

Abstract
A scale of relative gas-phase NO+ binding energies (BEs) has been constructed by evaluation of NO+-transfer equilibria L1NO+ + L2 ⇌ L2NO+ + L1 by Fourier-transform ion cyclotron resonance mass spectrometry and by application of the kinetic method, based on the metastable fragmentation of L1(NO+)L2 nitryl-ion bound dimers. The relative scale, anchored to the NO+ affinity of water, for 52 ligands, including alkyl halides, alkyl nitrates, alcohols, nitroalkanes, nitriles, aldehydes, ketones, and aromatic and heterocyclic compounds, led to an absolute NO+ affinity scale. The results are compared with those of an earlier study, and the apparent discrepancies are traced to a different choice of the absolute BE value used as the reference standard. The NO+ BEs fit a satisfactorily linear correlation when plotted versus the corresponding proton affinities (PAs). The NO+ BEs, while much lower than the PAs, are nevertheless higher than the corresponding BEs of the strictly related NO2+ cation, a result consistent with the experimental and theoretical results currently available on the structure and the stability of NO+ and NO2+ complexes. The NO+ BE vs. PA correlation allows one to estimate within 1–2 kcal·mol−1 the NO+ BE of the molecules included in the comprehensive PA compilations currently available. For example, the correlation gives the following NO+ affinities of the DNA bases, in kcal·mol−1 (1 kcal = 4.18 kJ): adenine, 40.3; cytosine, 40.4; guanine, 40.1; and thymine, 34.9. The experimental NO+ BE of thymine, the only one accessible to direct measurement, amounts to 35.6 ± 2 kcal·mol−1, which underlines the predictive value of the correlation. This study reports the second successful extension of the kinetic method to the evaluation of the absolute BEs of polyatomic cations, following our recent application to the strictly related NO2+ ion.
The chemistry of the nitryl ion, NO+, is the focus of active interest, recently heightened by the extraordinary multiplicity of roles currently attributed to NO. Nitryl ion, its salts and carriers, are long known as effective reagents in electrophilic nitrosation, whose study is the subject of continuing interest (refs. 1–4 and references cited in ref. 2), in particular as concerns the formation of nitrosoamines (5, 6). Passing to atmospheric chemistry, the low ionization potential of NO makes NO+ an effective “charge sink” in ionized air, and the promoter of a reaction chain eventually leading to hydrated-proton clusters (7, 8). As the consequence, complexation of NO+ by H2O and other atmospheric species (N2, CO2, O2) has a direct bearing on the chemistry of the ionospheric D region (9–11) and of the middle atmosphere (12). The biological and physiological significance of NO+ and of its complexes has received much attention, in connection with the recognized impact on human health of a variety of compounds important for dietary or environmental reasons, including nitrites, N-nitrosoamines, NOx oxides, etc. (13–15). For example, it has been suggested that NO+, or some NO+ sources present in biological systems, can be involved in the neurotoxic and neuroprotective actions of NO (16, 17) as well as in the nitrous acid-promoted crosslinking of DNA, a problem of considerable current interest (18, 19). Finally, the use of NO+ as a reagent has a considerable potential for trace gas analysis (20).
A quantitative knowledge of the interactions of NO+ with neutral ligands is central to the understanding and modelling of important problems in many research fields. This strongly suggests a systematic study of the binding energies (BEs) of the nitryl cation to representative ligands, to be carried preferably in the gas phase, to obtain results of general validity, unaffected by specific ion–solvent interactions and hence more directly comparable to those from theoretical approaches and more widely useful for modelling purposes. The interest of the problem was perceived as early as in 1980 by Reents and Freiser (21), who measured the NO+ BE to 28 ligands by ion cyclotron resonance (ICR) mass spectrometry. In view of the current upsurge of interest in the NO+ complexes, we have undertaken a systematic study aimed at the revision of the NO+ BE scale, taking into account the changes undergone in the meantime by certain reference standards used in the early study and the availability of new experimental tools. However, the principal motivation of this work is to be found in the attempt to extend the NO+ BE scale to a significantly larger number of ligands, including molecules of great biochemical relevance, such as the DNA bases. To this end, we have applied the ICR equilibrium method, largely used in gas-phase proton affinity (PA) measurements (22) and successfully extended to NO2+ transfer reactions (23), complemented by the kinetic method (24). The latter, so far restricted to monoatomic ions, at least as concerns the measurement of absolute BEs, has recently been extended to the construction of an absolute BE scale of a triatomic species, the NO2+ cation (23).
EXPERIMENTAL
All chemicals were research grade products obtained from Aldrich and were used without further purification. Methyl nitrite was synthesized and purified according to standard procedures. The gases were purchased from Matheson Gas Products with a stated purity in excess of 99.95 mol %. Mass analyzed ion kinetic energy (MIKE) spectra were recorded using a ZAB-2F mass spectrometer from VG Micromass. Typical operation conditions were as follows: electron energy 50 eV, repeller voltage 0 V, emission current 0.5 mA, accelerating voltage 8 kV. The chemical ionization spectra were recorded by utilizing a specially built cooling system, capable of thermostatting the source at temperatures not exceeding 50°C, upon ionization of gaseous mixtures of NO and the two ligands, whose composition was optimized to obtain the highest abundance of the L1(NO+)L2 dimers. The Fourier transform (FT)-ICR experiments were performed in a 47e APEX spectrometer from Bruker Spectrospin, equipped with an external ion source, operated at a total pressure not exceeding 7·10−5 torr (1 torr = 133 Pa). To prevent errors arising from the different response of the ionization manometer of the spectrometer to different compounds, premixed gaseous mixtures, obtained by weighed amounts of the reactants, were employed. When compounds of low volatility were used, the pressure readings were corrected for the different response of the ionization gauge to different gases according to the empirical method based on molecular polarizabilities (25). Each equilibrium was evaluated by at least three separate measurements.
RESULTS
The ICR Equilibrium Method.
Measurement of the equilibrium constants of the ligand-exchange reaction
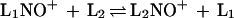 |
1 |
has allowed evaluation of the corresponding ΔG°1 changes. The nitrosating agent, generally nitrosated methyl nitrite (CH3O(NO)2)+, or protonated nitrous acid, (H2O-NO)+ in the case of low nitryl ion-affinity ligands, was produced in the external chemical ionization ion source of an FT-ICR mass spectrometer, driven into the resonance cell, isolated by selective-ejection techniques, and allowed to react with the ligands, contained in a known molar ratio in the cell, where equilibrium 1 was established. The NO+-transfer reaction was in most cases the only, and in all cases the predominant, reaction, the interference of undesired processes being much less vexing than in other equilibria—e.g., those involving NO2+ transfer. The main limitation of the equilibrium method arises from the low volatility and/or the lack of thermal stability of certain ligands which makes it difficult to evaluate their actual concentration from total-pressure measurements whose accuracy can be heavily affected by the presence of unknown amounts of volatile decomposition products.
The Kinetic Method.
In the case of interest, the application of the kinetic method is based on the mass analyzed ion kinetic energy (MIKE) spectrometry of L1(NO+)L2 nitryl ion-bound dimers obtained by NO chemical ionization of mixtures of the ligands. A systematic investigation has shown that the vast majority of the (L1, L2, NO)+ clusters undergo metastable fragmentation exclusively according to the competing processes
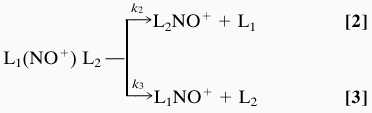 |
consistent with the L1(NO+)L2 nitryl ion-bound dimer structure. Under the assumptions customarily applied (24) one obtains the expression
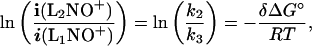 |
where i(L2NO+) and i(L1NO+) are the experimentally measured intensities of the fragments, δΔG° = ΔG°2 − ΔG°3 corresponds to the standard free energy change ΔG°1 of the nitryl ion-transfer reaction (1) and T represents the effective temperature of the dimers that undergo metastable dissociation in the time window accessible to observation. According to a well-established practice (24), T is deduced from the slope of a calibration plot based on a set of independently known ΔG° values. To this end, 12 pairs of ligands, henceforth denoted reference pairs (RPs), have been selected, whose L1(NO+)L2 dimers undergo extensive and clean metastable decompositions, and whose ΔG°1 changes had been measured independently by the equilibrium method. From the satisfactorily linear (correlation coefficient 0.9933) plot of ln(k2/k3) vs. the known δΔG° differences of the RPs one obtains T = 223 K, which has been used to calculate the ΔG°1 changes reported in Table 1, together with the corresponding values from the equilibrium method. The results from the two different methods are remarkably consistent, in that the discrepancies do not exceed 0.5 kcal·mol−1, their mean value being as low as 0.2 kcal·mol−1 (1 kcal = 4.18 kJ). Whereas neither the equilibrium method nor the kinetic method allows direct evaluation of δΔG° differences exceeding 2 kcal·mol−1, the results of the individual measurements can be combined to construct a ladder that spans the whole range investigated, more than 20 kcal·mol−1 (Fig. 1). Under the assumption that ΔG°1 ∼ ΔH°1, the free-energy scale from the fixed-temperature equilibrium and kinetic measurements approximates a relative BE scale that requires an independently known anchoring value to be converted into an absolute scale. We have adopted the H2O–NO+ BE of 18.5 ± 1.5 kcal·mol−1 from a direct measurement performed with a pulsed high-pressure ion source mass spectrometer (26). The choice has been suggested by the consideration that the results from this approach are independent of the possible changes undergone by the accepted values of certain reference data, such as the PA value of NH3, utilized in other indirect approaches. Furthermore, the value chosen agrees within 1 kcal·mol−1 with the BE reported from an ab initio calculation performed at the MP4(SDTQ)//6–311G**/MP2(FU)//6–31G** + ZPE (MP2(FU)/6–31G**) level of theory (27). Anchoring the relative scale of Fig. 1 to the H2O–NO+ BE, one obtains the absolute BE values listed in Table 2. The internal consistency of the ΔG°1 ladder, strengthened by multiple interlacing of its steps, is better than 0.3 kcal·mol−1. A larger uncertainty is introduced by taking ΔG°1 ≅ ΔH°1, as required to convert single-temperature equilibrium data into a BE scale, according to a practice largely adopted in the evaluation of gas-phase PAs (22). As discussed in the related study of NO2+ complexes (23), we conservatively estimate the uncertainty introduced by the above approximation to be generally below 0.8 kcal·mol−1, although there are reasons to believe that ΔG° changes closely approximate ΔH° changes in NO+ transfer equilibria (21). By combining the experimental scatter of the data with the uncertainty arising from taking ΔG° ≅ ΔH°, we estimate that the overall internal consistency of the scale is of the order of ± 1.2 kcal·mol−1. Evaluating the absolute accuracy of the BE values is more difficult. Apart from the error bar of the anchor, ±1.5 kcal·mol−1 (26), one is faced with the inherent difficulty of a rigorous analysis of the error propagation along the ladders of the scale, each interlaced with many others. Probably the only viable, and so far the only reported, criterion is that applied to the construction of a recent 77-ladders PA scale (28), namely the degree of agreement of the results with those from independent approaches, in particular high-level ab initio calculations. We note that in the five cases, well distributed along the scale, where comparison is possible the agreement with theoretically calculated NO+ BEs is better than ± 2 kcal·mol−1, which we take as an approximate measure of the absolute uncertainty of the data of Table 2.
Table 1.
ΔG° changes from the equilibrium and the kinetic method
| L1 | L2 | −ΔG°300,
kcal·mol−1
|
L1 | L2 | −ΔG°300,
kcal·mol−1
|
||
|---|---|---|---|---|---|---|---|
| Equilibrium method | Kinetic method | Equilibrium method | Kinetic method | ||||
| H2O | CH3Cl | ≅ | t-C4H9NO2 | C6H5NO2 | ≅ | ≅ | |
| CH3Cl | CH2(CN)2 | 1.1 | C6H5CN | C6H5NO2 | 0.7 | ||
| CH3Cl | C2H5Cl | 1.1 | t-C4H9CN | C6H5NO2 | 0.6 | ||
| H2O | C2H5Cl | 1.1 | i-C3H7NO2 | C6H5NO2 | 1.7 | ||
| C2H5Cl | CH3ONO2 | 1.3 | n-C4H9NO2 | C6H5NO2 | 1.8 | ||
| CH2(CN)2 | CH3ONO2 | 1.0 | C6H5NO2 | o-CH3C6H4NO2 | 1.2 | 0.9 | |
| CH3ONO2 | i-C3H7Cl | 0.6 | 0.4 | t-C4H9NO2 | o-CH3C6H4NO2 | 1.1 | |
| C2H5Cl | i-C3H7Cl | 2.1 | C6H5NO2 | CH3COCH3 | 1.4 | 1.5 | |
| i-C3H7Cl | C2H5ONO2 | 1.3 | RP | CH3COCH3 | C6H6 | ≅ | 0.3 |
| CH3ONO2 | C2H5ONO2 | 1.8 | 1.8 | C6H6 | CH3COOC2H5 | 0.3 | ≅ |
| C2H5ONO2 | CH3OH | 0.6 | CH3COCH3 | CH3COOC2H5 | 0.3 | RP | |
| CH3OH | i-C3H7ONO2 | 1.0 | 1.2 | C6H5NO2 | CH3COOC2H5 | 1.7 | |
| C2H5ONO2 | i-C3H7ONO2 | 1.6 | RP | CH3COOC2H5 | CH3COC2H5 | 0.7 | |
| i-C3H7ONO2 | CH3NO2 | 0.6 | 0.3 | C6H6 | CH3COC2H5 | 1.2 | 0.7 |
| CH3OH | CH3NO2 | 1.6 | CH3COCH3 | CH3COC2H5 | 1.0 | 1.0 | |
| CH3NO2 | CH3CN | 1.7 | o-CH3C6H4NO2 | 4-NO2m-xylene | 1.5 | ||
| i-C3H7ONO2 | CH3CN | 1.8 | CH3COC2H5 | (C2H5)2CO | 0.9 | 0.6 | |
| (CH2)2O | C2H5NO2 | 0.1 | RP | CH3COOC2H5 | (C2H5)2CO | 1.0 | |
| CH3CN | C2H5NO2 | 0.3 | 0.7 | CH3COC2H5 | i-C3H7COCH3 | 1.0 | RP |
| CH3NO2 | C2H5NO2 | 1.9 | RP | CH3COOC2H5 | i-C3H7COCH3 | 1.5 | |
| C2H5NO2 | C2H5CN | 0.5 | i-C3H7COCH3 | CH3COt-C4H9 | 0.6 | RP | |
| CH3CN | C2H5CN | 1.4 | 1.6 | (C2H5)2CO | CH3COt-C4H9 | 0.8 | 0.7 |
| C2H5CN | CH3CHO | ≅ | i-C3H7COCH3 | CH3COOt-C4H9 | 0.8 | 0.7 | |
| C2H5NO2 | CH3CHO | 0.7 | 0.5 | CH3COC2H5 | CH3COOt-C4H9 | 1.6 | RP |
| CH3CHO | n-C3H7NO2 | 0.0 | RP | CH3COOt-C4H9 | (t-C4H9)2CO | ≅ | |
| C2H5CN | n-C3H7NO2 | ≅ | CH3COt-C4H9 | (t-C4H9)2CO | ≅ | ||
| C2H5NO2 | n-C3H7NO2 | 0.8 | RP | i-C3H7COCH3 | (t-C4H9)2CO | 0.7 | |
| (CH2)2O | n-C3H7NO2 | 0.9 | RP | (t-C4H9)2CO | (i-C3H7)2CO | ≅ | |
| CH3CN | n-C3H7NO2 | 1.3 | 1.6 | CH3COOt-C4H9 | (i-C3H7)2CO | ≅ | |
| n-C3H7NO2 | n-C4H9NO2 | 0.2 | 0.2 | t-C4H9COCH3 | (i-C3H7)2CO | 0.7 | 0.3 |
| C2H5CN | n-C4H9NO2 | 0.1 | 0.5 | (i-C3H7)2CO | C6H5CH3 | 0.3 | |
| C2H5NO2 | n-C4H9NO2 | 1.0 | 1.3 | (t-C4H9)2CO | C6H5CH3 | 0.6 | |
| n-C4H9NO2 | n-C3H7CN | ≅ | CH3COOt-C4H9 | C6H5CH3 | 1.0 | 0.9 | |
| n-C3H7NO2 | n-C3H7CN | 0.3 | CH3COt-C4H9 | C6H5CH3 | 1.0 | 0.9 | |
| C2H5CN | n-C3H7CN | 0.3 | 0.4 | C6H5CH3 | C6H5COCH3 | 1.0 | 0.5 |
| n-C3H7CN | i-C3H7NO2 | ≅ | (i-C3H7)2CO | C6H5COCH3 | 0.7 | ||
| n-C4H9NO2 | i-C3H7NO2 | 0.4 | ≅ | (t-C4H9)2CO | C6H5COCH3 | 1.0 | |
| C2H5NO2 | i-C3H7NO2 | 1.6 | C6H5COCH3 | C6H5C2H5 | 0.2 | ≅ | |
| i-C3H7NO2 | i-C3H7CN | 0.3 | ≅ | (i-C3H7)2CO | C6H5C2H5 | 1.0 | |
| n-C3H7NO2 | i-C3H7CN | 0.9 | 0.6 | (t-C4H9)2CO | C6H5C2H5 | 1.2 | |
| CH3CHO | i-C3H7CN | 0.7 | CH3COOtC4H9 | C6H5C2H5 | 1.6 | 1.5 | |
| C2H5CN | i-C3H7CN | 0.8 | C6H5COCH3 | Thymine | 0.5 | ||
| i-C3H7CN | C6H5F | ≅ | C6H5C2H5 | (C3H5)2CO | 1.1 | 1.0 | |
| CH3CHO | C6H5F | 0.9 | C6H5COCH3 | (C3H5)2CO | 1.4 | ||
| (CH2)2O | C6H5F | 1.9 | (C3H5)2CO | CH3COC(OCH3)2CH3 | 0.5 | ||
| i-C3H7CN | t-C4H9CN | 0.8 | 0.5 | CH3COC(OCH3)2CH3 | 3-FC5H4N | 0.3 | |
| n-C3H7CN | t-C4H9CN | 1.2 | 0.9 | 3-FC5H4N | (C6H5)2CO | 0.8 | |
| t-C4H9CN | C6H5CN | ≅ | ≅ | CH3COC(OCH3)2CH3 | (C6H5)2CO | 1.4 | |
| i-C3H7CN | C6H5CN | 0.8 | 0.5 | (C3H5)2CO | (C6H5)2CO | 1.8 | |
| n-C3H7CN | C6H5CN | 0.8 | (C6H5)2CO | 4-ClC5H4N | 0.5 | ||
| n-C3H7NO2 | C6H5CN | 1.3 | 4-ClC5H4N | (C6H5CH2)2CO | 0.8 | ||
| C2H5CN | C6H5CN | 1.6 | (C6H5)2CO | (C6H5CH2)2CO | 1.2 | ||
| t-C4H9CN | t-C4H9NO2 | 0.3 | (C6H5CH2)2CO | CH3CON(CH3)2 | 0.2 | ||
| i-C3H7CN | t-C4H9NO2 | 1.3 | RP | 4-ClC5H4N | CH3CON(CH3)2 | 0.8 | |
| i-C3H7NO2 | t-C4H9NO2 | 1.6 | 1.2 | CH3CON(CH3)2 | C5H5N | 0.4 | |
| t-C4H9NO2 | o-CH3C6H4CN | ≅ | (C6H5CH2)2CO | C5H5N | 0.7 | ||
| C6H5CN | o-CH3C6H4CN | 0.5 | 0.4 | C5H5N | 3-CH3C5H4N | 1.0 | |
| t-C4H9CN | o-CH3C6H4CN | 0.3 | 3-CH3C5H4N | CH3CON(C2H5)2 | 0.4 | ||
| i-C3H7CN | o-CH3C6H4CN | 1.1 | C5H5N | CH3CON(C2H5)2 | 1.5 | ||
| o-CH3C6H4CN | C6H5NO2 | ≅ | |||||
RP denotes the reference pairs; i-, iso-. ΔG° differences below the experimental error are indicated by the notation ≅.
Figure 1.

ΔG° ladder for NO+ transfer reaction for the ligand pairs investigated. The symbol ≅ denotes ΔG° < 0.2 kcal·mol−1.
Table 2.
NO+ BE of selected ligands, L
| No. | L | BE, kcal·mol−1 |
|---|---|---|
| 1 | H2O | 18.5* |
| 2 | CH3Cl | 18.5 |
| 3 | CH2(CN)2 | 19.6 |
| 4 | C2H5Cl | 19.6 |
| 5 | CH3ONO2 | 20.7 |
| 6 | i-C3H7Cl | 21.4 |
| 7 | C2H5ONO2 | 22.7 |
| 8 | CH3OH | 23.3 |
| 9 | i-C3H7ONO2 | 24.3 |
| 10 | CH3NO2 | 24.9 |
| 11 | CH3CN | 26.4 |
| 12 | (CH2)2O | 26.9 |
| 13 | C2H5NO2 | 26.9 |
| 14 | C2H5CN | 27.6 |
| 15 | CH3CHO | 27.6 |
| 16 | n-C3H7NO2 | 27.7 |
| 17 | n-C4H9NO2 | 27.9 |
| 18 | n-C3H7CN | 28.0 |
| 19 | i-C3H7NO2 | 28.2 |
| 20 | i-C3H7CN | 28.4 |
| 21 | C6H5F | 28.6 |
| 22 | t-C4H9CN | 29.0 |
| 23 | C6H5CN | 29.0 |
| 24 | t-C4H9NO2 | 29.5 |
| 25 | o-CH3C6H4CN | 29.5 |
| 26 | C6H5NO2 | 29.7 |
| 27 | o-CH3C6H4NO2 | 30.7 |
| 28 | CH3COCH3 | 31.1 |
| 29 | C6H6 | 31.3 |
| 30 | CH3COOC2H5 | 31.4 |
| 31 | CH3COC2H5 | 32.1 |
| 32 | 4-NO2m-xylene | 32.2 |
| 33 | (C2H5)2CO | 32.7 |
| 34 | i-C3H7COCH3 | 33.0 |
| 35 | t-C4H9COCH3 | 33.5 |
| 36 | CH3COOt-C4H9 | 33.7 |
| 37 | (t-C4H9)2CO | 33.7 |
| 38 | (i-C3H7)2CO | 34.0 |
| 39 | C6H5CH3 | 34.5 |
| 40 | C6H5COCH3 | 35.1 |
| 41 | C6H5C2H5 | 35.3 |
| 42 | Thymine | 35.6 |
| 43 | (C3H5)2CO | 36.5 |
| 44 | CH3COC(OCH3)2CH3 | 37.0 |
| 45 | 3-FC5H4N | 37.3 |
| 46 | (C6H5)2CO | 38.3 |
| 47 | 4-ClC5H4N | 38.8 |
| 48 | (C6H5CH2)2CO | 39.5 |
| 49 | CH3CON(CH3)2 | 39.7 |
| 50 | C5H5N | 40.1 |
| 51 | 3-CH3C5H4N | 41.1 |
| 52 | CH3CON(C2H5)2 | 41.6 |
From ref. 26.
DISCUSSION
The discussion can usefully proceed from a brief survey of the information currently available on the structure of the NO+ complexes. The photodissociation experiments performed by Reents and Freiser characterize the adducts of nitryl ion with alcohols, ethers, ketones, and aromatic compounds as charge-transfer complexes (21), consistent with the results of a recent study based on the IR spectroscopy of (H2O)nNO+ clusters, which for n ≤ 3 have been assigned the structure of complexes containing H2O ligands bound to NO+ (29). Many related species, including (CO2)nNO+ clusters (30), protonated nitrous acid (27), oxirane–NO+ (31), acetaldehyde–NO+ (32), and alkane–NO+ adducts (33–35), have been characterized by theoretical methods as structured NO+ complexes. The picture outlined by the above studies points to a relatively distant and moderately intense coordination of the nitryl ion with the ligands, which is nevertheless closer than in the corresponding complexes formed by the strictly related nitronium ion. As an example, the latter forms with water a complex characterized by a larger separation of the monomers, 2.50 Å (36), vs. the H2O–NO+ separation of 2.204 Å (27).
Data Correlation and Analysis.
The BE scale of Table 2 differs from that reported by Reents and Freiser (21), since in all comparable cases their values are consistently larger by some 15 kcal·mol−1. However, the BE differences between ligands are generally very close in the two sets of data—i.e., the two relative BE scales practically coincide, pointing to a different choice of the reference BE used as the source of the discrepancy. The earlier study has utilized the NO+ BE of ethanol, indirectly evaluated from the PA of C2H5ONO, measured by the ICR “bracketing” technique (37), whose inherent accuracy is generally limited (22) and whose application is complicated, in the case of interest, by the propensity of (RONO)H+ ions to undergo NO+ transfer at a rate higher than H+ transfer (21). Moreover, the absolute PA of C2H5ONO, utilized by Reents and Freiser (21) to calculate the C2H5OH–NO+ BE, was based on a scale anchored to PA(NH3) taken equal to 208.5 kcal·mol−1, a value larger by 5 kcal·mol−1 than the currently accepted one (22, 28), which partially accounts for the discrepancy of the two BE scales. In conclusion, as recently suggested by other authors (38), it appears that the absolute anchor of the Reents and Freiser scale needs to be revised. On the basis of its direct, more reliable measurement (26), whose result agrees with that of a high-level theoretical approach (27), it appears that the choice of the H2O–NO+ BE as the absolute reference standard is to be preferred, and it is reassuring that it leads to NO+ BEs of various molecules in excellent agreement with those from high-level ab initio calculations. Thus, the NO+ BE of methanol has been computed to be 25.3 kcal·mol−1 (39), that of oxirane 27.3 kcal·mol−1 (31), and that of acetaldehyde 27.9 kcal·mol−1 (32). All these values, while consistent with those of Table 2—i.e., 23.3, 26.9, and 27.6 kcal·mol−1, respectively, are much lower than those of the earlier scale anchored to an NO+ BE of ethanol taken equal to 40.4 kcal·mol−1. It should be noted that, once adjusted to the anchor used in this work, the BE values reported by Reents and Freiser (21) become perfectly compatible with ours, and indeed can be used to advantage to complement those of Table 2 in the construction of an extended NO+ BE vs. PA correlation. The plot of Fig. 2 includes all ligands of known PA investigated, except those aromatic compounds which are known (21) to bind to NO+ giving π complexes, whereas protonation gives instead σ complexes (ref. 40 and references therein). The different nature of the adducts formed by NO+ and H+ prevents, in this case, meaningful correlation of the BEs of the two cations, which is possible instead if the molecule contains a nucleophilic center, other than the π system, to which NO+ binds, as in the case of aromatic aldehydes, ketones, nitro compounds, etc. that fit the general correlation
 |
a |
valid for all other ligands, where the energies are expressed in kcal·mol−1. The linearity of the general plot is satisfactory, its correlation coefficient being 0.9828. Those aromatic compounds that form π-type complexes with NO+ obey instead the following equation
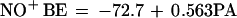 |
b |
whose correlation coefficient, 0.940 is appreciably lower, probably on account of the limited set of currently available data.
Figure 2.

General correlations between NO+ BEs and PA values from ref. 22. ○, Data from this work; •, data from ref. 21 (see text); ▪, data from ref. 30.
Comparison with Other Cations.
The NO+ BEs are considerably smaller than the corresponding PAs, consistent with the fact that at any given separation the electrostatic energy released upon charge expansion from the free ion to the ion-neutral complex is considerably smaller in the case of NO+ than of H+, which, in addition, is able to approach the nucleophilic center to a much shorter distance than NO+. The same arguments apply when contrasting NO+ with the closely related NO2+ cation. The NO+ BEs are in most cases considerably larger, owing again to the smaller size, the higher charge density, and the smaller ion-neutral separation that characterize NO+ complexes with respect to their NO2+ counterparts. Furthermore, at variance with NO2+, NO+ does not appear to undergo energetically unfavorable deformation upon association with the ligand—i.e., the N—O equilibrium bond length does not change appreciably in passing from free NO+ to the H2O–NO+ complex (27). The larger H2O–NO2+ separation (36) reflects the reluctance of NO2+ to form tightly bound complexes, where the close approach to the nucleophilic center entails the deformation of the cation. Another significant difference is that the BE vs. PA correlation displays a much better linearity in the case of NO+ than of NO2+, and satisfactorily fits a single linear plot, whereas the NO2+ correlation has a composite character, significantly changing for different classes of compounds (23).
Predictive Value of the NO+ BE vs. PA Correlation.
The satisfactory linearity and the wide range, spanning from nitrogen, PA = 118.2 kcal·mol−1, to 3-methylpyridine, PA = 224.1 kcal·mol−1, make correlation a useful from two different standpoints. First, comparison of the values predicted with those from independent approaches can be used to assess the validity of the present measurements. A significant example is the experimentally inaccessible NO+ BE of NH3, whose 31.7 ± 2 kcal·mol−1 value from very recent high-level calculations (41) compares well with the 33.0 ± 2 kcal·mol−1 from correlation a. More interesting, the connection can be used for predicting the NO+ affinity of a large variety of compounds, utilizing the comprehensive PA compilations currently available (22, 28). As an example of application to molecules of great biological interest, the gas-phase NO+ affinities of the DNA bases from correlation a are the following: adenine, 40.3 kcal·mol−1; cytosine, 40.4 kcal·mol−1; guanine, 40.1 kcal·mol−1; and thymine, 34.9 kcal·mol−1. Direct experimental measurement has been possible only in the case of thymine and its result, 35.6 kcal·mol−1, underlines the satisfactory accuracy, and hence the predictive value, of correlation a, although such a degree of agreement may be partially fortuitous, since, based on the combined uncertainties of the PA values taken from the National Institute of Standards and Technology database (22) and of correlation a, we attach an estimated error bar of ± 2 kcal·mol−1 to the indirectly calculated NO+ BE values.
Another diagnostic application concerns those aromatic molecules that contain, in addition to the π system, another nucleophilic center. Knowledge of the NO+ BE and PA allows one to ascertain whether correlation a or b applies, which in turn provides a criterion to decide whether a π-type or a n-type NO+ complex is formed, a useful information in the vast majority of cases where direct experimental evidence is lacking.
Extension of the Kinetic Methods to Polyatomic Ions.
Following the application to the strictly related nitronium ion (23), the present work provides another example of successful extension of the kinetic method to the evaluation of the absolute BE of a polyatomic ion. In fact, all previously published studies of dimers bound by polyatomic cations, including CH3+, CN+, OCNCO+, and NH4+ provide only the relative affinity order of the ligands investigated (24), rather than the absolute BE values required for general thermochemical applications and for modelling purposes. As in the case of NO2+, the kinetic method performed beyond expectations in the evaluation of the NO+ BE, giving results in excellent agreement with those from the ICR equilibrium method, and providing the only viable experimental approach in the numerous cases where application of the latter proved impossible.
Acknowledgments
The authors are indebted to F. Angelelli for FT-ICR measurements. Financial support from the Rome University “La Sapienza” and the Consiglio Nazionale delle Ricerche is gratefully acknowledged.
ABBREVIATIONS
- BE
binding energy
- ICR
ion cyclotron resonance
- PA
proton affinity
- FT
Fourier transform
- RP
reference pair
References
- 1.Patai S, editor. The Chemistry of Nitro and Nitroso Groups. New York: Wiley; 1970. Parts I and II. [Google Scholar]
- 2.Ridd J H. Adv Phys Org Chem. 1978;16:1–49. and references therein. [Google Scholar]
- 3.Joergensen K A, Lawesson S O. J Chem Soc Perkin Trans. 1985;2:231–235. [Google Scholar]
- 4.Williams D L H. In: Organic Reactivity, Physical and Biological Aspects. Golding B I, Griffin R J, Maskill H, editors. Cambridge, U.K.: R. Soc. Chem.; 1995. p. 320. [Google Scholar]
- 5.Magee P N, Barnes J N. Adv Cancer Res. 1967;10:163–246. doi: 10.1016/s0065-230x(08)60079-2. [DOI] [PubMed] [Google Scholar]
- 6.Aldred S E, Williams D L H, Gayley M. J Chem Soc Perkin Trans. 1982;2:777–782. [Google Scholar]
- 7.Fehsenfeld J C, Ferguson E E. J Geophys Res. 1969;74:2217–2222. [Google Scholar]
- 8.Puckett L J, Teague M W. J Chem Phys. 1971;54:2564–2572. [Google Scholar]
- 9.Ginzburg E I, Egenmuradova E A. Geomagn Aeron. 1991;31:699–704. [Google Scholar]
- 10.Yau A W, Whalen B A. Can J Phys. 1992;70:500–509. [Google Scholar]
- 11.Ginzburg E I, Egenmuradova E A. Geomagn Aeron. 1991;31:705–711. [Google Scholar]
- 12.Zadorozhnyi A M, Tucchov G A, Kikhtenko V N, Lastovicka J, Boska J, Novak A. J Atmos Terr Phys. 1992;54:183–192. [Google Scholar]
- 13.Committee on Nitrate and Alternative Curing Agents in Food and Health. Effects of Nitrate, Nitrite and N-Nitroso Compounds. Washington, DC: Natl. Acad. Press; 1981. [Google Scholar]
- 14.Lawely P D. In: Chemical Carcinogens, ACS Monograph 182. Searle C D, editor. Washington, DC: Am. Chem. Soc.; 1984. [Google Scholar]
- 15.Lijinsky W. Chemistry and Biology of N-nitroso Compounds. Cambridge, U.K.: Cambridge Univ. Press; 1982. [Google Scholar]
- 16.Lipton S A, Choi Y-B, Pan Z-H, Lel S Z, Chen H-S, Sucher N J, Loscalzo J, Singel D J, Stamler J S. Nature (London) 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 17.Stamler J S, Singel D J, Loscalzo J. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 18.Kirchner J J, Sigurdson S T, Hopkins P B. J Am Chem Soc. 1992;114:4020–4027. [Google Scholar]
- 19.Elcock A H, Lyne P D, Mulholland A J, Nandre A, Richards W G. J Am Chem Soc. 1995;117:4706–4707. [Google Scholar]
- 20.Spanel P, Smith D. J Chem Phys. 1996;104:1893–1899. [Google Scholar]
- 21.Reents W D, Freiser B S. J Am Chem Soc. 1981;103:2791–2797. [Google Scholar]
- 22.Lias, S. G., Bartmess, J. E., Liebman, J. F., Holmes, J. L., Levin, R. D. & Mallard, W. G. (1988) J. Phys. Chem. Ref. Data 17, Suppl. 1.
- 23.Cacace F, de Petris G, Pepi F, Angelelli F. Proc Natl Acad Sci USA. 1995;92:8635–8639. doi: 10.1073/pnas.92.19.8635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooks R G, Patrick J S, Kotiaho T, McLuckey S A. Mass Spectrom Rev. 1994;13:287–339. [Google Scholar]
- 25.Miller K J. J Am Chem Soc. 1990;112:8533–8542. [Google Scholar]
- 26.French M A, Hills L P, Kebarle P. Can J Chem. 1973;51:456–461. [Google Scholar]
- 27.de Petris G, Di Marzio A, Grandinetti F. J Phys Chem. 1991;95:9782–9787. [Google Scholar]
- 28.Szulejko J E, McMahon T B. J Am Chem Soc. 1993;115:7839–7848. [Google Scholar]
- 29.Cao Y, Choi J-H, Haas B-M, Okumura M. J Phys Chem. 1994;98:12176–12185. [Google Scholar]
- 30.Hiraoka K, Yamabe S. J Chem Phys. 1991;95:6800–6805. [Google Scholar]
- 31.Bernardi F, Robb M A, Rossi I, Venturini A. J Org Chem. 1993;58:7074–7078. [Google Scholar]
- 32.Cacace F, de Petris G, Pepi F, Rossi I, Venturini A. J Am Chem Soc. 1996;118:12719–12723. [Google Scholar]
- 33.Schreiner P R, Schleyer P v R, Schaefer H F., III J Am Chem Soc. 1993;115:9659–9666. [Google Scholar]
- 34.Schreiner P R, Schleyer P v R, Schaefer H F., III J Am Chem Soc. 1995;117:453–461. [Google Scholar]
- 35.Olah G A, Hartz N, Rasul G, Prakash G K S. J Am Chem Soc. 1995;117:1336–1343. [Google Scholar]
- 36.Lee T J, Rice J E. J Phys Chem. 1992;96:650–657. [Google Scholar]
- 37.Farid R, McMahon T B. Int J Mass Spectrom Ion Phys. 1978;27:163–183. [Google Scholar]
- 38.Ryzhov V, Klippenstein S J, Dunbar R C. J Am Chem Soc. 1996;118:5462–5468. [Google Scholar]
- 39.Aschi M, Grandinetti F. Chem Phys Lett. 1996;258:123–128. [Google Scholar]
- 40.Glukhovtsev, M. N., Pross, A., Nicolaides, A. & Radom, L. (1995) J. Chem. Soc. Chem. Commun. 2347–2348 and references therein.
- 41.Aschi, M. & Grandinetti, F. (1997) Chem. Phys. Lett., in press.