. Gangaidzo, R. S. Heyderman, J. Mielke, E. Mushangi, A. Taziwa, V. J. Robertson, P. Musvaire, and P. R. Mason. 2000. Impact of HIV infection on meningitis in Harare, Zimbabwe: a prospective study of 406 predominantly adult patients. AIDS 14:1401-1407. [DOI] [PubMed] [Google Scholar]
- 20.Holmskov, U., and J. C. Jensenius. 1993. Structure and function of collectins: humoral C-type lectins with collagenous regions. Behring Inst. Mitt. 93:224-235. [PubMed] [Google Scholar]
- 21.Ikeda, K., T. Sannoh, N. Kawasaki, T. Kawasaki, and I. Yamashina. 1987. Serum lectin with known structure activates complement through the classical pathway. J. Biol. Chem. 262:7451-7454. [PubMed] [Google Scholar]
- 22.Jensen, P. H., D. Weilguny, F. Matthiesen, K. A. McGuire, L. Shi, and P. Hojrup. 2005. Characterization of the oligomer structure of recombinant human mannan-binding lectin. J. Biol. Chem. 280:11043-11051. [DOI] [PubMed] [Google Scholar]
- 23.Kaur, S., V. K. Gupta, S. Thiel, P. U. Sarma, and T. Madan. 2007. Protective role of mannan-binding lectin in a murine model of invasive pulmonary aspergillosis. Clin. Exp. Immunol. 148:382-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lesage, G., and H. Bussey. 2006. Cell wall assembly in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 70:317-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lillegard, J. B., R. B. Sim, P. Thorkildson, M. A. Gates, and T. R. Kozel. 2006. Recognition of Candida albicans by mannan-binding lectin in vitro and in vivo. J. Infect. Dis. 193:1589-1597. [DOI] [PubMed] [Google Scholar]
- 26.Mansour, M. K., L. S. Schlesinger, and S. M. Levitz. 2002. Optimal T cell responses to Cryptococcus neoformans mannoprotein are dependent on recognition of conjugated carbohydrates by mannose receptors. J. Immunol. 168:2872-2879. [DOI] [PubMed] [Google Scholar]
- 27.Mombo, L. E., C. Y. Lu, S. Ossari, I. Bedjabaga, L. Sica, R. Krishnamoorthy, and C. Lapoumeroulie. 2003. Mannose-binding lectin alleles in sub-Saharan Africans and relation with susceptibility to infections. Genes Immun. 4:362-367. [DOI] [PubMed] [Google Scholar]
- 28.Neth, O., D. L. Jack, A. W. Dodds, H. Holzel, N. J. Klein, and M. W. Turner. 2000. Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect. Immun. 68:688-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Panepinto, J., L. Liu, J. Ramos, X. Zhu, T. Valyi-Nagy, S. Eksi, J. Fu, H. A. Jaffe, B. Wickes, and P. R. Williamson. 2005. The DEAD-box RNA helicase Vad1 regulates multiple virulence-associated genes in Cryptococcus neoformans. J. Clin. Investig. 115:632-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petersen, K. A., F. Matthiesen, T. Agger, L. Kongerslev, S. Thiel, K. Cornelissen, and M. Axelsen. 2006. Phase I safety, tolerability, and pharmacokinetic study of recombinant human mannan-binding lectin. J. Clin. Immunol. 26:465-475. [DOI] [PubMed] [Google Scholar]
- 31.Petrou, M. A., and D. C. Shanson. 2000. Susceptibility of Cryptococcus neoformans by the NCCLS microdilution and Etest methods using five defined media. J. Antimicrob. Chemother. 46:815-818. [DOI] [PubMed] [Google Scholar]
- 32.Reese, A. J., A. Yoneda, J. A. Breger, A. Beauvais, H. Liu, C. L. Griffith, I. Bose, M.-J. Kim, C. Skau, S. Yang, J. A. Sefko, M. Osumi, J.-P. Latge, E. Mylonakis, and T. L. Doering. 2007. Loss of cell wall alpha(1-3) glucan affects Cryptococcus neoformans from ultrastructure to virulence. Mol. Microbiol. 63:1385-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schelenz, S., R. Malhotra, R. B. Sim, U. Holmskov, and G. J. Bancroft. 1995. Binding of host collectins to the pathogenic yeast Cryptococcus neoformans: human surfactant protein D acts as an agglutinin for acapsular yeast cells. Infect. Immun. 63:3360-3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimizu, J., Y. Okumura, K. Yoda, and M. Yamasaki. 1997. A glutamine synthetase mutant of Saccharomyces cerevisiae shows defect in cell wall. J Gen. Appl. Microbiol. 43:157-162. [DOI] [PubMed] [Google Scholar]
- 35.Steenbergen, J. N., H. A. Shuman, and A. Casadevall. 2001. Cryptococcus neoformans interactions with amoebae suggest an explanation for its virulence and intracellular pathogenic strategy in macrophages. Proc. Natl. Acad. Sci. USA 98:15245-15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thiel, S., T. Vorup-Jensen, C. M. Stover, W. Schwaeble, S. B. Laursen, K. Poulsen, A. C. Willis, P. Eggleton, S. Hansen, U. Holmskov, K. B. M. Reid, and J. C. Jensenius. 1997. A second serine protease associated with mannan-binding lectin that activates complement. Nature 386:506-510. [DOI] [PubMed] [Google Scholar]
- 37.Toffaletti, D. L., T. H. Rude, S. A. Johnston, D. T. Durack, and J. R. Perfect. 1993. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J. Bacteriol. 175:1405-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tucker, M., R. R. Staples, M. A. Valencia-Sanchez, D. Muhlrad, and R. Parker. 2002. Ccr4p is the catalytic subunit of a Ccr4p/Pop2p/Notp mRNA deadenylase complex in Saccharomyces cerevisiae. EMBO J. 21:1427-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van de Wetering, J. K., L. M. G. van Golde, and J. J. Batenburg. 2004. Collectins. Players of the innate immune system. Eur. J. Biochem. 271:1229-1249. [DOI] [PubMed] [Google Scholar]
- 40.Vartivarian, S. E., G. H. Reyes, E. S. Jacobson, P. G. James, R. Cherniak, V. R. Mumaw, and M. J. Tingler. 1989. Localization of mannoprotein in Cryptococcus neoformans. J. Bacteriol. 171:6850-6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang, H., G. Zhou, L. Zhi, H. Yang, Y. Zhai, X. Dong, X. Zhang, X. Gao, Y. Zhu, and F. He. 2005. Association between mannose-binding lectin gene polymorphisms and susceptibility to severe acute respiratory syndrome coronavirus infection. J. Infect. Dis. 192:1355-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]