

Abstract
Nicotinic acetylcholine receptors (nAChRs) are widely expressed throughout the central nervous system and participate in a variety of physiological functions. Recent advances have revealed roles of nAChRs in the regulation of synaptic transmission and synaptic plasticity, particularly in the hippocampus and midbrain dopamine centers. In general, activation of nAChRs causes membrane depolarization and directly and indirectly increases the intracellular calcium concentration. Thus, when nAChRs are expressed on presynaptic membranes their activation generally increases the probability of neurotransmitter release. When expressed on postsynaptic membranes, nAChR-initiated calcium signals and depolarization activate intracellular signaling mechanisms and gene transcription. Together, the presynaptic and postsynaptic effects of nAChRs generate and facilitate the induction of long-term changes in synaptic transmission. The direction of hippocampal nAChR-mediated synaptic plasticity –either potentiation or depression – depends on the timing of nAChR activation relative to coincident presynaptic and postsynaptic electrical activity, and also depends on the location of cholinergic stimulation within the local network. Therapeutic activation of nAChRs may prove efficacious in the treatment of neuropathologies where synaptic transmission is compromised, as in Alzheimer’s or Parkinson’s disease.
Keywords: Nicotine, Synaptic Plasticity, LTP, Development, Hippocampus, Ventral Tegmental Area
Neuronal nicotinic acetylcholine receptors
Receptor structure
Nicotinic acetylcholine receptors (nAChRs) are widely expressed throughout the central nervous system [1]. Each receptor/channel consists of five subunits surrounding a water-filled, cation-permeable pore [2, 3] (Fig. 1). Each nAChR subunit has a similar general linear structure and transmembrane topology. The nAChR subunits have a large extracellular N-terminal domain that contributes to ligand binding, followed by three hydrophobic transmembrane regions (M1 to M3), a large intracellular loop, a fourth transmembrane region (M4), and finally a short extracellular C-terminus (Fig. 1A) [4]. The M2 transmembrane segment in each subunit provides the main lining of the ionic pore with some contribution from M1. The M1, M3 and M4 segments separate the pore-lining region from the hydrophobic membrane [5]. Subunits α2 through α10 have been cloned and are homologous to the α1 subunit identified in muscle [6–9]. The neuronal non-α subunits, β2 through β4, have also been identified [6–9]. Many neuronal nAChRs are assembled as αβ hetero-oligomers with a typical α:β stoichiometry of 2:3. In brain, the most common hetero-oligomeric nAChRs are assembled from α4 and β2 subunits (Fig. 1B) [3, 10, 11]. More complex hetero-oligomeric compositions are also possible, such as the α4α6β2 nAChRs commonly distributed within midbrain dopamine areas. Homo-oligomeric nAChRs are only formed by α7, α8 or α9 subunits, with α7 homo-oligomers being the only ones widely distributed in the brain (Fig. 1B) [10, 12, 13]. Accumulating evidence reveals that α7-containing (α7*) and β2-containing (β2*) nAChRs are expressed at preterminal, axonal, somatic and dendritic locations [10, 14–20].
Fig. 1.
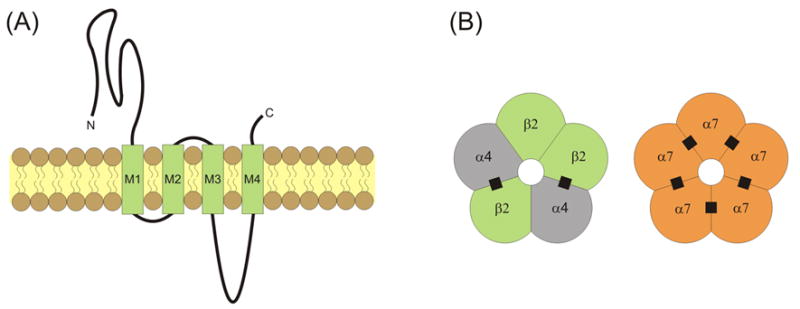
Transmembrane topology and pentameric structure of nAChRs. (A) nAChRs consist of four transmembrane domains (M1 through M4) with extracellular C- and N-termini. (B) Subunits are assembled into pentamers that include a water-filled cation-permeable pore. The most common nAChRs in the brain are hetero-oligomeric α4β2 nAChRs and homo-oligomeric α7 nAChRs. The recognized ACh binding sites are indicated by filled black squares.
Functional characteristics
Nicotinic AChRs transition between three principal states: closed, open and desensitized [21]. In the closed state the channel is non-conducting, but the receptor sites can bind ligands. Upon binding agonist (usually 2) the channel is open and conducts cations. In the desensitized state the channel is non-conducting and the receptor is unresponsive to ligands.
Ionic flow through the open channel, particularly with respect to the proportion of calcium ions, depends on the nAChR subunit composition. For instance, the ratio of Ca2+ to Na+ permeability is α7* > β2* > muscle nAChRs [18, 19, 22–25]. The marked permeability of α7 nAChRs to Ca2+ has significant ramifications for synaptic transmission and plasticity. The α7* and β2* nAChRs are further distinguished by their fast and slow rates of desensitization, fast and slow tau of recovery from desensitization, and their low and high affinity for ACh, respectively [8, 26–28].
Neuronal nicotinic AChR subtypes are activated by nicotine and ACh, and inhibited by mecamylamine (MEC) [7]. The α7* nAChRs are activated by choline, which is produced at signaling concentrations as a metabolite of ACh hydrolysis [29, 30]. Selective inhibitors of the α7* nAChRs include α-bungarotoxin (α-BTX) and low concentrations (≤ 5 nM) of methyllycaconitine (MLA) [19, 31]. MLA at 50 nM begins to inhibit other nAChR subtypes significantly [32]. In electrophysiological studies of rodents, dihydro-β-erythroidine (DHβE) inhibits β2* nAChRs [33], and low concentrations of epibatidine activate those receptors [34]. Epibatidine binding studies indicate a higher affinity for rat β2* than for β4* nAChRs [35], however, binding studies in primate brain tissue indicate that epibatidine displays less selectivity between β2* and β4* nAChRs [36]. The agonist, A-85380, more selectively activates β2* nAChRs [37]. For all activators and inhibitors, selectivity for one subunit over another is restricted to defined concentration ranges [28, 38–40].
Extensive and complex expression patterns of nAChRs are complemented by the prominent innervations of cholinergic afferents throughout the brain [41]. Fast, synaptic nAChR-mediated EPSPs are relatively rare, but they have been detected in the hippocampus, the supraoptic nucleus, and in the cortex [42–45]. Given the number of neuronal types in which nAChR currents have been measured following local puffs of ACh, this list likely underestimates the extent of synaptic nAChR activity in the brain. Cholinergic signaling at synapses is brief and intense. Within the cleft of the neuromuscular junction, ACh reaches a relatively high concentration (~1 mM for ~1 ms) before being hydrolyzed by acetylcholinesterase [46]. A combination of rapid delivery and then breakdown of ACh minimizes the desensitization of nAChRs. Synaptically released ACh can also diffuse to non-synaptic sites, and evidence suggests that significant cholinergic signaling in the CNS is mediated via such volume transmission [9, 47, 48].
Neuronal nAChRs facilitate presynaptic neurotransmitter release
Neurotransmitters are released from presynaptic terminals, a process that typically requires an action potential to invade and depolarize the terminal. Depolarization activates voltage-gated Ca2+ channels (VGCCs), and the intra-terminal Ca2+ increase triggers neurotransmitter release. Vesicular release can also occur stochastically in an action potential-independent manner. Because nAChRs are expressed presynaptically and because the cationic current through nAChRs can both depolarize membranes and raise intracellular Ca2+ levels, nAChRs influence neurotransmitter release.
Presynaptic nAChRs
Early experiments conducted at mossy fiber–CA3 glutamatergic synapses in the rat hippocampus [49] and at medial habenula–interpeduncular nucleus glutamatergic synapses in the chick brain [50] provided evidence identifying a presynaptic function for nAChRs. First, in the presence of TTX to eliminate action potentials arriving at the presynaptic terminals, nicotine application increased the frequency of spontaneous (miniature) EPSCs. Nicotine did not affect the sensitivity of neurotransmitter detection, as inferred by an absence of change in EPSC amplitudes. Second, the amplitudes of stimulus-evoked responses were increased with a decreased incidence of synaptic failures, suggesting an increase in the probability of vesicle release. Third, the amount of transmitter released by nicotine stimulation was enhanced by increasing the concentration of extracellular Ca2+ and decreased by reducing the concentration of extracellular Ca2+. Under some conditions, Ca2+ signaling initiated by nAChRs was sufficient to trigger neurotransmitter release, as the Ca2+ influx through VGCCs was not obligatory. Later work showed that nAChRs not only mediated a direct Ca2+ signal, they also activated VGCCs and indirectly stimulated release from intracellular stores [51–54]. Fourth, nicotine robustly increased the intra-terminal Ca2+ concentration. Fifth, the effects of nicotine were mediated through multiple nAChR subtypes, often including the α7* nAChRs that have a high Ca2+-permeability. Taken together, those findings provided electrophysiological evidence for a presynaptic function for nAChRs in intact neurons. Those results complement and extend biochemical assays of nicotine-evoked neurotransmitter and ion release completed on synaptosomes prepared from a variety of brain regions [55–57]. In the sections below we focus our attention on the nicotinic mechanisms contributing to synaptic transmission and plasticity as determined by electrophysiological experiments.
Nicotinic AChRs regulate the release of multiple neurotransmitters
Activation of presynaptic nicotinic receptors facilitates the release of a variety of neurotransmitters throughout the brain [9, 58–61]. Nicotinic receptor activity directly and indirectly initiates an intracellular Ca2+ signal that contributes to neurotransmitter release. In many cases the Ca2+ influx through nAChRs (often α7*) is sufficient to evoke neurotransmitter release [49, 62]. However, the depolarization caused by the cation influx through nAChRs also can trigger neurotransmitter release indirectly by activating VGCCs [52, 63–66]. Given that CaMKII blocks facilitation by nicotine of VGCCs, it is reasonable to propose that the Ca2+ influx through nAChRs may regulate VGCCs via CaMKII-dependent, Ca2+-dependent mechanisms [67]. In addition, there are other intracellular events, such as Ca2+-induced Ca2+ release from intracellular stores that can be indirectly activated by nAChR activity [68].
In paired-pulse facilitation experiments, two stimuli of equal size are delivered to presynaptic fibers in rapid succession, and the amount of neurotransmitter released by the second stimulus is greater than the amount released by the first (priming) stimulus. Because the paired stimuli are identical in size, each stimulus provides a comparable depolarization to the terminal. The increase in intracellular Ca2+ from each stimulus has a half-decay time of a few hundred msec and, thus, intra-terminal Ca2+ can accumulate when two stimuli are closely paired [69]. Because the probability of transmitter release varies approximately with the fourth power of the Ca2+ concentration [69], a modest increase in intra-terminal Ca2+ from the second stimulus results in a robust increase in neurotransmitter release (e.g., paired-pulse facilitation). As nicotine, and likely endogenous ACh, acts through presynaptic nAChRs to elevate intra-terminal Ca2+, nAChR activation may thus function as a ‘priming’ stimulus to augment the efficacy of incoming APs.
Nicotinic AChRs regulate hippocampal synaptic plasticity in vitro
The hippocampus is central to learning, memory, and attention mechanisms, and nAChRs contribute to these functions [6, 70–77]. For instance, local application of nAChR antagonists impairs working but not reference memory [76], whereas working memory deficits are reversed by nicotine following cholinergic denervation of the hippocampus [77]. The hippocampus receives cholinergic input from the septum-diagonal band complex, whose fibers project to all regions including the dentate gyrus, CA3 and CA1, and to most cell types, including pyramidal cells, granule cells, interneurons, and hilar neurons [78, 79]. Targeted neurons robustly express nAChRs, in particular α7* nAChRs, but also β2* nAChRs [10, 16, 18–20]. Small nicotinic receptor-mediated currents have been measured in pyramidal neurons and substantially greater nicotinic currents have been measured in interneurons [6, 25, 42, 44, 80–84], with volume transmission also present [47]. In fact, α7* nAChRs are commonly found at both presynaptic and postsynaptic sites in the hippocampus CA1 region as identified with immunogold labeling and electron microscopy [16]. Remarkably, the density of nAChRs is similar to the densities of both NMDARs and AMPARs at these synapses [85].
Synaptic plasticity is a cellular correlate of memory and is modulated by nAChR activation
A cellular correlate to learning and memory in the hippocampus and elsewhere is a change in synaptic efficacy between neurons, such as STP, LTP and LTD [86–88]. These types of changes can be evoked by pairing presynaptic stimulation at specific frequencies with coincident postsynaptic depolarization. Changes in synaptic strength are interpreted from the change in amplitude or slope of the EPSP or EPSC, or amplitude of the population spike (a synchronized discharge of multiple neurons measured with extracellular ‘field’ recordings). For instance, if the ratio of EPSP amplitude (post-pairing protocol)/EPSP amplitude (pre-pairing protocol) is greater than 1, then synaptic potentiation has occurred.
Numerous studies have now shown that nAChR activation facilitates the induction of LTP in vitro. Such facilitation may be operationally defined as nicotine boosting STP to LTP, and facilitation occurs in acute brain slices prepared from naïve animals, animals chronically-treated with nicotine, and even slices from aged animals in which LTP is difficult to induce [81, 89–92]. Furthermore, after establishing the maximum possible LTP via high-frequency stimulation, subsequent application of nicotine can increase the amplitude of LTP to a new maximum unattainable by stimulation alone [93].
Timing of nAChR activation regulates hippocampal synaptic plasticity
In one set of experiments, STP was induced at Schaffer collateral–CA1 synapses in acutely prepared hippocampal slices while varying the timing of nicotine application relative to afferent stimulation (Fig. 2A–F) [92]. STP was induced by 1 sec of 100 Hz stimulation paired with 100 pA depolarization of CA1 pyramidal neurons and persisted for ~20 min. Brief (0.5 to 1 sec) puffs of ACh (1 mM, applied in the presence of atropine to block muscarinic AChRs) were delivered to the pyramidal cells and evoked action potential (AP) discharge. One of three outcomes was obtained that depended on the timing of the last ACh-induced AP relative to afferent stimulation [92]. First, if the last ACh-induced AP preceded HFS by more than 5 sec (Fig. 2B, F), or trailed HFS (Fig. 2E, F), then STP was unaltered. Second, if the last ACh-induced AP preceded HFS by 1–5 sec, LTP was produced that persisted for at least one hour (Fig. 2C, F). Third, if the last ACh-induced AP occurred <1 sec prior to HFS, then LTD resulted (Fig. 2D, F). Mimicking ACh’s postsynaptic depolarization with direct current injection failed to boost STP to LTP, revealing an obligatory requirement for nAChR activation.
Fig. 2.

Temporal-dependence of nAChR activation for hippocampal synaptic plasticity. (A) Illustration of the experimental setup for experiments shown in panels (B) through (E). Whole-cell patch recordings were obtained from CA1 pyramidal somata, with activation of Schaffer collateral afferents in stratum radiatum. A puffer pipette delivered brief pulses of ACh (in the presence of atropine) to the pyramidal neuron’s dendrites. All postsynaptic potentials (PSPs) were normalized to baseline. (B) ACh-induced APs that preceded HFS of the Schaffer collaterals by ≥ 10 sec did not affect STP. (C) In contrast, ACh-induced APs that terminated 1–5 sec prior to HFS boosted STP to LTP. (D) If the APs terminated < 1 sec before HFS, then LTD resulted. (E) If the ACh-evoked APs followed HFS then there was no effect on STP. (F) Summary of the findings in (B) through (E). The normalized PSPs from the last 15 min of post-HFS recording were averaged and then plotted against the time between the end of ACh application and the onset of HFS. Negative time values correspond to the interval between the last ACh-induced AP and the onset of HFS. Positive time values correspond to the interval between the end of HFS and the onset of the first ACh-induced AP. The curve drawn through the data points indicates the general trend of the data. Arrows indicate timing of HFS for all panels. Adapted with permission from [92] (copyright 2005 by the Society for Neuroscience).
The temporal profile revealed in Figure 2F differs substantially from reports of spike timing dependent plasticity (STDP), which have identified intervals on the order of a few msec over which single EPSPs paired with single APs can change synaptic weights [94, 95]. Cholinergic regulation of plasticity, which emphasizes pairing intervals on the order of seconds, provides a broad window over which the outcome of presynaptic information can be transmitted and influence synaptic strength.
Boosting STP to LTP requires the activation of mainly postsynaptic α7* nAChRs (with some β2* nAChRs) in the rat hippocampus [81, 89, 90, 92, 93, 96]. Despite the fast desensitization of α7 nAChRs, brief activation of these receptors in rapid succession (e.g., <200 msec inter-stimulus intervals) actually potentiates their currents [97]. During this imposed protocols, activation of α7* nAChRs is only required for the induction of LTP, and not its expression or maintenance, as blocking α7 nAChRs has no effect once LTP is established [98].
Location of nAChR activation regulates hippocampal synaptic plasticity
In another set of experiments, STP was induced at Schaffer collateral–CA1 synapses, while varying the location of agonist application (Fig. 3A–C) [81]. For these experiments a more intense stimulation protocol (3 × 1 sec, 100Hz stimulus trains) was used to produce robust LTP [81]. When nAChRs were activated by puffing ACh onto neighboring GABAergic interneurons, which have been shown to inhibit CA1 pyramidal neurons directly [81, 84, 99, 100], this protocol produced only STP. Thus nicotinic effects in the hippocampus on synaptic transmission are mediated to a significant extent by the direct activation of inhibitory GABAergic interneurons, which serves to decrease the net output of pyramidal cells, or negate plasticity mechanisms in pyramidal cells. Interestingly, for the cellular combination of interneuron-interneuron-pyramidal cell, nicotinic activation of the first GABAergic neuron can inhibit the second interneuron, which then disinhibits the pyramidal cell, thereby potentially increasing pyramidal cell output [84]. In situ the nicotinic influences via GABAergic networks are generally stronger than direct action at glutamatergic pyramidal neurons because nAChRs are more densely expressed on the GABAergic interneurons [84, 99, 101, 102].
Fig. 3.

Spatial-dependence of nAChR activation for hippocampal synaptic plasticity. (A) Illustration of the experimental setup for experiments shown in panels (B) and (C). The puffer pipette applies ACh to a GABAergic interneuron neighboring the pyramidal neuron. All PSPs were normalized to baseline. (B, C) HFS-evoked LTP (B) was prevented by activating inhibitory interneurons with ACh (C). Arrows indicate timing of HFS for all panels. Adapted with permission from [81] (copyright 2001 by Cell Press).
nAChR-mediated synaptic plasticity and membrane properties
Synaptic plasticity can be modulated by active membrane events like action potentials (APs), but also by passive membrane properties. Na+-dependent APs are usually initiated in the axon initial segment or first node of Ranvier and the APs propagate down the axon [103–105]. For many cell types, including CA1 pyramidal cells, APs also propagate backwards into the dendrites where they can contribute to synaptic plasticity [106, 107]. Back-propagating APs depolarize dendrites, which removes the Mg2+ block of NMDARs and also inactivates low threshold A-type K+ channels. Together, these processes make synaptic integration and plasticity more permissive [108]. Back-propagation of APs is also important for nAChR-mediated synaptic plasticity, as plasticity is blocked by loading CA1 pyramidal neurons with the Na+ channel blocker QX-314 [92]. By virtue of their depolarizing influence, nAChRs may also directly regulate the availability of voltage-dependent ion channels, decreasing the availability of the aforementioned A-type K+ channels via inactivation, and increasing the activation of ion channels linked with propagating excitation like Na+ and Ca2+ channels. In some cells, due to the expression of Ca2+-activated K+ channels, the Ca2+ influx through nAChRs could alternatively contribute to the stabilization or hyperpolarization of the membrane potential.
Nicotinic AChRs may also modify the passive (electrotonic) characteristics of neurons. For instance, opening nAChR channels provides an effective shunt by increasing membrane conductance (decreasing impedance). This shunt will change the membrane time and length constants. Such changes may affect the electrotonic filtering of the amplitudes and time courses of synaptic inputs. Overall these nicotinic properties could alter the likelihood that synaptic potentials can integrate, conduct to the soma, depolarize the neuron to AP threshold, or trigger synaptic plasticity [9, 21].
Intracellular signaling mechanisms in nAChR-mediated synaptic plasticity
The mechanisms through which nAChRs modulate plasticity resemble known mechanisms for non-cholinergic regulation of synaptic plasticity [92]. In particular, there is evidence for a requirement for NMDA receptors in nAChR-mediated boosting of STP to LTP [93, 96, 98]. Because nAChRs have a voltage-dependent-rectification [24, 109], they are suited to supply a substantial Ca2+ influx to neurons at hyperpolarized membrane potentials, where the driving force on Ca2+ ions is high. Thus, when considering that NMDARs readily pass Ca2+ ions only at depolarized voltages, the Ca2+ influx through nAChRs may extend the voltage range over which Ca2+-dependent intracellular processes are initiated. Interestingly, in cultured hippocampal neurons, nicotinic simulation may in fact reduce some NMDAR currents in a calmodulin-dependent manner. This modulation may provide a cholinergic mechanism under which some synapses can convey information via AMPARs without any long-term change in synaptic efficacy [110]. Additionally, loading neurons with a high affinity Ca2+ chelator prevented the STP to LTP transition, revealing the importance of intracellular Ca2+ signaling to ACh-induced plasticity. In this regard, it has been shown that Ca2+ influx through nAChR can further increase the concentration of intracellular Ca2+ by activating Ca2+-induced Ca2+ release from internal stores [68, 111], which may also contribute to synaptic plasticity.
In a recent study, field recordings obtained from the dentate gyrus in acute slices revealed that, consistent with previous studies, nicotine enhances stimulation-induced LTP in an α7* nAChR- and NMDAR-dependent manner [98]. However, unlike the LTP induced by high-frequency stimulation (HFS) alone, nAChR-augmented LTP required activation of mGluR5, Ca2+ influx through L-type VGCCs, and Ca2+-induced Ca2+ release from ryanodine-sensitive intracellular Ca2+ stores. Interestingly, following two weeks of twice-daily nicotine injections, HFS elicited a robust LTP in acute slices prepared from these animals. The magnitude of LTP measured in these slices was comparable to that obtained for the acute combination of HFS and bath application of nicotine. Blocking mGluR5 or ryanodine-sensitive Ca2+ stores reduced this LTP to the level normally expected for the HFS protocol alone, revealing that acute in vitro or chronic in vivo nicotine administration may function through similar pathways.
Modification of transcription by nAChR activation
In addition to second messenger pathways, long-term changes in neuronal function often require regulation at the gene level, and nicotine, at least in differentiated PC12 cells, activates transcription [112]. In addition, it has been shown that 1 μM nicotine applied for 25 min to cultured hippocampal neurons activates the transcription factor CREB, which in turn activates the immediate early gene c-fos. This transcriptional activation requires both CaMKII/IV and MAP kinases, as well as Ca2+ release from internal stores [113]. This combination of events immediately increases phosphorylated CREB (pCREB), whose levels remain elevated for at least one hour. Interestingly, pCREB levels are reduced by ~1/2 if glutamate receptors are blocked. Thus, nAChR-mediated facilitation of presynaptic glutamate release may act in tandem with postsynaptic nAChR-mediated gene transcription to effect long-term changes in neural function. CREB, and in particular pCREB, have been implicated in various forms of learning and memory [114, 115]. The results suggest that changes in CREB activation via nAChRs represent a means by which nicotine may modify neural function over very long periods.
Nicotinic AChRs regulate hippocampal synaptic plasticity in vivo
Our understanding of nAChR regulation of synaptic transmission and plasticity has for the most part been gleaned from in vitro studies, which offer a high level of experimental control to probe mechanistic questions. However, potential drawbacks, such as the severing or remodeling of network connections and changes in cellular physiology that may impact plasticity [116], suggest the need for systems-level approaches with in vivo strategies.
Systemic administration of very high doses of nicotine has been shown to evoke LTP at perforant path–dentate gyrus synapses in anesthetized mice [34, 117]. In the dentate gyrus, chemical LTP evoked by 3 mg/kg nicotine gradually develops over the first ~10 min post-injection and reaches a stable >2-fold increase over baseline that persists for at least 2 hours [117]. In contrast to in vitro cases, where nicotine boosts STP to LTP, in the intact anesthetized animal very high doses of nicotine actually induce LTP in the absence of the tetanization of afferent inputs. Interestingly, nicotine-induced LTP and perforant path HFS-induced LTP are similar in time course and amplitude, and both are blocked by pre-administration of mecamylamine, suggesting that tetanus-induced LTP requires the endogenous activation of cholinergic inputs and nAChRs. Neither nicotine-induced nor HFS-induced LTP is blocked by mecamylamine delivered after nicotine injection or following HFS. As seen in vitro, this result indicates that the induction, but not the expression or maintenance of LTP, is dependent on nAChRs. This same study also showed that the α7* nAChR agonist choline elicited a dose-dependent LTP in vivo, and that the maximum LTP evoked by choline could be further increased by nicotine [117]. In a complementary experiment, these authors showed that epibatidine, a somewhat specific β2* agonist, also produced chemical LTP [118]. Like choline-induced LTP, epibatidine-induced LTP was sub-maximal and was increased to maximum values by nicotine. The amount of potentiation evoked by choline or epibatidine individually reveals that α7* and β2* receptors, respectively, each contribute about 50% to the LTP evoked by nicotine [117, 118]. At Schaffer collateral–CA1 synapses in anesthetized rats, α7* nAChRs contribute approximately one-third of the HFS-induced LTP [119].
Although these results are intriguing, there are a number of issues to consider. The antagonist mecamylamine in wake rodents also weakly inhibits NMDARs [120] and inhibits ACh synthesis [121]. Thus, work with more specific blockers that exclude alternative conclusions will be necessary. In addition, the rodents were anesthetized and LTP was evoked by very high concentrations of agonists: concentrations of nicotine that would induce seizures in wake mice [122]. The use of anesthesia further confounds the nicotinic role because some anesthetics inhibit nAChRs [123]. In addition, nAChRs often influence circuit events by a predominant action upon GABAergic inhibitory interneurons [84, 101], and GABAergic signaling is important during the induction of nicotine-evoked plasticity [81, 124]. Given the propensity for anesthetics to modulate GABAergic activity, GABARs, and nAChRs [123, 125], additional studies using freely-moving animals and physiological concentrations of nicotine are going to be extremely important to understand the biological implications of nicotine influences over plasticity in vivo.
Nicotinic AChRs contribute to hippocampal plasticity during early postnatal development
During the first few weeks of postnatal life, the rodent brain undergoes extraordinary development. As noted for adult animals, nAChR-mediated plasticity appears to play important roles during these times as well. In early (<1 week) postnatal rats, α7* nAChRs regulate the frequency of GABA-mediated giant depolarizing potentials (GDPs) in CA3 [126]. As pairing GDPs with mossy fiber input is known to strengthen mossy fiber–CA3 synapses [127], early nicotinic regulation may contribute to the maturation of these neuronal connections.
In the developing hippocampus, nAChRs provide a powerful control over glutamate release probability, converting low probability synapses to high probability synapses [128] and vice versa [129]. The effect is sufficiently dramatic that nicotine can convert ‘silent’ synapses to functional synapses [128]. In the adult, ‘silent’ synapses do not express AMPARs in the postsynaptic membrane and, thus, cannot detect glutamate release at hyperpolarized potentials [130]. In contrast, in the early postnatal brain, ‘silent’ synapses express postsynaptic glutamate receptors, but the probability of presynaptic neurotransmitter release is near zero. As noted in adult neurons, nicotine application to juvenile neurons markedly increases the probability of release from Schaffer collateral fibers onto CA1 pyramidal neurons in an α7* nAChR-dependent manner. Presynaptic nAChR activity increases the frequency, but not the amplitude, of spontaneous synaptic currents. Importantly, stimulating cholinergic fibers within juvenile slices mimics the effect of nicotine, suggesting that the switch from low to high probability synapse is mediated by endogenous ACh. For those rare cases when synapses with a high probability of release were identified, nicotine application decreased release probability, an effect that required both α7* and β2* nAChRs [129]. Taken together, these results suggest that activation of nAChRs may function as a bidirectional switch on release probability in developing hippocampal neurons.
Interestingly, the transition of GABA as an excitatory neurotransmitter in the juvenile brain to an inhibitory neurotransmitter in the adult brain is also regulated by nAChR signaling [131]. In juvenile neurons, expression of the Cl− transporter NKCC1 maintains a high intracellular Cl− concentration, and thus GABA-mediated channel openings favor an outward flow of negative current that is depolarizing. In adult neurons the Cl− transporter KCC2 is more robustly expressed, and it maintains a high extracellular Cl− concentration that favors hyperpolarization when the Cl− channels are opened [132, 133]. During the second postnatal week NKCC1 expression is reduced and KCC2 expression is increased, rendering GABA inhibitory. These events were recently shown to involve α7* nAChR-mediated signaling, albeit through an unidentified transduction mechanism [131].
Nicotinic AChRs regulate synaptic plasticity in midbrain dopaminergic neurons
Nicotinic receptors are known to regulate synaptic plasticity in areas that play a critical role in reward and addiction, such as the neurons of the ventral midbrain. Of particular significance are the dopaminergic neurons of the ventral tegmental area (VTA), which respond to rewarding stimuli or reward-predicting stimuli [134]. VTA neurons broadcast reward and salience information to areas of the brain underlying emotion and decision making, including the prefrontal cortex, amygdala, striatum, and nucleus accumbens [41]. The VTA dopaminergic neurons receive excitatory input from numerous sources, including the prefrontal cortex, and inhibitory inputs from afferent projections and from local GABAergic interneurons. Cholinergic inputs arrive from the laterodorsal and the pedunculopontine tegmental nuclei and project to both dopaminergic and GABAergic cells in this region. The ventral midbrain neurons express many nAChR subunits, with α7* and β2* nAChRs predominating [135, 136]. Nicotine robustly activates this system, resulting in the acquisition of behaviors that are reinforced by drug use [137–139]. In fact, a single exposure to nicotine elevates dopamine levels in the nucleus accumbens for more than two hours in rats (Fig. 4A) [140, 141]. The question of how a drug such as nicotine triggers long-term changes in the midbrain dopamine system has been explored from the hypothesis that nicotine, acting through nAChRs, modulates short- and long-term plasticity.
Fig. 4.

Synaptic plasticity in the VTA. (A) Dopamine levels are elevated in the nucleus accumbens of rats for more than two hours following a single i.p. injection of 0.6 mg/kg nicotine. (B) Bath applied nicotine increases the frequency of AP discharge in dopaminergic neurons mainly via β2* nAChRs. (C) Bath applied nicotine increases the frequency of spontaneous EPSCs (sEPSCs) mainly via presynaptic α7* nAChRs. The coincidence of postsynaptic and presynaptic activation by nicotine is sufficient to induce LTP of the glutamatergic synapses. (D) The frequency of spontaneous IPSCs (sIPSCs) first increases, and then decreases below baseline following desensitization of β2* nAChRs on the somata of GABAergic neurons. The decrease in IPSC frequency indirectly enhances the glutamatergic excitation of dopamine neurons. Adapted with permission from [140, 144] (copyright 1997 by Nature Publishing Group [144] and copyright 2004 by Cold Spring Harbor Laboratory Press [140]).
nAChRs facilitate glutamate release and evoke postsynaptic depolarization to initiate VTA LTP
Low concentrations of nicotine that approximate the concentration delivered by smoking a cigarette [142] produce LTP in dopaminergic neurons [137, 143]. Bath application of nicotine produces a substantial postsynaptic depolarization that markedly increases the frequency of AP output from VTA dopaminergic neurons via (mainly) β2* nAChRs (Fig. 4B) [143–146]. Nicotine further acts via presynaptic (mainly) α7* nAChRs to increase the frequency of spontaneous EPSCs (Fig. 4C) [140, 146]. This presynaptic facilitation persists during the nicotine application (e.g., 25 min), revealing that the α7* nAChRs are available for activation as long as nicotine is present [136, 140]. Furthermore, the amplitude of synaptically-evoked events is significantly increased by nicotine, collectively suggesting an increase in the probability of glutamate release [140, 143, 146]. The increased delivery of glutamate activates postsynaptic AMPARs, which in combination with the nAChR-mediated postsynaptic depolarization, provides a robust voltage change reducing the Mg2+ blockade on NMDARs. Together the effects of nicotine on presynaptic and postsynaptic membranes produce LTP. This process is in contrast to the hippocampus, where nAChR activation in vitro must be combined with coincident presynaptic tetanization and postsynaptic depolarization by the recording electrode to boost STP to LTP.
nAChRs inhibit interneuron output to strengthen LTP
Occurring simultaneously with the strengthening of excitatory connections onto VTA dopaminergic neurons is a decrease in the strength of inhibitory connections. Application of nicotine to midbrain slices initially results in an increase in the frequency and amplitude of inhibitory events due to the activation of presumably preterminal or somatic (mainly) β2* nAChRs (Fig. 4D) [140, 147, 148]. However, the β2* nAChRs rapidly desensitize [136], and the frequency of inhibitory events falls to below pre-nicotine levels, suggesting a tonic cholinergic excitatory drive onto these neurons. This reduction in inhibitory tone disinhibits the dopaminergic neurons, further strengthening the nicotine-induced enhancement of excitatory inputs.
α7* and β2* nAChRs differentially regulate dopamine neuron AP output in vivo
Midbrain dopamine neurons in vivo generate APs in regular- and burst-firing patterns [149, 150]. Both the frequency of regular firing, and the incidence of bursting, can be increased by nicotine administration [151]. In an effort to identify the specific nAChR subunits that mediate the effects of nicotine on dopamine neuron output, in one study AP output was first subdivided into four principal modes: 1) low frequency regular-firing, low-bursting (LFLB), 2) low frequency regular-firing, high-bursting (LFHB), 3) high frequency regular-firing, low-bursting (HFLB), and 4) high frequency regular-firing, high-bursting (HFHB) [152]. Using an elegant combination of molecular and genetic tools, these authors showed that activation of β2* nAChRs switch dopamine neurons from the ‘resting’ mode (LFLB) to any of the ‘excited’ modes [152]. Once β2* nAChRs shift the neuron into an excited mode, activation of α7* nAChRs fine tunes the pattern of AP output by switching the neuron to other excited output modes (e.g., from HFHB to LFHB or from HFHB to HFLB). Interestingly, selective re-expression of β2* nAChRs into midbrain dopamine neurons of β2 knockout mice is sufficient to restore nicotine-evoked changes in electrophysiological properties, nicotine-evoked dopamine release and nicotine self-administration [153]. These thoughtful experiments revealed fundamental roles for β2* nAChRs in midbrain dopamine neurons.
Comparison of nAChR LTP mechanisms between hippocampus and VTA
The contributions of nAChRs to synaptic plasticity in the hippocampus can be summarized as follows: 1) nAChRs increase presynaptic glutamate release, 2) nAChRs contribute a small postsynaptic membrane depolarization, which presumably increases the open probability of NMDARs via the voltage-dependent relief of Mg2+ blockade, 3) nAChRs directly and indirectly contribute an intracellular Ca2+ signal in the postsynaptic cell that may activate plasticity-evoking Ca2+-dependent signaling and gene transcription, 4) presynaptic activity and nAChR activation may be temporally coordinated to influence synaptic transmission and plasticity, 5) nAChR activation may be spatially coordinated to enhance GABAergic interneuron signaling to block LTP induction on pyramidal neurons, and 6) nAChR signaling can awaken or silence synapses during early postnatal development and control the transition of GABA from an excitatory to an inhibitory neurotransmitter. Taken together, the data support the view that nAChRs exert a temporally- and spatially-dependent bidirectional control over hippocampal synaptic plasticity, both in vitro and in vivo.
In contrast to the facilitory (modulatory) role for nAChRs during LTP induction in the in vitro hippocampal slice, nAChR activity induces LTP in the midbrain slice containing the VTA. Within the VTA, nicotine contributes to LTP of glutamatergic synapses onto dopamine neurons via three major mechanisms: 1) transient activation of postsynaptic β2* nAChRs to depolarize the dopaminergic neurons, 2) sustained activation of presynaptic α7* nAChRs to augment glutamate transmission onto dopaminergic neurons, and 3) activation and desensitization of β2* receptors on GABAergic interneurons to decrease inhibition onto dopaminergic neurons. Within the VTA, nicotinic cholinergic input could conceivably act as a coincidence mechanism, simultaneously recruiting both presynaptic and postsynaptic elements. The fact that nAChR activation in the hippocampus in vivo produces de novo LTP in the absence of afferent tetanization suggests that the hippocampal slice preparation may lack all the long-range connections and the network properties of the in vivo situation. Thus, nAChRs modulate, regulate and likely induce synaptic plasticity in both the hippocampus and the VTA.
Potential therapeutic roles for nAChRs in pathological states
The importance of nAChRs to synaptic transmission and synaptic plasticity throughout the brain is evident from pharmacological studies and investigations with nAChR-subunit knockout mice [152, 154–57]}. Any changes to the expression levels of these receptors, or changes to the afferents that supply these receptors with ACh, could influence the physiology and behavior. A corollary is that some forms of pathology that include compromised synaptic transmission or plasticity could potentially benefit from exogenous stimulation of nAChRs to boost transmission or plasticity capabilities closer to normal levels.
Cholinergic signaling and nAChRs are implicated in many pathological changes to the brain, including Alzheimer’s disease, Parkinson’s disease, schizophrenia, and addiction [137, 158–160]. For instance, in Alzheimer’s disease, the numbers of nAChRs decrease, particularly in the hippocampus and cortex [158, 161]. Evidence indicates that augmenting nAChR activity allosterically or inhibiting acetylcholinesterase may improve attention and rates of learning [75, 162–164], and these effects also may slow neurodegeneration in models of Alzheimer’s disease [165, 166]. Other evidence has shown that, in rats and nonhuman primates, nicotine is neuroprotective against loss of nigrostriatal dopamine neurons [167]. Given the deficit of dopaminergic neurotransmission in Parkinson’s disease, therapeutic activation of nAChRs to facilitate dopamine release onto its target cells may benefit those patients as well. For example, Alzheimer’s drugs, such as donepezil and galantamine, were found to enhance nicotinic function and indirectly influence dopamine release in the striatum [168].
Improved treatments will require the development of new nAChR agonists and antagonists that target nAChRs locally in some cases and broadly in other cases [169, 170]. In addition, the development of new allosteric modulators [171] – those compounds that do not directly activate the receptor but augment function of the receptor when the ligand is present –will also be valuable. Because allosteric modulators do not themselves activate receptors, they will operate with the same spatial and temporal profile as endogenous cholinergic signals. The development of novel compounds, as well as new molecular tools like siRNA and conditional/inducible knock-out or knock-in mice, will also be a benefit to basic research. Presently the nicotinic field has not extensively benefited from the combination of molecular tools and powerful, high-resolution in vivo recordings from freely-moving animals. Those techniques will open up the black-box observations of behavioral studies. The application of these powerful approaches will provide new kinds of data and open additional avenues for research and provide new targets for drug development. These combinations of tools and approaches could be used to determine exactly how and when specific nAChR subunits contribute to processes such as synaptic transmission, synaptic plasticity, normal development, and dysfunction as well as higher order functions such as cognition and consciousness.
Acknowledgments
Work from the laboratory is supported by the NIH’s NINDS and NIDA. BEM is supported by fellowships from the Natural Sciences and Engineering Research Council of Canada, and the Alberta Heritage Foundation for Medical Research. ANP is supported by a training fellowship from the NINDS.
Abbreviations
- ACh
acetylcholine
- AP
action potential
- AMPAR
α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor
- CaMK
Ca2+/calmodulin-dependent protein kinase
- CNS
central nervous system
- CREB
cAMP response element binding protein
- EPSC
excitatory postsynaptic current
- EPSP
excitatory postsynaptic potential
- GABA
gamma-amino butyric acid
- GDP
giant depolarizing potential
- HFS
high frequency stimulation
- IPSC
inhibitory postsynaptic current
- LTD
long-term depression
- LTP
long-term potentiation
- MAP kinase
mitogen activated protein kinase
- mGluR
metabotropic glutamate receptor
- nAChR
nicotinic acetylcholine receptor
- NMDAR
N-methyl-D-aspartate receptor
- PSP
postsynaptic potential
- sEPSC
spontaneous EPSC
- sIPSC
spontaneous IPSC
- STDP
spike timing dependent plasticity
- STP
short-term potentiation
- VGCC
voltage-gated Ca2+ channel
- VTA
ventral tegmental area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–76. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 2.Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- 3.Cooper E, Couturier S, Ballivet M. Pentameric structure and subunit stoichiometry of a neuronal nicotinic acetylcholine receptor. Nature. 1991;350:235–8. doi: 10.1038/350235a0. [DOI] [PubMed] [Google Scholar]
- 4.Lindstrom JM. Acetylcholine receptors and myasthenia. Muscle Nerve. 2000;23:453–77. doi: 10.1002/(sici)1097-4598(200004)23:4<453::aid-mus3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 5.Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–55. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- 6.Jones S, Sudweeks S, Yakel JL. Nicotinic receptors in the brain: correlating physiology with function. Trends in neurosciences. 1999;22:555–61. doi: 10.1016/s0166-2236(99)01471-x. [DOI] [PubMed] [Google Scholar]
- 7.McGehee DS, Role LW. Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol. 1995;57:521–46. doi: 10.1146/annurev.ph.57.030195.002513. [DOI] [PubMed] [Google Scholar]
- 8.Role LW, Berg DK. Nicotinic receptors in the development and modulation of CNS synapses. Neuron. 1996;16:1077–85. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- 9.Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- 10.Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, et al. Distribution of alpha 2, alpha 3, alpha 4, and beta 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol. 1989;284:314–35. doi: 10.1002/cne.902840212. [DOI] [PubMed] [Google Scholar]
- 11.Karlin A. Emerging structure of the nicotinic acetylcholine receptors. Nature reviews. 2002;3:102–14. doi: 10.1038/nrn731. [DOI] [PubMed] [Google Scholar]
- 12.Couturier S, Bertrand D, Matter JM, Hernandez MC, Bertrand S, Millar N, et al. A neuronal nicotinic acetylcholine receptor subunit (alpha 7) is developmentally regulated and forms a homo-oligomeric channel blocked by alpha-BTX. Neuron. 1990;5:847–56. doi: 10.1016/0896-6273(90)90344-f. [DOI] [PubMed] [Google Scholar]
- 13.Chen D, Patrick JW. The alpha-bungarotoxin-binding nicotinic acetylcholine receptor from rat brain contains only the alpha7 subunit. J Biol Chem. 1997;272:24024–9. doi: 10.1074/jbc.272.38.24024. [DOI] [PubMed] [Google Scholar]
- 14.Lena C, Changeux JP, Mulle C. Evidence for “preterminal” nicotinic receptors on GABAergic axons in the rat interpeduncular nucleus. J Neurosci. 1993;13:2680–8. doi: 10.1523/JNEUROSCI.13-06-02680.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zarei MM, Radcliffe KA, Chen D, Patrick JW, Dani JA. Distributions of nicotinic acetylcholine receptor alpha7 and beta2 subunits on cultured hippocampal neurons. Neuroscience. 1999;88:755–64. doi: 10.1016/s0306-4522(98)00246-2. [DOI] [PubMed] [Google Scholar]
- 16.Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, et al. Ultrastructural distribution of the alpha7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci. 2001;21:7993–8003. doi: 10.1523/JNEUROSCI.21-20-07993.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu J, Zhu Y, Heinemann SF. Identification of sequence motifs that target neuronal nicotinic receptors to dendrites and axons. J Neurosci. 2006;26:9780–93. doi: 10.1523/JNEUROSCI.0840-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castro NG, Albuquerque EX. alpha-Bungarotoxin-sensitive hippocampal nicotinic receptor channel has a high calcium permeability. Biophys J. 1995;68:516–24. doi: 10.1016/S0006-3495(95)80213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khiroug L, Giniatullin R, Klein RC, Fayuk D, Yakel JL. Functional mapping and Ca2+ regulation of nicotinic acetylcholine receptor channels in rat hippocampal CA1 neurons. J Neurosci. 2003;23:9024–31. doi: 10.1523/JNEUROSCI.23-27-09024.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hille B. Ion Channels of Excitable Membranes. Sunderland: Sinauer Associates, Inc.; 2001. [Google Scholar]
- 22.Vernino S, Amador M, Luetje CW, Patrick J, Dani JA. Calcium modulation and high calcium permeability of neuronal nicotinic acetylcholine receptors. Neuron. 1992;8:127–34. doi: 10.1016/0896-6273(92)90114-s. [DOI] [PubMed] [Google Scholar]
- 23.Bertrand D, Galzi JL, Devillers-Thiery A, Bertrand S, Changeux JP. Mutations at two distinct sites within the channel domain M2 alter calcium permeability of neuronal alpha 7 nicotinic receptor. Proc Natl Acad Sci U S A. 1993;90:6971–5. doi: 10.1073/pnas.90.15.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haghighi AP, Cooper E. A molecular link between inward rectification and calcium permeability of neuronal nicotinic acetylcholine alpha3beta4 and alpha4beta2 receptors. J Neurosci. 2000;20:529–41. doi: 10.1523/JNEUROSCI.20-02-00529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fayuk D, Yakel JL. Ca2+ permeability of nicotinic acetylcholine receptors in rat hippocampal CA1 interneurones. J Physiol. 2005;566:759–68. doi: 10.1113/jphysiol.2005.089789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corringer PJ, Bertrand S, Bohler S, Edelstein SJ, Changeux JP, Bertrand D. Critical elements determining diversity in agonist binding and desensitization of neuronal nicotinic acetylcholine receptors. J Neurosci. 1998;18:648–57. doi: 10.1523/JNEUROSCI.18-02-00648.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA. Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci. 1997;17:5747–59. doi: 10.1523/JNEUROSCI.17-15-05747.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luetje CW, Patrick J. Both alpha- and beta-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J Neurosci. 1991;11:837–45. doi: 10.1523/JNEUROSCI.11-03-00837.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Papke RL, Bencherif M, Lippiello P. An evaluation of neuronal nicotinic acetylcholine receptor activation by quaternary nitrogen compounds indicates that choline is selective for the alpha 7 subtype. Neurosci Lett. 1996;213:201–4. doi: 10.1016/0304-3940(96)12889-5. [DOI] [PubMed] [Google Scholar]
- 30.Alkondon M, Albuquerque EX. Subtype-specific inhibition of nicotinic acetylcholine receptors by choline: a regulatory pathway. The Journal of pharmacology and experimental therapeutics. 2006;318:268–75. doi: 10.1124/jpet.106.103135. [DOI] [PubMed] [Google Scholar]
- 31.Alkondon M, Pereira EF, Wonnacott S, Albuquerque EX. Blockade of nicotinic currents in hippocampal neurons defines methyllycaconitine as a potent and specific receptor antagonist. Mol Pharmacol. 1992;41:802–8. [PubMed] [Google Scholar]
- 32.Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, Wonnacott S. Methyllycaconitine is a potent antagonist of alpha-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. The Journal of pharmacology and experimental therapeutics. 2002;302:197–204. doi: 10.1124/jpet.302.1.197. [DOI] [PubMed] [Google Scholar]
- 33.Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. The Journal of pharmacology and experimental therapeutics. 1993;265:1455–73. [PubMed] [Google Scholar]
- 34.Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. III. Agonist actions of the novel alkaloid epibatidine and analysis of type II current. The Journal of pharmacology and experimental therapeutics. 1995;274:771–82. [PubMed] [Google Scholar]
- 35.Parker MJ, Harvey SC, Luetje CW. Determinants of agonist binding affinity on neuronal nicotinic receptor beta subunits. The Journal of pharmacology and experimental therapeutics. 2001;299:385–91. [PubMed] [Google Scholar]
- 36.Kulak JM, Musachio JL, McIntosh JM, Quik M. Declines in different beta2* nicotinic receptor populations in monkey striatum after nigrostriatal damage. The Journal of pharmacology and experimental therapeutics. 2002;303:633–9. doi: 10.1124/jpet.102.039347. [DOI] [PubMed] [Google Scholar]
- 37.Sullivan JP, Donnelly-Roberts D, Briggs CA, Anderson DJ, Gopalakrishnan M, Piattoni-Kaplan M, et al. A-85380 [3-(2(S)-azetidinylmethoxy) pyridine]: in vitro pharmacological properties of a novel, high affinity alpha 4 beta 2 nicotinic acetylcholine receptor ligand. Neuropharmacology. 1996;35:725–34. doi: 10.1016/0028-3908(96)84644-2. [DOI] [PubMed] [Google Scholar]
- 38.Chavez-Noriega LE, Crona JH, Washburn MS, Urrutia A, Elliott KJ, Johnson EC. Pharmacological characterization of recombinant human neuronal nicotinic acetylcholine receptors h alpha 2 beta 2, h alpha 2 beta 4, h alpha 3 beta 2, h alpha 3 beta 4, h alpha 4 beta 2, h alpha 4 beta 4 and h alpha 7 expressed in Xenopus oocytes. The Journal of pharmacology and experimental therapeutics. 1997;280:346–56. [PubMed] [Google Scholar]
- 39.McIntosh JM, Plazas PV, Watkins M, Gomez-Casati ME, Olivera BM, Elgoyhen AB. A novel alpha-conotoxin, PeIA, cloned from Conus pergrandis, discriminates between rat alpha9alpha10 and alpha7 nicotinic cholinergic receptors. J Biol Chem. 2005;280:30107–12. doi: 10.1074/jbc.M504102200. [DOI] [PubMed] [Google Scholar]
- 40.Smith JW, Mogg A, Tafi E, Peacey E, Pullar IA, Szekeres P, et al. Ligands selective for alpha4beta2 but not alpha3beta4 or alpha7 nicotinic receptors generalise to the nicotine discriminative stimulus in the rat. Psychopharmacology (Berl) 2007;190:157–70. doi: 10.1007/s00213-006-0596-8. [DOI] [PubMed] [Google Scholar]
- 41.Parent A. Carpenter’s Human Neuroanatomy. Baltimore: Williams & Wilkins; 1996. [Google Scholar]
- 42.Frazier CJ, Buhler AV, Weiner JL, Dunwiddie TV. Synaptic potentials mediated via alpha-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal interneurons. J Neurosci. 1998;18:8228–35. doi: 10.1523/JNEUROSCI.18-20-08228.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hatton GI, Yang QZ. Synaptic potentials mediated by alpha 7 nicotinic acetylcholine receptors in supraoptic nucleus. J Neurosci. 2002;22:29–37. doi: 10.1523/JNEUROSCI.22-01-00029.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alkondon M, Pereira EF, Albuquerque EX. alpha-bungarotoxin- and methyllycaconitine-sensitive nicotinic receptors mediate fast synaptic transmission in interneurons of rat hippocampal slices. Brain Res. 1998;810:257–63. doi: 10.1016/s0006-8993(98)00880-4. [DOI] [PubMed] [Google Scholar]
- 45.Roerig B, Nelson DA, Katz LC. Fast synaptic signaling by nicotinic acetylcholine and serotonin 5-HT3 receptors in developing visual cortex. J Neurosci. 1997;17:8353–62. doi: 10.1523/JNEUROSCI.17-21-08353.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuffler SW, Yoshikami D. The number of transmitter molecules in a quantum: an estimate from iontophoretic application of acetylcholine at the neuromuscular synapse. J Physiol. 1975;251:465–82. doi: 10.1113/jphysiol.1975.sp011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Umbriaco D, Garcia S, Beaulieu C, Descarries L. Relational features of acetylcholine, noradrenaline, serotonin and GABA axon terminals in the stratum radiatum of adult rat hippocampus (CA1) Hippocampus. 1995;5:605–20. doi: 10.1002/hipo.450050611. [DOI] [PubMed] [Google Scholar]
- 48.Descarries L, Gisiger V, Steriade M. Diffuse transmission by acetylcholine in the CNS. Prog Neurobiol. 1997;53:603–25. doi: 10.1016/s0301-0082(97)00050-6. [DOI] [PubMed] [Google Scholar]
- 49.Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–6. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- 50.McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–6. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- 51.Dajas-Bailador FA, Mogg AJ, Wonnacott S. Intracellular Ca2+ signals evoked by stimulation of nicotinic acetylcholine receptors in SH-SY5Y cells: contribution of voltage-operated Ca2+ channels and Ca2+ stores. Journal of neurochemistry. 2002;81:606–14. doi: 10.1046/j.1471-4159.2002.00846.x. [DOI] [PubMed] [Google Scholar]
- 52.Dickinson JA, Hanrott KE, Mok MH, Kew JN, Wonnacott S. Differential coupling of alpha7 and non-alpha7 nicotinic acetylcholine receptors to calcium-induced calcium release and voltage-operated calcium channels in PC12 cells. Journal of neurochemistry. 2007;100:1089–96. doi: 10.1111/j.1471-4159.2006.04273.x. [DOI] [PubMed] [Google Scholar]
- 53.Rathouz MM, Vijayaraghavan S, Berg DK. Acetylcholine differentially affects intracellular calcium via nicotinic and muscarinic receptors on the same population of neurons. J Biol Chem. 1995;270:14366–75. doi: 10.1074/jbc.270.24.14366. [DOI] [PubMed] [Google Scholar]
- 54.Rathouz MM, Vijayaraghavan S, Berg DK. Elevation of intracellular calcium levels in neurons by nicotinic acetylcholine receptors. Mol Neurobiol. 1996;12:117–31. doi: 10.1007/BF02740649. [DOI] [PubMed] [Google Scholar]
- 55.Grady S, Marks MJ, Wonnacott S, Collins AC. Characterization of nicotinic receptor-mediated [3H]dopamine release from synaptosomes prepared from mouse striatum. Journal of neurochemistry. 1992;59:848–56. doi: 10.1111/j.1471-4159.1992.tb08322.x. [DOI] [PubMed] [Google Scholar]
- 56.Marks MJ, Farnham DA, Grady SR, Collins AC. Nicotinic receptor function determined by stimulation of rubidium efflux from mouse brain synaptosomes. The Journal of pharmacology and experimental therapeutics. 1993;264:542–52. [PubMed] [Google Scholar]
- 57.Quik M, McIntosh JM. Striatal alpha6* nicotinic acetylcholine receptors: potential targets for Parkinson’s disease therapy. The Journal of pharmacology and experimental therapeutics. 2006;316:481–9. doi: 10.1124/jpet.105.094375. [DOI] [PubMed] [Google Scholar]
- 58.Wonnacott S. Presynaptic nicotinic ACh receptors. Trends in neurosciences. 1997;20:92–8. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- 59.Engelman HS, MacDermott AB. Presynaptic ionotropic receptors and control of transmitter release. Nature reviews. 2004;5:135–45. doi: 10.1038/nrn1297. [DOI] [PubMed] [Google Scholar]
- 60.Vizi ES, Lendvai B. Modulatory role of presynaptic nicotinic receptors in synaptic and non-synaptic chemical communication in the central nervous system. Brain Res Brain Res Rev. 1999;30:219–35. doi: 10.1016/s0165-0173(99)00016-8. [DOI] [PubMed] [Google Scholar]
- 61.MacDermott AB, Role LW, Siegelbaum SA. Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci. 1999;22:443–85. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- 62.Lena C, Changeux JP. Role of Ca2+ ions in nicotinic facilitation of GABA release in mouse thalamus. J Neurosci. 1997;17:576–85. doi: 10.1523/JNEUROSCI.17-02-00576.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang BW, Liao WN, Chang CT, Wang SJ. Facilitation of glutamate release by nicotine involves the activation of a Ca2+/calmodulin signaling pathway in rat prefrontal cortex nerve terminals. Synapse. 2006;59:491–501. doi: 10.1002/syn.20267. [DOI] [PubMed] [Google Scholar]
- 64.Tredway TL, Guo JZ, Chiappinelli VA. N-type voltage-dependent calcium channels mediate the nicotinic enhancement of GABA release in chick brain. J Neurophysiol. 1999;81:447–54. doi: 10.1152/jn.1999.81.2.447. [DOI] [PubMed] [Google Scholar]
- 65.Soliakov L, Wonnacott S. Voltage-sensitive Ca2+ channels involved in nicotinic receptor-mediated [3H]dopamine release from rat striatal synaptosomes. Journal of neurochemistry. 1996;67:163–70. doi: 10.1046/j.1471-4159.1996.67010163.x. [DOI] [PubMed] [Google Scholar]
- 66.Kulak JM, McIntosh JM, Yoshikami D, Olivera BM. Nicotine-evoked transmitter release from synaptosomes: functional association of specific presynaptic acetylcholine receptors and voltage-gated calcium channels. Journal of neurochemistry. 2001;77:1581–9. doi: 10.1046/j.1471-4159.2001.00357.x. [DOI] [PubMed] [Google Scholar]
- 67.Lee A, Zhou H, Scheuer T, Catterall WA. Molecular determinants of Ca(2+)/calmodulin-dependent regulation of Ca(v)2.1 channels. Proc Natl Acad Sci U S A. 2003;100:16059–64. doi: 10.1073/pnas.2237000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharma G, Vijayaraghavan S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc Natl Acad Sci U S A. 2001;98:4148–53. doi: 10.1073/pnas.071540198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu LG, Saggau P. Presynaptic calcium is increased during normal synaptic transmission and paired-pulse facilitation, but not in long-term potentiation in area CA1 of hippocampus. J Neurosci. 1994;14:645–54. doi: 10.1523/JNEUROSCI.14-02-00645.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Levin ED, Christopher NC, Briggs SJ, Rose JE. Chronic nicotine reverses working memory deficits caused by lesions of the fimbria or medial basalocortical projection. Brain Res Cogn Brain Res. 1993;1:137–43. doi: 10.1016/0926-6410(93)90021-v. [DOI] [PubMed] [Google Scholar]
- 71.Levin ED, Conners CK, Silva D, Hinton SC, Meck WH, March J, et al. Transdermal nicotine effects on attention. Psychopharmacology (Berl) 1998;140:135–41. doi: 10.1007/s002130050750. [DOI] [PubMed] [Google Scholar]
- 72.Picciotto MR, Zoli M, Lena C, Bessis A, Lallemand Y, Le Novere N, et al. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–7. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- 73.Newhouse PA, Potter A, Levin ED. Nicotinic system involvement in Alzheimer’s and Parkinson’s diseases. Implications for therapeutics Drugs Aging. 1997;11:206–28. doi: 10.2165/00002512-199711030-00005. [DOI] [PubMed] [Google Scholar]
- 74.Potter AS, Newhouse PA. Effects of acute nicotine administration on behavioral inhibition in adolescents with attention-deficit/hyperactivity disorder. Psychopharmacology (Berl) 2004;176:182–94. doi: 10.1007/s00213-004-1874-y. [DOI] [PubMed] [Google Scholar]
- 75.White HK, Levin ED. Four-week nicotine skin patch treatment effects on cognitive performance in Alzheimer’s disease. Psychopharmacology (Berl) 1999;143:158–65. doi: 10.1007/s002130050931. [DOI] [PubMed] [Google Scholar]
- 76.Ohno M, Yamamoto T, Watanabe S. Blockade of hippocampal nicotinic receptors impairs working memory but not reference memory in rats. Pharmacol Biochem Behav. 1993;45:89–93. doi: 10.1016/0091-3057(93)90091-7. [DOI] [PubMed] [Google Scholar]
- 77.Grigoryan GA, Mitchell SN, Hodges H, Sinden JD, Gray JA. Are the cognitive-enhancing effects of nicotine in the rat with lesions to the forebrain cholinergic projection system mediated by an interaction with the noradrenergic system? Pharmacol Biochem Behav. 1994;49:511–21. doi: 10.1016/0091-3057(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 78.Frotscher M, Leranth C. Cholinergic innervation of the rat hippocampus as revealed by choline acetyltransferase immunocytochemistry: a combined light and electron microscopic study. J Comp Neurol. 1985;239:237–46. doi: 10.1002/cne.902390210. [DOI] [PubMed] [Google Scholar]
- 79.Woolf NJ. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol. 1991;37:475–524. doi: 10.1016/0301-0082(91)90006-m. [DOI] [PubMed] [Google Scholar]
- 80.Hefft S, Hulo S, Bertrand D, Muller D. Synaptic transmission at nicotinic acetylcholine receptors in rat hippocampal organotypic cultures and slices. J Physiol. 1999;515 ( Pt 3):769–76. doi: 10.1111/j.1469-7793.1999.769ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ji D, Lape R, Dani JA. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron. 2001;31:131–41. doi: 10.1016/s0896-6273(01)00332-4. [DOI] [PubMed] [Google Scholar]
- 82.Jones S, Yakel JL. Functional nicotinic ACh receptors on interneurones in the rat hippocampus. J Physiol. 1997;504 ( Pt 3):603–10. doi: 10.1111/j.1469-7793.1997.603bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Alkondon M, Pereira EF, Eisenberg HM, Albuquerque EX. Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J Neurosci. 1999;19:2693–705. doi: 10.1523/JNEUROSCI.19-07-02693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ji D, Dani JA. Inhibition and disinhibition of pyramidal neurons by activation of nicotinic receptors on hippocampal interneurons. J Neurophysiol. 2000;83:2682–90. doi: 10.1152/jn.2000.83.5.2682. [DOI] [PubMed] [Google Scholar]
- 85.Racca C, Stephenson FA, Streit P, Roberts JD, Somogyi P. NMDA receptor content of synapses in stratum radiatum of the hippocampal CA1 area. J Neurosci. 2000;20:2512–22. doi: 10.1523/JNEUROSCI.20-07-02512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- 87.Malenka RC, Nicoll RA. Long-term potentiation--a decade of progress? Science. 1999;285:1870–4. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 88.Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–7. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- 89.Fujii S, Ji Z, Morita N, Sumikawa K. Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Res. 1999;846:137–43. doi: 10.1016/s0006-8993(99)01982-4. [DOI] [PubMed] [Google Scholar]
- 90.Hamid S, Dawe GS, Gray JA, Stephenson JD. Nicotine induces long-lasting potentiation in the dentate gyrus of nicotine-primed rats. Neurosci Res. 1997;29:81–5. doi: 10.1016/s0168-0102(97)00074-6. [DOI] [PubMed] [Google Scholar]
- 91.Fujii S, Sumikawa K. Acute and chronic nicotine exposure reverse age-related declines in the induction of long-term potentiation in the rat hippocampus. Brain Res. 2001;894:347–53. doi: 10.1016/s0006-8993(01)02057-1. [DOI] [PubMed] [Google Scholar]
- 92.Ge S, Dani JA. Nicotinic acetylcholine receptors at glutamate synapses facilitate long-term depression or potentiation. J Neurosci. 2005;25:6084–91. doi: 10.1523/JNEUROSCI.0542-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rosato-Siri M, Cattaneo A, Cherubini E. Nicotine-induced enhancement of synaptic plasticity at CA3-CA1 synapses requires GABAergic interneurons in adult anti-NGF mice. J Physiol. 2006;576:361–77. doi: 10.1113/jphysiol.2006.114587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–5. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- 95.Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–72. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mann EO, Greenfield SA. Novel modulatory mechanisms revealed by the sustained application of nicotine in the guinea-pig hippocampus in vitro. J Physiol. 2003;551:539–50. doi: 10.1113/jphysiol.2003.045492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Klein RC, Yakel JL. Paired-pulse potentiation of alpha7-containing nAChRs in rat hippocampal CA1 stratum radiatum interneurones. J Physiol. 2005;568:881–9. doi: 10.1113/jphysiol.2005.096081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Welsby P, Rowan M, Anwyl R. Nicotinic receptor-mediated enhancement of long-term potentiation involves activation of metabotropic glutamate receptors and ryanodine-sensitive calcium stores in the dentate gyrus. Eur J Neurosci. 2006;24:3109–18. doi: 10.1111/j.1460-9568.2006.05187.x. [DOI] [PubMed] [Google Scholar]
- 99.Alkondon M, Pereira EF, Eisenberg HM, Albuquerque EX. Nicotinic receptor activation in human cerebral cortical interneurons: a mechanism for inhibition and disinhibition of neuronal networks. J Neurosci. 2000;20:66–75. doi: 10.1523/JNEUROSCI.20-01-00066.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Buhler AV, Dunwiddie TV. alpha7 nicotinic acetylcholine receptors on GABAergic interneurons evoke dendritic and somatic inhibition of hippocampal neurons. J Neurophysiol. 2002;87:548–57. doi: 10.1152/jn.00316.2001. [DOI] [PubMed] [Google Scholar]
- 101.Alkondon M, Pereira EF, Barbosa CT, Albuquerque EX. Neuronal nicotinic acetylcholine receptor activation modulates gamma-aminobutyric acid release from CA1 neurons of rat hippocampal slices. The Journal of pharmacology and experimental therapeutics. 1997;283:1396–411. [PubMed] [Google Scholar]
- 102.Zhu PJ, Chiappinelli VA. Nicotine modulates evoked GABAergic transmission in the brain. J Neurophysiol. 1999;82:3041–5. doi: 10.1152/jn.1999.82.6.3041. [DOI] [PubMed] [Google Scholar]
- 103.Clark BA, Monsivais P, Branco T, London M, Hausser M. The site of action potential initiation in cerebellar Purkinje neurons. Nat Neurosci. 2005;8:137–9. doi: 10.1038/nn1390. [DOI] [PubMed] [Google Scholar]
- 104.Monsivais P, Clark BA, Roth A, Hausser M. Determinants of action potential propagation in cerebellar Purkinje cell axons. J Neurosci. 2005;25:464–72. doi: 10.1523/JNEUROSCI.3871-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Khaliq ZM, Raman IM. Axonal propagation of simple and complex spikes in cerebellar Purkinje neurons. J Neurosci. 2005;25:454–63. doi: 10.1523/JNEUROSCI.3045-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stuart G, Spruston N, Sakmann B, Hausser M. Action potential initiation and backpropagation in neurons of the mammalian CNS. Trends in neurosciences. 1997;20:125–31. doi: 10.1016/s0166-2236(96)10075-8. [DOI] [PubMed] [Google Scholar]
- 107.Johnston D, Christie BR, Frick A, Gray R, Hoffman DA, Schexnayder LK, et al. Active dendrites, potassium channels and synaptic plasticity. Philos Trans R Soc Lond B Biol Sci. 2003;358:667–74. doi: 10.1098/rstb.2002.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Migliore M, Hoffman DA, Magee JC, Johnston D. Role of an A-type K+ conductance in the back-propagation of action potentials in the dendrites of hippocampal pyramidal neurons. J Comput Neurosci. 1999;7:5–15. doi: 10.1023/a:1008906225285. [DOI] [PubMed] [Google Scholar]
- 109.Forster I, Bertrand D. Inward rectification of neuronal nicotinic acetylcholine receptors investigated by using the homomeric alpha 7 receptor. Proc Biol Sci. 1995;260:139–48. doi: 10.1098/rspb.1995.0071. [DOI] [PubMed] [Google Scholar]
- 110.Fisher JL, Dani JA. Nicotinic receptors on hippocampal cultures can increase synaptic glutamate currents while decreasing the NMDA-receptor component. Neuropharmacology. 2000;39:2756–69. doi: 10.1016/s0028-3908(00)00102-7. [DOI] [PubMed] [Google Scholar]
- 111.Shoop RD, Chang KT, Ellisman MH, Berg DK. Synaptically driven calcium transients via nicotinic receptors on somatic spines. J Neurosci. 2001;21:771–81. doi: 10.1523/JNEUROSCI.21-03-00771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Greenberg ME, Ziff EB, Greene LA. Stimulation of neuronal acetylcholine receptors induces rapid gene transcription. Science. 1986;234:80–3. doi: 10.1126/science.3749894. [DOI] [PubMed] [Google Scholar]
- 113.Hu M, Liu QS, Chang KT, Berg DK. Nicotinic regulation of CREB activation in hippocampal neurons by glutamatergic and nonglutamatergic pathways. Mol Cell Neurosci. 2002;21:616–25. doi: 10.1006/mcne.2002.1202. [DOI] [PubMed] [Google Scholar]
- 114.Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol. 2003;71:401–37. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 115.Mizuno K, Giese KP. Hippocampus-dependent memory formation: do memory type-specific mechanisms exist? J Pharmacol Sci. 2005;98:191–7. doi: 10.1254/jphs.crj05005x. [DOI] [PubMed] [Google Scholar]
- 116.Ho OH, Delgado JY, O’Dell TJ. Phosphorylation of proteins involved in activity-dependent forms of synaptic plasticity is altered in hippocampal slices maintained in vitro. Journal of neurochemistry. 2004;91:1344–57. doi: 10.1111/j.1471-4159.2004.02815.x. [DOI] [PubMed] [Google Scholar]
- 117.Matsuyama S, Matsumoto A, Enomoto T, Nishizaki T. Activation of nicotinic acetylcholine receptors induces long-term potentiation in vivo in the intact mouse dentate gyrus. Eur J Neurosci. 2000;12:3741–7. doi: 10.1046/j.1460-9568.2000.00259.x. [DOI] [PubMed] [Google Scholar]
- 118.Matsuyama S, Matsumoto A. Epibatidine induces long-term potentiation (LTP) via activation of alpha4beta2 nicotinic acetylcholine receptors (nAChRs) in vivo in the intact mouse dentate gyrus: both alpha7 and alpha4beta2 nAChRs essential to nicotinic LTP. J Pharmacol Sci. 2003;93:180–7. doi: 10.1254/jphs.93.180. [DOI] [PubMed] [Google Scholar]
- 119.Freir DB, Herron CE. Nicotine enhances the depressive actions of A beta 1–40 on long-term potentiation in the rat hippocampal CA1 region in vivo. J Neurophysiol. 2003;89:2917–22. doi: 10.1152/jn.00996.2002. [DOI] [PubMed] [Google Scholar]
- 120.O’Dell TJ, Christensen BN. Mecamylamine is a selective non-competitive antagonist of N-methyl-D-aspartate- and aspartate-induced currents in horizontal cells dissociated from the catfish retina. Neurosci Lett. 1988;94:93–8. doi: 10.1016/0304-3940(88)90276-5. [DOI] [PubMed] [Google Scholar]
- 121.Elrod K, Buccafusco JJ. Correlation of the amnestic effects of nicotinic antagonists with inhibition of regional brain acetylcholine synthesis in rats. The Journal of pharmacology and experimental therapeutics. 1991;258:403–9. [PubMed] [Google Scholar]
- 122.Salas R, Cook KD, Bassetto L, De Biasi M. The alpha3 and beta4 nicotinic acetylcholine receptor subunits are necessary for nicotine-induced seizures and hypolocomotion in mice. Neuropharmacology. 2004;47:401–7. doi: 10.1016/j.neuropharm.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 123.Gentry CL, Lukas RJ. Local anesthetics noncompetitively inhibit function of four distinct nicotinic acetylcholine receptor subtypes. The Journal of pharmacology and experimental therapeutics. 2001;299:1038–48. [PubMed] [Google Scholar]
- 124.Couey JJ, Meredith RM, Spijker S, Poorthuis RB, Smit AB, Brussaard AB, et al. Distributed Network Actions by Nicotine Increase the Threshold for Spike-Timing-Dependent Plasticity in Prefrontal Cortex. Neuron. 2007;54:73–87. doi: 10.1016/j.neuron.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 125.Hara K, Harris RA. The anesthetic mechanism of urethane: the effects on neurotransmitter-gated ion channels. Anesthesia and analgesia. 2002;94:313–8. doi: 10.1097/00000539-200202000-00015. table of contents. [DOI] [PubMed] [Google Scholar]
- 126.Maggi L, Sher E, Cherubini E. Regulation of GABA release by nicotinic acetylcholine receptors in the neonatal rat hippocampus. J Physiol. 2001;536:89–100. doi: 10.1111/j.1469-7793.2001.00089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kasyanov AM, Safiulina VF, Voronin LL, Cherubini E. GABA-mediated giant depolarizing potentials as coincidence detectors for enhancing synaptic efficacy in the developing hippocampus. Proc Natl Acad Sci U S A. 2004;101:3967–72. doi: 10.1073/pnas.0305974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Maggi L, Le Magueresse C, Changeux JP, Cherubini E. Nicotine activates immature “silent” connections in the developing hippocampus. Proc Natl Acad Sci U S A. 2003;100:2059–64. doi: 10.1073/pnas.0437947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Maggi L, Sola E, Minneci F, Le Magueresse C, Changeux JP, Cherubini E. Persistent decrease in synaptic efficacy induced by nicotine at Schaffer collateral-CA1 synapses in the immature rat hippocampus. J Physiol. 2004;559:863–74. doi: 10.1113/jphysiol.2004.067041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–26. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 131.Liu Z, Neff RA, Berg DK. Sequential interplay of nicotinic and GABAergic signaling guides neuronal development. Science. 2006;314:1610–3. doi: 10.1126/science.1134246. [DOI] [PubMed] [Google Scholar]
- 132.Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, et al. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–5. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 133.Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nature reviews. 2002;3:728–39. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- 134.Schultz W. Behavioral dopamine signals. Trends in neurosciences. 2007 doi: 10.1016/j.tins.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 135.Charpantier E, Barneoud P, Moser P, Besnard F, Sgard F. Nicotinic acetylcholine subunit mRNA expression in dopaminergic neurons of the rat substantia nigra and ventral tegmental area. Neuroreport. 1998;9:3097–101. doi: 10.1097/00001756-199809140-00033. [DOI] [PubMed] [Google Scholar]
- 136.Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci. 2003;23:3176–85. doi: 10.1523/JNEUROSCI.23-08-03176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dani JA, Ji D, Zhou FM. Synaptic plasticity and nicotine addiction. Neuron. 2001;31:349–52. doi: 10.1016/s0896-6273(01)00379-8. [DOI] [PubMed] [Google Scholar]
- 138.Balfour DJ, Wright AE, Benwell ME, Birrell CE. The putative role of extra-synaptic mesolimbic dopamine in the neurobiology of nicotine dependence. Behav Brain Res. 2000;113:73–83. doi: 10.1016/s0166-4328(00)00202-3. [DOI] [PubMed] [Google Scholar]
- 139.Dani JA, Heinemann S. Molecular and cellular aspects of nicotine abuse. Neuron. 1996;16:905–8. doi: 10.1016/s0896-6273(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 140.Pidoplichko VI, Noguchi J, Areola OO, Liang Y, Peterson J, Zhang T, et al. Nicotinic cholinergic synaptic mechanisms in the ventral tegmental area contribute to nicotine addiction. Learn Mem. 2004;11:60–9. doi: 10.1101/lm.70004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Di Chiara G. Role of dopamine in the behavioural actions of nicotine related to addiction. Eur J Pharmacol. 2000;393:295–314. doi: 10.1016/s0014-2999(00)00122-9. [DOI] [PubMed] [Google Scholar]
- 142.Henningfield JE, Stapleton JM, Benowitz NL, Grayson RF, London ED. Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug Alcohol Depend. 1993;33:23–9. doi: 10.1016/0376-8716(93)90030-t. [DOI] [PubMed] [Google Scholar]
- 143.Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53:606–17. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- 144.Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–4. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- 145.Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, et al. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–7. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- 146.Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–57. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- 147.Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–19. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 148.Yin R, French ED. A comparison of the effects of nicotine on dopamine and non-dopamine neurons in the rat ventral tegmental area: an in vitro electrophysiological study. Brain Res Bull. 2000;51:507–14. doi: 10.1016/s0361-9230(00)00237-9. [DOI] [PubMed] [Google Scholar]
- 149.Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci. 1984;4:2877–90. doi: 10.1523/JNEUROSCI.04-11-02877.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984;4:2866–76. doi: 10.1523/JNEUROSCI.04-11-02866.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Grenhoff J, Aston-Jones G, Svensson TH. Nicotinic effects on the firing pattern of midbrain dopamine neurons. Acta physiologica Scandinavica. 1986;128:351–8. doi: 10.1111/j.1748-1716.1986.tb07988.x. [DOI] [PubMed] [Google Scholar]
- 152.Mameli-Engvall M, Evrard A, Pons S, Maskos U, Svensson TH, Changeux JP, et al. Hierarchical control of dopamine neuron-firing patterns by nicotinic receptors. Neuron. 2006;50:911–21. doi: 10.1016/j.neuron.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 153.Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, et al. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–7. doi: 10.1038/nature03694. [DOI] [PubMed] [Google Scholar]
- 154.De Biasi M. Nicotinic receptor mutant mice in the study of autonomic function. Curr Drug Targets CNS Neurol Disord. 2002;1:331–6. doi: 10.2174/1568007023339148. [DOI] [PubMed] [Google Scholar]
- 155.Champtiaux N, Changeux JP. Knockout and knockin mice to investigate the role of nicotinic receptors in the central nervous system. Prog Brain Res. 2004;145:235–51. doi: 10.1016/s0079-6123(03)45016-4. [DOI] [PubMed] [Google Scholar]
- 156.Picciotto MR, Caldarone BJ, Brunzell DH, Zachariou V, Stevens TR, King SL. Neuronal nicotinic acetylcholine receptor subunit knockout mice: physiological and behavioral phenotypes and possible clinical implications. Pharmacol Ther. 2001;92:89–108. doi: 10.1016/s0163-7258(01)00161-9. [DOI] [PubMed] [Google Scholar]
- 157.Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–9. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]
- 158.Paterson D, Nordberg A. Neuronal nicotinic receptors in the human brain. Prog Neurobiol. 2000;61:75–111. doi: 10.1016/s0301-0082(99)00045-3. [DOI] [PubMed] [Google Scholar]
- 159.Dani JA, Harris RA. Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat Neurosci. 2005;8:1465–70. doi: 10.1038/nn1580. [DOI] [PubMed] [Google Scholar]
- 160.Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci U S A. 1997;94:587–92. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Perry E, Martin-Ruiz C, Lee M, Griffiths M, Johnson M, Piggott M, et al. Nicotinic receptor subtypes in human brain ageing, Alzheimer and Lewy body diseases. Eur J Pharmacol. 2000;393:215–22. doi: 10.1016/s0014-2999(00)00064-9. [DOI] [PubMed] [Google Scholar]
- 162.Levin ED, Rezvani AH. Development of nicotinic drug therapy for cognitive disorders. Eur J Pharmacol. 2000;393:141–6. doi: 10.1016/s0014-2999(99)00885-7. [DOI] [PubMed] [Google Scholar]
- 163.Maelicke A, Samochocki M, Jostock R, Fehrenbacher A, Ludwig J, Albuquerque EX, et al. Allosteric sensitization of nicotinic receptors by galantamine, a new treatment strategy for Alzheimer’s disease. Biol Psychiatry. 2001;49:279–88. doi: 10.1016/s0006-3223(00)01109-4. [DOI] [PubMed] [Google Scholar]
- 164.Papke RL, Meyer E, Nutter T, Uteshev VV. alpha7 receptor-selective agonists and modes of alpha7 receptor activation. Eur J Pharmacol. 2000;393:179–95. doi: 10.1016/s0014-2999(00)00009-1. [DOI] [PubMed] [Google Scholar]
- 165.Wang HY, Lee DH, D’Andrea MR, Peterson PA, Shank RP, Reitz AB. beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J Biol Chem. 2000;275:5626–32. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- 166.Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, et al. alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J Biol Chem. 2001;276:13541–6. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- 167.Quik M, O’Neill M, Perez XA. Nicotine neuroprotection against nigrostriatal damage: importance of the animal model. Trends Pharmacol Sci. 2007;28:229–35. doi: 10.1016/j.tips.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 168.Zhang L, Zhou FM, Dani JA. Cholinergic drugs for Alzheimer’s disease enhance in vitro dopamine release. Mol Pharmacol. 2004;66:538–44. doi: 10.1124/mol.104.000299. [DOI] [PubMed] [Google Scholar]
- 169.Buccafusco JJ. Neuronal Nicotinic Receptor Subtypes: defining therapeutic targets. Molecular interventions. 2004;4:285–95. doi: 10.1124/mi.4.5.8. [DOI] [PubMed] [Google Scholar]
- 170.Dani JA, De Biasi M, Liang Y, Peterson J, Zhang L, Zhang T, et al. Potential applications of nicotinic ligands in the laboratory and clinic. Bioorg Med Chem Lett. 2004;14:1837–9. doi: 10.1016/j.bmcl.2003.07.036. [DOI] [PubMed] [Google Scholar]
- 171.Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–8. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]