

Abstract
Human hepatic peroxisomal AGT (alanine:glyoxylate aminotransferase) is a PLP (pyridoxal 5′-phosphate)-dependent enzyme whose deficiency causes primary hyperoxaluria Type I, a rare autosomal recessive disorder. To acquire experimental evidence for the physiological function of AGT, the Keq,overall of the reaction, the steady-state kinetic parameters of the forward and reverse reactions, and the pre-steady-state kinetics of the half-reactions of the PLP form of AGT with L-alanine or glycine and the PMP (pyridoxamine 5′-phosphate) form with pyruvate or glyoxylate have been measured. The results indicate that the enzyme is highly specific for catalysing glyoxylate to glycine processing, thereby playing a key role in glyoxylate detoxification. Analysis of the reaction course also reveals that PMP remains bound to the enzyme during the catalytic cycle and that the AGT–PMP complex displays a reactivity towards oxo acids higher than that of apoAGT in the presence of PMP. These findings are tentatively related to possible subtle rearrangements at the active site also indicated by the putative binding mode of catalytic intermediates. Additionally, the catalytic and spectroscopic features of the naturally occurring G82E variant have been analysed. Although, like the wild-type, the G82E variant is able to bind 2 mol PLP/dimer, it exhibits a significant reduced affinity for PLP and even more for PMP compared with wild-type, and an altered conformational state of the bound PLP. The striking molecular defect of the mutant, consisting in the dramatic decrease of the overall catalytic activity (∼0.1% of that of normal AGT), appears to be related to the inability to undergo an efficient transaldimination of the PLP form of the enzyme with amino acids as well as an efficient conversion of AGT–PMP into AGT–PLP. Overall, careful biochemical analyses have allowed elucidation of the mechanism of action of AGT and the way in which the disease causing G82E mutation affects it.
Keywords: alanine:glyoxylate aminotransferase, disease-causing mutation, primary hyperoxaluria, pyridoxal 5′-phosphate, pyridoxamine 5′-phosphate
Abbreviations: AGT, alanine:glyoxylate aminotransferase; PLP, pyridoxal 5′-phosphate; PMP, pyridoxamine 5′-phosphate; PH1, primary hyperoxaluria Type I
INTRODUCTION
PH1 (primary hyperoxaluria Type I) is a rare autosomal recessive disorder caused by a deficiency of the hepatic enzyme AGT (alanine:glyoxylate aminotransferase). AGT deficiency results in the oxidation of glyoxylate rather than in its transamination to glycine. In this pathology, accumulated glyoxylate oxidizes to oxalate, excretion of which leads to the chronic deposition of insoluble calcium oxalate in the kidneys and urinary tract. AGT is encoded by the AGXT gene and is normally peroxisomal in human liver. Two main polymorphic variants have been identified: the ‘minor’ allele differs from the ‘major’ allele by the presence of P11L and I340M amino acid substitutions in the former. In addition, the minor AGXT allele contains a 74 bp duplication in intron 1 [1]. The I340M replacement and intron 1 duplication appear to have little or no effect on the properties of AGT, whereas the P11L replacement seems to cause some, but not detrimental, effects on the functionality of the enzyme. However, the latter substitution plays an important role in PH1 in that a number of missense mutations cause significantly adverse effects on the AGT function only in the presence of the P11L polymorphism. Decrease of catalytic activity, inhibition of dimerization, aggregation and rapid degradation, defect of PLP binding or on the import of AGT into peroxisomes have been described as the major effects found in the most common naturally occurring amino acids substitutions [2,3]. Apart from the synergistic effects of the P11L polyphormism and the above-mentioned mutations, another mutation which segregates with the major AGXT allele is known to be responsible for PH1. The sequence of AGT cDNA from the liver of PH1 patients having normal levels of hepatic peroxisomal immunoreactive AGT protein, but no AGT catalytic activity, revealed the presence of a single point mutation (G→A at cDNA nucleotide 367) which is predicted to cause a glycine to glutamate substitution at residue 82 of the AGT protein [4].
AGT is a PLP (pyridoxal 5′-phosphate)-containing enzyme that catalyses the reaction shown in Scheme 1. As in many other aminotransferases, in the first half-transamination reaction, L-alanine reacts with the pyridoxal form of the enzyme (AGT–PLP) to yield the oxo acid (pyruvate) and the pyridoxamine form of the enzyme (AGT–PMP, where PMP is pyridoxamine 5′-phosphate). In the second half-transamination, glyoxylate reacts with AGT–PMP to reform AGT–PLP and glycine (Scheme 2). Although the serious consequences of peroxisomal AGT deficiency allow one to expect a prominent role of hepatic detoxification for the transamination catalysed by AGT using glyoxylate as an amino acceptor, at present no kinetic and equilibrium data for the reactions catalysed by normal AGT supporting this view are available. The X-ray structure at 2.5 Å (1 Å=0.1 nm) of normal human AGT in complex with the competitive inhibitor amino-oxyacetic acid has been solved [5]. The enzyme exists as a dimer composed of two identical subunits, each of which can be divided into an N-terminal extension of about 20 amino acid residues, a large domain of about 260 residues containing most of the active site and the dimerization interface, and a smaller domain of about 110 residues of uncertain function. The analysis of this structure has permitted the rationalization in terms of enzyme folding, dimerization and stability of the effects of the most important missense mutations in the AGXT gene. However, the molecular basis of these effects is far from being elucidated. Presently, a detailed picture of the mechanism of action of the wild-type enzyme is needed to advance our understanding of this important protein. Therefore the objective of our present work was to study the wild-type enzyme using kinetic, spectroscopic and computational methods. We show that the overall transamination reaction strongly favours glyoxylate detoxification. Evidence is also provided that (i) the high affinity of PMP allows it to remain bound to the enzyme during the catalytic cycle, and (ii) the AGT–PMP complex displays a reactivity towards oxo acids higher than that of apoAGT in the presence of PMP. Spectroscopic and bioinformatic studies suggest the occurrence of subtle rearrangements at the active site associated with some identified catalytic intermediates. In addition, the G82E mutant has been constructed, expressed and purified. The kinetic and spectroscopic properties of the G82E mutant reveal the molecular mechanism underlying the dramatic loss of activity due to substitution of the glycine residue at position 82 by a glutamate residue.
Scheme 1. Reaction catalysed by AGT.

Scheme 2. Mechanism of the reaction catalyzed by AGT.

The double arrows represent transaldimination steps.
EXPERIMENTAL
Materials
PLP, PMP, L- and D-alanine, glycine, L-aspartic acid, L-glutamic acid, L-phenylalanine, L-arginine, L-serine, glyoxylate, pyruvate, rabbit muscle L-lactic dehydrogenase and isopropyl-β-D-thiogalactoside were all purchased from Sigma. All other chemicals were of the highest purity available.
Site-directed mutagenesis
The G82E mutant form of human AGT was made on the wild-type construct pAGT-His [2] using the QuikChange™ site-directed mutagenesis kit from Stratagene (see Supplementary Materials at http://www.BiochemJ.org/bj/408/bj4080039add.htm).
Expression and purification of recombinant wild-type and G82E mutant AGT-His tagged
Escherichia coli JM109 cells transformed with the plasmid pAGT-His or pAGT[G82E]-His were grown using standard techniques. Details of the preparation of AGT in the wild-type and G82E mutant forms are given in the Supplementary Materials (at http://www.BiochemJ.org/bj/408/bj4080039add.htm). The wild-type and mutant enzymes were stored at −20 °C without apparent loss of activity for at least 1 month. The typical yields were approx. 13 mg/litre of culture. The apparent molar absorption coefficient at 280 nm was determined as described by Pace et al. [6] and was equal to 9.54×104 M−1·cm−1.
Enzyme activity assays
During enzyme purification, assays were carried out as described previously [7], except that the AGT assay was modified by using the following concentrations: 150 mM L-alanine, 10 mM glyoxylate and 150 μM PLP, and a reaction time of 10 min. To determine the kinetic parameters of the transaminase activity, the production of pyruvate or glyoxylate was determined using an assay based on measuring the amount of dinitrophenylhydrazine derivative by HPLC, using a fixed amount of wild-type enzyme (0.1 μM) in the presence of exogenous PLP (150 μM) or apomutant (10 μM pre-incubated for 1 h at 25 °C with 1 mM PLP) [8]. Substrate saturation curves were fitted using eqn (1). Initial velocity data were fitted using eqn (2), and dead-end inhibition data were fitted using eqns (3), (4) or (5) for competitive, uncompetitive or mixed inhibition respectively.
 |
(1) |
 |
(2) |
 |
(3) |
 |
(4) |
 |
(5) |
where v is the initial velocity, V is maximum velocity, A and B are substrate concentrations, Ka and Kb are the Km for substrates A and B respectively, and Kis and Kii are slope and intercept constants.
The detection and quantification of PLP and PMP during the reactions were performed using the HPLC procedure reported previously [9].
Equilibrium dissociation constant for wild-type or mutant AGT
The apparent equilibrium constant for dissociation of PLP from wild-type AGT (0.1 μM), KD(PLP), was determined by measuring the quenching of the intrinsic fluorescence of apoenzyme in the presence of PLP at concentrations ranging from 0.005 to 5 μM. The KD(PLP) value of the wild-type–coenzyme complex was obtained using eqn (6):
 |
(6) |
where [E]t and [PLP]t represent the total concentrations of AGT dimer and PLP respectively, Y refers to the intrinsic fluorescence quenching change at the PLP concentration [PLP], and Ymax refers to the intrinsic fluorescence quenching when all enzyme molecules are complexed with coenzyme.
Coenzymes association and dissociation kinetics
Association rate constants for PLP combining with wild-type apoAGT were measured under pseudo-first-order conditions with excess of coenzyme. The progress of the interaction was monitored by the increase in the 429 nm CD signal as a function of time at various coenzyme concentrations. The data were fitted to eqn (7):
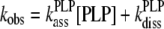 |
(7) |
Where kPLPass and kPLPdiss represent the association and dissociation rate constants respectively of apo-wild-type AGT for PLP.
Equilibrium constant measurements of the overall transamination
Holo wild-type AGT (6 μM) was incubated with L-alanine (100 mM), pyruvate (10 mM), glycine (10 mM) and glyoxylate (100 mM), and aliquots were removed at time points from 0 to 24 h. The observed equilibrium concentrations were obtained at 7 h and did not vary during the next 17 h. The equilibrium constant (Keq,overall) of the overall transamination catalysed by AGT was determined directly by measurements of pyruvate and glyoxylate by means of HPLC analysis of their dinitrophenylhydrazine derivatives at equilibrium. The experiments were performed in triplicate and the S.E.M. was less than 5% of the mean value.
Preparation of AGT in the apoform and in the PMP form (AGT–PMP)
AGT in the apo form was prepared in the following manner: holo wild-type or mutant (10–20 μM) was incubated with 500 mM L-alanine for a few min at room temperature (25 °C) in a final volume of 2 ml. The reaction mixture, concentrated to 0.2 ml with the aid of a Centricon-30 tube, was first washed twice with 2 ml of 1 M potassium phosphate buffer (pH 6), and then 4–5 times with 100 mM potassium phosphate buffer (pH 7.4). AGT–PMP, i.e. PMP bound to apoenzyme, was prepared in two ways. In the first method, the holo form (10–20 μM) was incubated with 500 mM L-alanine as described above. The reaction mixture was then transferred to a Centricon-30 device and extensively washed with 100 mM potassium phosphate buffer (pH 7.4) at 4 °C. In the second method, AGT–PMP was prepared by incubating apoAGT with a saturating concentration (100 μM) of PMP and then washed to eliminate unbound PMP.
Pre-steady-state and steady-state analysis by UV-visible spectrophotometry
The reaction of wild-type holoAGT (7 μM) with various concentrations of L-alanine (5–500 mM), glycine (20–500 mM), L-serine (10–100 mM), L-phenylalanine (10–100 mM) or L-arginine (20-250 mM) as well as the reaction of AGT–PMP (7 μM) with various concentrations of glyoxylate (0.1–5 mM) or pyruvate (0.1–2.5 mM) were carried out in 100 mM potassium phosphate buffer (pH 7.4) at 25 °C in a total volume of 160 μl. The reaction of the G82E mutant, pre-incubated with 100 μM PLP, with 500 mM L-alanine was also investigated. In each case, the absorbance spectra (500) from 250 to 550 nm were recorded on a J&M Tidas 16256 diode array detector (Molecular Kinetics) using a BioLogic SFM300 instrument. The dead-time was 3.6 ms at a flow velocity of 12 ml/s. The rate constants measured by following the changes at 423 and 330 nm were determined by non-linear regression to eqn (8):
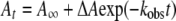 |
(8) |
where At is the absorbance at time t, ΔA is the amplitude of the phase, kobs is the observed rate constant and A is the final absorbance. Data were analysed using the Biokine 4.01 (Biologic) software provided with the instrument. The kmax and apparent Km values for the half-reactions were determined by plotting the observed rate constants versus substrate concentrations and fitting the data to eqn (9):
 |
(9) |
where kr is a term that represents the contribution of the reverse reaction. When the kr value is not significant, data were fitted to eqn (9) in which the kr term was omitted.
Reaction of wild-type holoAGT (7 μM) with L-glutamate (50–500 mM) or L-aspartate (50–200 mM) was performed in 100 mM potassium phosphate buffer, pH 7.4, at 25 °C in a total volume of 250 μl. The decrease in the 423 nm absorbance band or the increase in the 330 nm absorbance band was measured in a Jasco V-550 spectrophotometer as a function of time. The kobs values for the 330 nm increase or the 423 nm decrease were fit to a first-order curve using Origin® 7.03 (OriginLab).
HPLC detection of PLP and PMP formation in the G82E transamination half-reactions
G82E in the apo form was incubated for 1 h at room temperature with various concentrations of PLP (0.1–1 mM) or PMP (0.02–10 mM) to ensure binding of coenzymes to the mutant had come to equilibrium. After incubation, L-alanine (500 mM) was added to the reaction mixture containing the mutant and PLP, while pyruvate (1 mM) or glyoxylate (5 mM) was added to the reaction mixtures containing the mutant and PMP. At appropriate times, a 250 μl aliquot was removed and quenched with 10% trichloroacetic acid. The precipitated enzyme was removed by centrifugation (16000 g at 25 °C for 5 min). PLP or PMP content in the supernatants was determined using the previously described HPLC method [9]. The rate of non-enzymatic transamination between PLP and L-alanine and between PMP and pyruvate or glyoxylate under the same experimental conditions was measured and subtracted from the rate of total transamination seen in the enzymatic samples. The amount of PMP formed at different reaction times (8–600 s) during the L-alanine transamination half-reaction was plotted against time and fitted to eqn (10):
 |
(10) |
where PMPt represents the PMP concentrations at increasing time t, A is the amplitude of the fast phase, kfast and kslow are the rate constants of the fast and the slow phase respectively, and E0 is the total apoenzyme.
Spectroscopic measurements
Absorption spectra were made with a Jasco V-550 spectrophotometer. The enzyme solution was drawn through a 0.2 μm filter to reduce light scattering from the small amount of precipitate. Fluorescence spectra were taken with a FP750 Jasco spectro-fluorometer using 5 nm bandwidths on both sides at protein concentrations varying from 1 to 10 μM. Spectra blanks, i.e. spectra of samples containing all components except AGT, were taken immediately prior to measurements of samples containing enzyme, and subtracted from spectra of samples containing AGT. CD spectra were obtained with a Jasco J-710 spectropolarimeter with a thermostatically controlled cell compartment at 25 °C. For near-UV and visible wavelengths, protein concentrations between 5 and 10 μM were used in a cuvette with a 1 cm path length. Routinely, four spectra were recorded at a scan speed of 50 nm/min with a bandwidth of 2 nm and averaged automatically except where indicated. For far-UV measurements, the protein concentration was 0.1 mg/ml with a path length of 0.1 cm.
Data analysis
The kinetic experiments were performed at least in duplicate, and in each case the S.E.M. was less than 10%. All data analysis was performed by non-linear curve fitting using Origin® 7.03 (OriginLab), and the errors indicated result from fitting to the appropriate equations.
Molecular modelling studies
The three-dimensional co-ordinates of the human AGT (PDB code: 1H0C; [5]) were used as a starting point to generate the PMP and PLP–L-alanine bound forms of the enzyme by means of energy minimization. The details and the minimization protocol adopted are given in the Supplementary Materials (at http://www.BiochemJ.org/bj/408/bj4080039add.htm). The PLP and PMP forms of AGT were compared with the corresponding forms of BtrR [10], an aminotransferase involved in butirosin synthesis. The rationale for this choice is based on the fact that BtrR is the closest homologue of AGT (VAST E-value 10e-13.3) [11] displaying aminotransferase activity for which the crystal structures of the PLP- and PMP-bound forms have been solved. An initial pairwise structural alignment between AGT and BtrR was calculated with the program CE [12]. The alignment obtained was visually inspected and, where necessary, manually refined to optimize the matching of the secondary elements, functionally conserved residues known to interact with the PLP moiety and hydrophobic regions.
RESULTS AND DISCUSSION
Spectroscopic features of AGT
Recombinant holoAGT binds 2 moles of PLP per dimer and displays, in 100 mM potassium phosphate buffer (pH 7.4), in addition to an absorption maximum at 280 nm, an absorbance maximum centred at 420 nm and a shoulder at about 340 nm, probably corresponding to the oxoenamine and enolimine tautomers of the internal aldimine respectively (Figure 1A). The ratio A280/A420 is equal to ∼8. The CD spectrum of holoAGT in the visible region has a positive dichroic band at 429 nm and a small negative dichroic signal in the 340 nm region (Figure 1B). It is also shown here that the enzyme possesses positive dichroic bands in the aromatic region at 285 and 290 nm and a negative dichroic band in the 256–266 nm region, which would indicate the asymmetry of certain aromatic amino acids, most probably associated with the active site. Reduction of holoAGT with either NaBH4 or NaCNBH3 causes the disappearance of the 420 nm absorbance and of the 429 nm dichroic signal with the concomitant appearance of a 330 nm absorbance band and a 340 nm positive dichroic band. This result is consistent with the reduction of the internal Schiff base. In addition, reduction results in the appearance of a positive dichroic signal at 260 nm. On excitation of holoAGT at 280 nm, a characteristic emission maximum at 336 nm is observed (Figure 1C). When the holoenzyme is excited at 420 or 340 nm, a weak broad emission is observed at about 500 and 420 nm respectively (results not shown). The apoenzyme does not exhibit either absorbance or dichroic bands in the visible region or PLP emission. However, the emission spectrum of the apoenzyme upon excitation at 280 nm has an emission band 1-nm blue-shifted and an intensity approx. 4-fold higher than that of the holoenzyme.
Figure 1. Absorption, CD and fluorescence spectral changes of AGT–PLP alone and in the presence of substrates or substrate analogue.

Absorption (A) and CD (B) spectra of 7 μM AGT–PLP (–) and in the presence of 500 mM L-alanine (- - -), 500 mM glycine (········) or 500 mM D-alanine (-·-·-·). (C) Fluorescence emission spectra (excitation at 280 nm) of 1.7 μM AGT–PLP (–) and in the presence of 500 mM D-alanine (-·-·-·). In each case, the buffer was 100 mM potassium phosphate (pH 7.4).
Binding of PLP to apoAGT
The observed rate constant for the association of PLP with apoen-zyme increases linearly with PLP concentration over the range 100–840 μM under pseudo-first-order conditions with excess coenzyme. The data were fitted to eqn (7) yielding a kPLPdiss value of nearly zero and a kPLPass value equal to 41±1 M−1·s−1 (results not shown).
From titration analysis of the apoenzyme with PLP the data for quenching of intrinsic fluorescence versus PLP concentration fitted to eqn (6) yielded a KD(PLP) value for the apoAGT–PLP complex equal to 275±32 nM. Therefore the kPLPdiss could be estimated to be ∼1×10−5 s−1. It should be noted that the apoAGT–PLP complex has an intrinsic emission spectrum identical with that of the holoenzyme.
Kinetic and equilibrium studies
Double-reciprocal plots of the steady-state kinetic data for the forward and reverse AGT-catalysed transamination were consistently sets of parallel lines (results not shown). Data were best fitted using eqn (2), which adheres to a ping-pong or double-displacement kinetic mechanism, yielding the kinetic parameters reported in Table 1. Inhibition experiments of the forward reaction have been carried out: D-alanine, a dead-end analogue of L-alanine, is competitive versus L-alanine and uncompetitive versus glyoxylate, whereas pyruvate, a product of the reaction, is a competitive inhibitor versus glyoxylate, and a mixed-type inhibitor versus L-alanine (Table 2). Thus similar to other aminotransferases, a ping-pong kinetic mechanism for AGT is suggested by the parallel initial velocity and dead-end inhibition patterns.
Table 1. Steady-state kinetic parameters of wild-type and the G82E mutant AGT.
| Substrate | Cosubstrate | kcat (s−1) | Km (mM) | kcat/Km (mM−1·s−1) |
|---|---|---|---|---|
| Wild-type | ||||
| L-Alanine | Glyoxylate | 45±2 | 31±4 | 1.4±0.2 |
| Glyoxylate | L-Alanine | 45±3 | 0.23±0.05 | 196±44 |
| Glycine | Pyruvate | 10.33±0.03 | 22±2 | 0.015±0.002 |
| Pyruvate | Glycine | 0.36±0.03 | 0.21±0.02 | 1.7±0.2 |
| G82E mutant | ||||
| L-Alanine | Glyoxylate | 0.070±0.02 | 15±2 | 0.0047±0.0006 |
| Glyoxylate | L-Alanine | 0.068±0.003 | 0.15±0.06 | 0.45±0.18 |
Table 2. Dead-end inhibition patterns for the alanine:glyoxylate amino-transferase reaction.
| Varied substrate | Fixed substrate | Inhibitor | Pattern | Kis (mM) | Kii (mM) |
|---|---|---|---|---|---|
| L-Alanine | Glyoxylate | D-Alanine | Competitive | 14.3±4.3 | – |
| Glyoxylate | L-alanine | D-Alanine | Uncompetitive | – | 28.7±15.6 |
| L-Alanine | Glyoxylate | Pyruvate | Mixed | 15.8±4.5 | 22.8±1.8 |
| Glyoxylate | L-alanine | Pyruvate | Competitive | 2.3±1.2 | – |
Although the serious consequences of peroxisomal AGT deficiency allow one to suggest that the enzyme has a prominent role in hepatic glyoxylate detoxification, to date no information indicating that AGT is highly specific for catalysing glyoxylate to glycine processing is available. Therefore as a first step to address this question, equilibrium studies of the peroxisomal AGT have been carried out. The equilibrium constant for the overall transamination reaction,
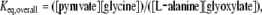 |
was found to be ∼9400. This value is in excellent agreement with the value of 9700, calculated by the following Haldane relation
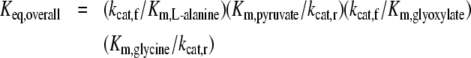 |
where kcat,f and kcat,r are the kcat values for the forward and reverse reactions respectively.
Thus the reaction catalysed by AGT is largely shifted towards the products. This clearly indicates the role of AGT in the conversion of glyoxylate into glycine. The following lines of evidence support this view: (i) the kcat of the overall transamination of the pair alanine/glyoxylate is about 100-fold higher than that of the pair glycine/pyruvate, and (ii) unlike pyruvate, glyoxylate, at least up to 100 mM, does not exert inhibitory action on the forward reaction.
Spectral changes during the half-transamination reactions
Upon addition of L-alanine to holoAGT (also indicated as AGT–PLP), changes in the absorbance and dichroic bands in the visible region immediately occur, as shown in Figures 1(A) and 1(B). Mixing of AGT–PLP with L-alanine also resulted in the immediate appearance of a positive dichroic signal at 260 nm (Figure 1B). After total denaturation, the reaction mixture was subjected to centrifugation at 16000 g for 10 min. The HPLC analysis of the supernatant revealed that almost all of the original PLP content was converted into PMP. Thus the absorbance and CD changes describe the conversion of AGT from the PLP into the PMP form. The complete conversion of PLP into PMP also occurs upon addition of aspartate, glutamate, arginine, serine and phenylalanine in their L-form to the enzyme.
Unlike the amino acids mentioned above, glycine (500 mM) added to the enzyme (7 μM) at pH 7.4 causes an immediate 6 nm blue-shift of both the absorbance and the dichroic band of the internal Schiff base as well as a modest increase in the 340 nm absorbance band. In addition, a positive 260 nm dichroic band could be seen. These spectral bands, which remain unchanged with time, are possibly indicative of an equilibrium mixture of an external aldimine and a ketimine species (Figures 1A and 1B). Indeed, after denaturation of the reaction mixture, the HPLC analysis of the supernatant reveals that approx. 30% of the original PLP content was transformed into PMP. To verify whether this equilibrium could be altered by continuous removal of glyoxylate, aliquots were withdrawn at various times from a reaction mixture containing AGT and glycine in the presence of lactate dehydrogenase (0.5 mg/ml) and NADH (200 μM) and subjected to HPLC analysis after total denaturation. The analysis revealed that the original PLP was gradually converted into PMP reaching a conversion of approx. 95% within 1 h. The fractional changes in the 414 nm absorbance as a function of glycine concentration were used to measure the KD for external Schiff base formation. The apparent KD for glycine is 32±5 mM (results not shown).
As shown in Figures 1(A) and 1(B), the addition of D-alanine, an unproductive substrate analogue, to AGT–PLP resulted in changes of the absorbance and dichroic band in the visible region as well as in the appearance of a positive dichroic band at 260 nm. On excitation at 280 nm, the AGT–D-alanine complex displayed an emission maximum at 338 nm, 2-nm red-shifted with respect to that of AGT–PLP alone (Figure 1C). These spectral features, which remain unaltered with time, are presumably indicative of the formation of the external aldimine with D-alanine that cannot go on to form products because of the inability of the enzyme to abstract the α proton. The α proton would probably be oriented improperly for abstraction in D-alanine.
After 5 min of reaction of AGT–PLP (10 μM) with L-alanine (500 mM) in 100 mM potassium phosphate buffer (pH 7.4), the solution was filtered through a Centricon-30 device. Then, the filtrate and the retentate were subjected to HPLC analysis. Pyruvate (∼18 μM) was found in the filtrate but not in the retentate. The retentate contained an enzymatic form which exhibited absorbance, dichroic and intrinsic fluorescence features that are shown in Figures 2(A), 2(B) and 2(C) respectively. After denaturation of the retentate, HPLC analysis of the supernatant revealed the presence of PMP in an amount corresponding with that of the original PLP content of AGT–PLP. If the solution of the retentate was diluted to a concentration of 100 nM, and then filtered through a Centricon-30 tube, no detectable PMP was found in the filtrate. All these data indicate that the enzymatic species present in the retentate is PMP tightly bound to the protein. This species is named AGT–PMP.
Figure 2. Absorption, CD and fluorescence spectra of AGT–PMP alone and in the presence of glyoxylate.

(A) Absorption spectra of 9 μM AGT–PMP (–) and in the presence of 5 mM glyoxylate (- - -). (B) CD spectra of AGT–PMP (–), AGT–PMP in the presence of 5 mM glyoxylate (- - -) and apoAGT after 15 h incubation with 100 μM PMP (········). (C) Fluorescence emission spectrum (excitation at 280 nm) of 1.6 μM apoAGT (········) and AGT–PMP (–). In each case, the buffer was 100 mM potassium phosphate pH 7.4
Upon addition of glyoxylate or pyruvate to AGT–PMP, a recovery of the absorbance and dichroic features of the original holoenzyme was immediately seen. This transition was accompanied by the disappearance of the 260 nm positive dichroic band (Figures 2A and 2B). As expected, the recovery of the absorbance and dichroic bands typical of AGT–PLP was also obtained when apoAGT was incubated with PMP (100 μM) in the presence of glyoxylate (5 mM). However, under these conditions the rate of this event followed by visible CD measurements occured slowly, with a rate constant of 0.007±0.001 min−1 (Figure 3). In order to define the step which slows down this reaction, association of apoenzyme with 100 μM PMP was monitored by collecting CD spectra as a function of time. The observed rate constant, measured by the increase of the dichroic signal at 340 nm, was equal to 0.0048±0.0003 min−1, a value similar to that measured for the reaction of apoAGT in the presence of PMP and glyoxylate. Thus the rate-determining step of the latter process is the binding of PMP to AGT. Unfortunately, the characterization of the dependence of this event on PMP concentration is seriously hindered by the small signal changes and light-scattering effects. The species resulting for association of apoAGT to PMP exhibits dichroic bands in the near-UV region (Figure 2B) and emission intrinsic fluorescence spectra identical with those of AGT–PMP. After addition of glyoxylate, even at substoichiometric concentrations, this enzymatic species is immediately converted into the AGT–PLP form.
Figure 3. Time-dependent CD spectral changes occurring on incubation of apoAGT with PMP in the presence of glyoxylate.

ApoAGT (········) (5 μM) was incubated with 100 μM PMP in the presence of 5 mM glyoxylate in 100 mM potassium phosphate buffer (pH 7.4) at 25 °C. CD spectra were recorded at 0.5, 7.5, 28, 51, 81, 120, 146, 171 and 184 s.
All together, these data underline the structural and functional role of the AGT–PMP complex in catalysis. The higher affinity of PMP compared with that of PLP (at least 3-fold) for apoenzyme allows PMP to remain bound during the catalytic cycle. This finding, together with the higher reactivity of AGT–PMP toward oxo acids compared with that of apoAGT and PMP in the presence of an oxoacid, is relevant for subsequent catalysis. Furthermore, the comparison between the spectroscopic features of AGT–PLP and AGT–PMP reveals that, in addition to the typical differences in their absorbance and dichroic bands in the visible region as well as in their coenzyme emission fluorescence maxima, AGT–PMP displays a positive dichroic signal at 260 nm and an emission maximum of intrinsic fluorescence 2-nm red-shifted with respect to that of AGT–PLP. Identical spectroscopic differences with respect to AGT–PLP have also been detected either in the external aldimine of AGT with D-alanine or in AGT–PLP after reduction with NaBH4. It is not easy to envisage the structural basis of these differences since the ellipticity band could arise from either the binding of PLP derivatives and/or reorientation of aromatic active site residues. It is known that bound PLP derivatives displaying different absorbance bands could also be optically active or inactive in the near-UV region. Notably, slight modifications in their chemical structure give rise to different signals [13]. In AGT, various coenzyme-bound species (external aldimine, phosphopyridoxylysine, PMP) cause the appearance of the same 260 nm CD signal, thus making it difficult to attribute a same signal to different chemical species. Although this possibility cannot be ruled out, we have investigated, by means of a bioinformatic approach, whether binding of PLP derivatives could induce slight rearrangements of aromatic active-site residues responsible for the dichroic signal.
Molecular modelling
Unfortunately, although several structures of AGT from various sources are available [5,14,15], none has been solved in the external aldimine or PMP form. Based on this observation, we decided to design a docking model indicative of the putative binding mode of PMP and of the PLP–L-alanine complex to AGT. The putative position of the manually docked molecules and of their neighbouring residues was relaxed by energy minimization means. Compared with the AGT–PLP structure, the AGT–PMP complex tilts by approx. 15° with respect to PLP towards Trp108 and Asp183 (Figure 4). The predicted movement is probably due to the need to accommodate the amino group of PMP and to avoid steric clashes with Lys209. Consequently, the Trp108 ring undergoes a slight flip around Cα–Cβ, and the side-chains of Asp183, Lys209 and Tyr260 undergo minor adjustments, in order to better accommodate PMP.
Figure 4. Modelling of the active site of the PMP- and L-alanine–PLP-bound forms of AGT.

The AGT active site residues that are involved in cofactor binding are illustrated and labelled according to the sequence position of human AGT. The docked structures of AGT–PMP (magenta sticks) and AGT–PLP in complex with L-alanine (blue sticks) are shown. The structure of AGT–PLP complexed with aminooxyacetic acid (AOA) [5] is also shown for reference, as grey sticks. Oxygen atoms are coloured red, nitrogen atoms blue, and phosphorus orange. This figure was created using pyMOL [28].
Modelling of the L-alanine binding mode indicates that the simulated external aldimine has a high degree of structural similarity to the AGT–PMP form (Figure 4).
The structural superposition between AGT and BtrR [rmsd (root mean square deviation) 2.4 Å], both in the PLP form, shows that several residues are conserved, including the active-site residues Trp108, Asp183 and Lys209 (Trp92, Asp163 and Lys192 in BtrR; numbering refers to the crystal structure of BtrR from Bacillus circulans). The comparison between the PLP- and PMP-bound forms of BtrR reveals that, even if these structures are very similar, they display subtle differences in the position and orientation of either the pyridine ring or the side-chains of Trp92, Tyr342 and Asp163 located at the active site. In particular, upon PLP into PMP conversion, the Trp92 side-chain undergoes a remarkable conformational change, flipping by approx. 45° around the Cα–Cβ bond. Thus, from a qualitative viewpoint, the reorientation of the pyridine moiety and the movements of a limited number of active site residues associated to the PLP– PMP transition predicted for AGT seems to be similar, but not identical, to those observed in BtrR. In fact, the significant conformational change involving Trp92 observed in BtrR would be energetically impaired in AGT–PMP by the presence of Ile107 and Leu351 residues. It should be considered that these residues are in a favourable position (as assessed by rotameric analysis) and should undergo major structural modifications in order to better accommodate a putative ∼45° flip of Trp108.
In conclusion, bioinformatic analyses allow us to suggest that the transition between the PLP and the PMP form of AGT is accompanied by minimal movements of active-site residues and tilting of the coenzymatic form. However, it should be taken into account that energy minimization is able to find only local energy minima and does not allow us to precisely quantify the conformational transition of Trp108. Nevertheless, the predicted subtle movements of aromatic residues at the active site support the observed experimental data, and could account for the observed changes in the near-UV CD and intrinsic fluorescence spectra of AGT in the PMP and in the external aldimine forms in comparison with AGT–PLP. Following this view, it can be suggested that the changes associated with the transaldimination process could be a determinant for tight PMP binding and subsequent catalysis. Similar adjustments could also occur in the reduced holoAGT, the latter resembling the PMP form.
Although the molecular basis of the above postulated reorientation of PMP and/or aromatic active-site residues remains at present elusive, it is of interest that similar slight rearrangements have been observed by comparing not only the PLP and the PMP forms of BtrR from B. circulans [10], but also those of ArnB from Salmonella typhimurium [20] and histidinol phosphate aminotransferase from E. coli [21]. It is noteworthy that, as for AGT, for these three aminotransferases the physiological function is the conversion of the proper oxo compound into its amino derivative performed by the aminated form of the coenzyme.
In addition, the docked conformation of the L-alanine–PLP complex into the active site of AGT highlights the residues implicated in the interaction with the substrate (Figure 4), and supports the reliability of our bioinformatic approach. In fact, the α-carboxylate moiety of L-alanine forms an ion-pair interaction with the guanidinium group of Arg360 in an orientation similar to that observed for the amino-oxyacetic acid, which was co-crystallized with AGT [5]. The Cβ carbon of L-alanine is stabilized by the hydrophobic interaction with the side-chains of Tyr260 and Leu351. Thus the model indicates that the position of L-alanine is optimal for the transamination reaction; on the other hand, glycine, lacking the stabilizing interaction with Tyr260 and Leu351, could result in a misplaced conformation.
Kinetic parameters of half-transamination reactions
The finding that, unlike for other transaminases [16–19] so far studied, the Keq,overall for AGT is >1 leads us to a detailed analysis of the half-transamination reactions of AGT–PLP with L-alanine or glycine and AGT–PMP with pyruvate or glyoxylate. The time-dependent spectral changes occurring during the reaction of holoAGT with L-alanine have been recorded by rapid scanning stopped-flow analysis and are shown in Figure 5. The apparent first-order rate constant, kobs, determined by measuring the rate of appearance of the 330 nm absorbance band, showed a hyperbolic dependence on L-alanine concentration (inset of Figure 5). For comparison with L-alanine, other amino acids were tested for their reactivity with AGT–PLP. Whereas the reaction of the enzyme with serine, phenylalanine and arginine has been followed by stopped-flow experiments, the reaction with glutamate or aspartate has been recorded by conventional spectrophotometry. In contrast with the reaction with alanine, serine, arginine and glutamate, which exhibited saturation kinetics, reactions with phenylalanine and aspartate showed a linear dependence of the kobs on substrate concentration within the concentration range studied (up to 100 mM). Under this situation ([S]≪Kappm), the Michaelis–Menten equation can be transformed into
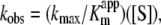 |
where kmax/Kappm represents the specificity constant for these non-saturating substrates. The kmax, Kappm and kmax/Kappm values thus obtained for each substrate are summarized in Table 3. It should be noted that a meaningful kr value has been obtained for L-alanine only.
Figure 5. Rapid scanning stopped-flow spectra obtained upon reaction of AGT–PLP with L-alanine at 25 °C.

The first 20 spectra were collected at 0.001, 0.007, 0.013, 0.019, 0.025, 0.031, 0.037, 0.043, 0.049, 0.055, 0.061, 0.067, 0.073, 0.079, 0.085, 0.091, 0.097, 0.106, 0.118 and 0.13 s, the last 30 spectra were recorded from 0.142 to 0.85 s. AGT–PLP (7 μM) was mixed with L-alanine (100 mM) in 100 mM potassium phosphate buffer (pH 7.4) at 25 °C. The inset shows the dependence of the kobs for the increase at 330 nm absorbance as a function of L-alanine concentration. The points are the experimental data, whereas the curve is from a fit to eqn (9).
Table 3. Transamination half-reaction kinetic parameters.
*No saturation detected at [L-phenylalanine] and [L-aspartate] up to 100 mM. †Data can be fitted equally well to eqn (9) without the term kr.
| kmax (s−1) | Kappm(mM) | kmax/Kappm (mM−1·s−1) | kr (s−1) | |
|---|---|---|---|---|
| L-Alanine | 49±5 | 38±9 | 1.3±0.3 | 6±2 |
| L-Serine | 15±3 | 213±76 | 0.070±0.03 | 0.3±0.1† |
| L-Phenylalanine* | – | – | 0.005±0.001 | 0.1±0.05† |
| L-Arginine | 0.134±0.005 | 51±6 | 2.6×10−3±3×10−4 | – |
| L-Glutamate | 0.032±0.003 | 409±91 | 8×10−5±2×10−5 | – |
| L-Aspartate* | – | – | 1×10−9±1×10−10 | – |
| Glyoxylate | 49±1 | 0.22±0.01 | 220±11 | 0.4±0.3† |
| Pyruvate | 15±1 | 0.3±0.06 | 50±10 | – |
The interaction of AGT–PLP with glycine has been monitored by stopped-flow spectrophotometry. The transient kinetic traces of AGT–PLP at 420 nm exhibit biphasic behaviour (consisting of an increase followed by a decrease), suggesting two kinetically distinguishable events (Figure 6). The rate constants for the first and the second phase both display a hyperbolic dependence on glycine concentration (inset of Figure 6). The first process, characterized by a kmax=2.1±0.3 s−1, an apparent dissociation constant of 95±35 mM, and a kr of 1.2±0.6 s−1, can be attributed to external aldimine formation. The second process is consistent with the conversion of the external aldimine into ketimine, considering that the rate of the decrease of the 420 nm absorbance parallels that of the increase at 330 nm. The resulting kmax and Kappm were 1.12±0.05 s−1 and 35±7 mM respectively. The latter value is in excellent agreement with the KD value for external Schiff base formation (see above).
Figure 6. Transient kinetic analysis of the reaction of AGT–PLP with glycine.

Plot of the absorbance at 420 nm with time for the reaction of 7 μM AGT–PLP with 500 mM glycine. The dotted line represents a two-exponential fit. Inset: dependence of the rate constants (kobs) for increase (■) or decrease (●) at 420 nm as a function of glycine concentration. The solid lines represent the data fitted to eqn (9).
The glyoxylate or pyruvate half-transamination process has been followed by rapid scanning stopped-flow analysis by monitoring the increase at 420 nm with time after addition of various concentrations of glyoxylate (Figure 7) or pyruvate to AGT–PMP. The rate of AGT–PLP formation, kobs, was dependent on the concentration of the oxo acid, and the concentration dependency fitted well to eqn (9) (inset of Figure 7). The kmax, Kappm and kmax/Kappm thus obtained for glyoxylate and pyruvate are reported in Table 3.
Figure 7. Single-wavelength stopped-flow measurements of the reaction of AGT–PMP with glyoxylate at 25 °C.

The reaction of AGT–PMP (7 μM) with various concentrations of glyoxylate in potassium phosphate buffer (pH 7.4) at 25 °C. Time courses at 420 nm are shown. The dotted lines are from a fit to eqn (8). The inset shows the dependence of the kobs for the increase of the intensity at 420 nm as a function of glyoxylate concentration. The points shown are the experimental values, while the curve is from the data fitted to eqn (9).
On the basis of all these kinetic data, the following conclusions could be drawn. The catalytic efficiencies of the half-reaction L-alanine↔ pyruvate were ∼1.3 mM−1·s−1 in the L-alanine→pyruvate direction and 50 mM−1·s−1 in the pyruvate→L-alanine direction. This implies a Keq of this half-reaction value of approx. 40. It is worth noting that the catalytic efficiency for L-alanine measured under pre-steady-state conditions is similar to the value of kcat/Km for L-alanine measured under steady-state conditions, thus indicating that the rate-limiting step of this process is the ketimine formation or its hydrolysis. On the other hand, the catalytic efficiency for pyruvate measured under pre-steady state conditions is considerably higher than that measured under steady state conditions (1.7 mM−1·s−1), thus indicating that the kcat is affected by some other slower step(s). The analysis of the half-reaction glycine↔ glyoxylate is more complex. Glycine→glyoxylate conversion under pre-steady-state conditions is characterized by the formation of the external aldimine followed by its partial conversion into ketimine. Unlike for L-alanine and other amino acid substrates (Table 3), the transaldimination process is detectable and proceeds with a kon (kmax/Kappm) and a koff (kr) equal to 0.021 mM−1·s−1 and 1 s−1 respectively. The koff term could probably reflect a stabilization of the external aldimine of AGT with glycine lower than that with L-alanine, as can also be deduced by the putative binding mode of L-alanine or glycine in the active site of AGT. The kmax of the conversion of AGT–PLP-glycine into AGT–PMP-glyoxylate is calculated as ∼1 s−1. However, since this value does not represent a microscopic constant, being largely affected by the reverse reaction, it is impossible to measure the catalytic efficiency in the direction glycine→glyoxylate. The catalytic efficiency of the glyoxylate→glycine conversion (∼200 mM−1·s−1) measured under pre-steady-state conditions highly reflects the kcat/Km value for glyoxylate (Table 1) obtained by steady-state measurements. The kinetic behaviour of AGT towards glycine prevents the evaluation of the Keq for the half reaction glycine↔ glyoxylate. Nevertheless, it can be inferred that the high value of the Keq,overall is mainly due to the equilibrium glycine↔ glyoxylate largely shifted towards glycine.
Spectroscopic and kinetic properties of the G82E mutant
The G82E mutant, obtained following the expression and purification procedure in the absence of PLP (see Supplementary Material at http://www.BiochemJ.org/bj/408/bj4080039add.htm), does not display detectable absorbance and dichroic bands in the visible region, and exhibits dichroic signals in the near-UV similar to those of the wild-type. Furthermore, to test the structural integrity of the mutant, we measured its CD spectrum in the far-UV region. No differences were observed between the spectra of the mutant and the wild-type AGT, which indicates that the mutation does not affect the overall secondary structure of the enzyme. Again, like the wild-type, the mutant elutes as a dimer on a Sephacryl S-300 column (GE Healthcare) down to a 5 μM concentration, thus indicating that the mutation does not alter the associative process of monomers into dimers.
Absorbance differential and CD spectra of the apomutant (10 μM) in the presence of 500 μM PLP (a higher PLP concentration is hampered by light-scattering effects) showed a slow (requiring about 1 h to reach equilibrium) binding process (kobs=0.28±0.03 min−1) of the coenzyme giving rise to the absorbance and dichroic bands shown in Figures 8(A) and 8(B) respectively. Although the wild-type AGT displays an optical activity (millidegrees per absorbance unit) value of 97 millidegrees/A420, the mutant shows an optical activity of 9 millidegrees/A432. This suggests that the microenvironment of the internal aldimine in the G82E mutant is greatly different from that of wild-type. Reduction of the reconstituted mutant with NaBH4 followed by extensive washing through a Centricon-30 device resulted in a species which, although having absorbance and dichroic bands in the visible region identical with those of the reduced wild-type, did not exhibit the positive dichroic band at 260 nm (Figure 8B).
Figure 8. Absorption and CD spectra of the G82E mutant.

(A) Differential absorption spectra of 13 μM apomutant G82E in the presence of 500 μM PLP at the indicated times. (B) CD spectra of 10 μM apomutant G82E incubated for 90 min with 500 μM PLP before (–) and after (- - -) NaBH4 reduction. In each case, the buffer was 100 mM potassium phosphate (pH 7.4).
Steady-state kinetic parameters for the alanine/glyoxylate substrate pair have been measured using 10 μM mutant pre-incubated with 1 mM PLP. The G82E mutation resulted in a ∼650-fold decrease in kcat (0.070±0.02 s−1), while the Km values were not significantly changed (Table 1). Kinetic parameters for the glycine/pyruvate substrate pair are difficult to obtain because of the very slow reaction rate in this direction. In fact, when 15 μM apomutant pre-incubated with 1 mM PLP was allowed to react with 0.2 M glycine and 1 mM pyruvate, glyoxylate was linearly produced with time, reaching a value after 4 h of about 140 μM, corresponding to an initial rate of 6.5×10−4 s−1. Assuming that this value is close to the kcat value of the reverse transamination, the rate of this reaction in G82E is ∼550-fold slower than the corresponding reaction in wild-type. Therefore replacement of Gly82 with a glutamate residue seems to dramatically affect both the forward and the reverse reactions to a similar extent.
In order to ascribe the catalytic consequence of this amino acid substitution to a particular reaction step, the effects of the mutation on the reaction of the mutant in either PLP or PMP form with amino acids or oxo acids has been investigated by means of kinetic analyses. The G82E mutant (8.5 μM), pre-incubated for 1 h with 1 mM PLP, was allowed to react with 500 mM L-alanine. Aliquots were removed from this reaction mixture at various times, and, after denaturation, the PMP content of the supernatants was measured by HPLC. The results reported in Figure 9 indicate that the L-alanine transamination half-reaction in the G82E mutant displayed a fast phase followed by a slow linear phase. A fit of the data to eqn (10) gives a y-intercept value of 13±1 μM PMP, a kslow of 0.002±0.0004 s−1 and a kfast of 0.1±0.02 s−1, with the latter value being in agreement with the kcat of 0.07 s−1 obtained in steady-state kinetic measurements under the same experimental conditions. When this experiment was carried out at various PLP concentrations, the y-intercept values showed a hyperbolic dependence on PLP concentration, giving a ymax-intercept of 16±1 μM PMP and a value of 198±50 μM PLP corresponding to ½ymax. On the other hand, the kslow was linearly dependent on PLP concentration. A set of conclusions can be drawn from the fast phase of the above experiment: (i) the kfast probably represents the rate of transamination catalysed by the PLP bound to the mutant; (ii) at saturating PLP concentration the G82E mutant is able to bind ∼2 mol PLP/mol of enzyme; and (iii) the PLP concentration value corresponding to ½ymax could probably reflect the apparent equilibrium dissociation constant of the mutant for PLP.
Figure 9. PMP formation during the half-transamination reaction of L-alanine catalysed by the G82E mutant.

L-Alanine (500 mM) was added to apoG82E mutant pre-incubated with 1 mM PLP (■); 1 mM PLP was added to a mixture containing apoG82E and 500 mM L-Alanine (○); 1 mM PLP and 500 mM L-alanine were incubated at 25 °C for 30 min, and then added to apomutant (▲). Aliquots were removed from the reaction mixtures at the indicated times and the PMP content was measured by HPLC as described in the Experimental section. In each case, the mutant concentration was 8.5 μM, and the buffer was 100 mM potassium phosphate (pH 7.4).
Up to now, the PH1 disease associated with substitution of Gly82 by a glutamate residue has been related to the inability of the variant to bind PLP [2]. Recently, Danpure [22] proposed that the mutant glutamate side-chain could fill the space that should be occupied by the coenzyme, thus preventing the internal Schiff base formation. Gly82 is located at the beginning of helix 81–94 in the large N-terminal domain, and its peptide NH provides one of the hydrogen bonds for the phosphate of PLP. The interaction of the phosphate of PLP with a glycine loop is a common feature of PLP enzymes of fold types I, II and III [23]. Mutation of glycine in serine dehydratase [24] and aminolevulinate synthase [25] causes a decrease in their PLP-binding affinity. Thus it is not surprising that substitution of Gly82 with glutamate results in an ∼700-fold increase in the equilibrium dissociation constant for PLP. It can be inferred, also considering that mutation does not affect the quaternary structure of the enzyme, that the decreased affinity of the mutant for PLP could be ascribed to the loss of the positively charged end of the 81–94 helix dipole that interacts with the phosphate moiety of PLP. However, it is of interest to note that glutamate at position 82 gives rise to a variant that is able to bind ∼2 moles of PLP/mol enzyme, even if its internal aldimine seems to be characterized by a microenvironment different from that of the wild-type. This result is in apparent contrast with that previously reported by Lumb and Danpure [2]. These authors state that G82E mutant did not bind its cofactor despite the presence of saturating amounts of PLP throughout the purification procedure [2]. This apparent contradiction can be understood by considering that the absorbance spectra of the G82E mutant were recorded by these authors at an enzyme concentration of 10 μM, ∼20-fold lower than the apparent KD(PLP) value.
Another aspect of the half-reaction of the G82E mutant with L-alanine to be considered is the meaning of the slow phase. When 1 mM PLP was added to a mixture containing 8.5 μM apomutant and 500 mM L-alanine, a linear plot of PMP formed against time was obtained. The slope of the linear fit passing through the origin was found to be 0.002±0.0002 s−1, a value identical with that of kslow obtained when the mutant was pre-incubated with the coenzyme (Figure 9). Thus the kslow of the reaction of the G82E mutant possibly represents the rate of transamination of the mutant not previously bound to PLP and appears to be at least partially limited by PLP binding. It is of interest that if PLP (1 mM) and L-alanine (500 mM) were incubated at 25 °C for 30 min (a time sufficient to convert free PLP into PLP–L-alanine Schiff base), and then apomutant (8.5 μM) was added, the resulting rate of PMP formation was approx. 2-fold higher than that measured without pre-incubation of coenzyme with L-alanine (Figure 9). Thus binding of a preformed external aldimine to apo G82E mutant seems to improve, even to a limited extent, the rate of PMP formation.
The finding that G82E, at nearly saturating PLP concentration, displays an overall transaminase activity as low as 0.1% that of the normal AGT deserves attention. Since Gly82 plays no obvious catalytic role in transamination, one possible explanation for the drastic reduction in the catalytic power of the mutant is that the PLP binding mode in the normal AGT, besides providing an anchoring point for the coenzyme, may be of structural importance in the catalytic pathway of the enzyme.
To establish whether the kfast is governed by step(s) preceding the aldimine→ketimine conversion, the reaction of 15 μM AGT G82E in the presence of 100 μM PLP with various concentrations of L-alanine was followed by monitoring the 420 nm increase by stopped-flow spectrophotometry. This spectral change cannot be monitored at higher PLP concentration, as free PLP absorbs in this region and obscures the expected increase at 420 nm that occurs due to the formation of external aldimine. kobs values obtained as a function of L-alanine concentration were fitted to a hyperbola yielding kmax and Kapp values of 0.12±0.01 s−1 and 202±48 mM respectively. The kmax value was similar to the value of kfast obtained for PMP formation and to the kcat measured under steady-state conditions. Thus it can be inferred that, in contrast with normal AGT, for which the rate-determining step is the ketimine hydrolysis or the product release, the rate-determining step for the mutant is the formation of external aldimine.
It is also of interest that addition of L-alanine to the G82E mutant (10 μM) pre-incubated with 200 μM PLP causes the disappearance of the dichroic signal at 420 nm but does not determine the appearance of the positive 260 nm dichroic signal observed for the wild-type enzyme. In addition, if the reaction mixture of mutant with L-alanine was subjected to filtration through a Centricon-30 device, analysis of the filtrate by HPLC revealed the presence not only of pyruvate but also of PMP. This indicates that, unlike for wild-type, the PMP formed during the half-transamination reaction does not remain bound to the mutant enzyme, thus suggesting that in the mutant the slight adjustments at the active site, postulated above for wild-type, cannot occur.
The finding that even a prolonged time of incubation of apomutant with PMP (up to 5 mM) does not result in a dichroic signal at 340 nm, as seen for wild-type, confirms that in the G82E mutant PMP binding is greatly affected, and indicates that, unlike for wild-type, the affinity of PMP for the apomutant is lower than that of PLP. Indeed, replacement of the glycine residue in position 82 with a glutamate residue reduces the affinity of PLP by ∼700-fold, whereas it reduces the affinity of PMP by at least 50000-fold. However, a conversion of AGT mutant–PMP into AGT mutant–PLP could be observed if pyruvate or glyoxylate to a final concentration of 1 or 5 mM respectively, was added to reaction mixtures containing apomutant and different PMP concentrations. In both cases, the G82E mutant is not saturated with PMP up to 9 mM, as seen by the linear dependence of the rate of PLP formation on PMP concentration. These data yield the following lower limits: the apparent dissociation constant of PMP from apomutant is >9 mM and the maximal rate of conversion of PMP into PLP in the presence of glyoxylate or pyruvate is >0.027 or >0.010 min−1 respectively. Thus the oxo acid half-transamination takes place when PMP and oxo acid are added to the apomutant but with a rate slower than that catalysed by the wild-type. A possible explanation for this result could be the destabilization of the PMP form in the mutant due to the loss of an undetermined interaction between the side-chain of Gly82 and PMP. The resolution of the AGT–PMP crystal structure of wild-type would probably provide some insight into the PMP binding mode to normal AGT and therefore on the structural role that Gly82 may play in PMP binding. Studies to address this are in progress.
Taken together, all the above data suggest that, although Gly82 does not play a direct catalytic role in the half-transamination reactions, the binding states of PLP and PMP appear to be appreciably affected by the mutation and, as a consequence, the mutation results in an extensive decrease of the kcat values of both transamination reactions.
One classic therapy for PH1 patients is vitamin B6 supplementation [22,26]. However, clinical studies indicate that only a minority (10–30%) of PH1 patients are responsive to pharmacological doses of pyridoxine [27]. Our investigation improves the understanding of the correlation between the genotype and the enzymatic phenotype, thus allowing us to foresee the response to pyridoxine in patients carrying the G82E mutation. Administration of pyridoxine to these patients is not sufficient to counteract the disease in that the molecular defect of this variant does not appear to be the intrinsic inability to bind PLP, more a serious perturbation of its binding site and a drastic reduction in PMP binding. In fact, the dramatic drop of the overall transaminase activity of the G82E variant with respect to that of the wild-type lies on its impairment to undergo an efficient transaldimination of the PLP form of the enzyme with amino acids as well as an efficient conversion of AGT–PMP into AGT–PLP. The administration of a preformed external aldimine rather than pyridoxine could perhaps be a better approach in that at least it would allow bypass of the transaldimination step.
In conclusion, the use of a variety of biochemical techniques has allowed us to gain an in-depth understanding of the characteristics of normal AGT, to transfer this information into the G82E mutant, to unravel the effects of this mutation on the catalytic activity, and to advance a proposal for the treatment of patients bearing this mutation.
Online data
Acknowledgments
We are grateful to Professor Christopher J. Danpure (University College London, London, U.K.) for providing us with the plasmid pAGT-His. This work was supported by funding from the Italian Ministero dell'Istruzione, dell'Università e della Ricerca (PRIN 2005) and Consorzio Interuniversitario Biotecnologie (to C. B. V.).
References
- 1.Danpure C. J., Fryer P., Griffiths S., Guttridge K. M., Jennings P. R., Allsop J., Moser A. B., Naidu S., Moser H. W., MacCollin M., et al. Cytosolic compartmentalization of hepatic alanine:glyoxylate aminotransferase in patients with aberrant peroxisomal biogenesis and its effect on oxalate metabolism. J. Inherit. Metab. Dis. 1994;17:27–40. doi: 10.1007/BF00735393. [DOI] [PubMed] [Google Scholar]
- 2.Lumb M. J., Danpure C. J. Functional synergism between the most common polymorphism in human alanine:glyoxylate aminotransferase and four of the most common disease-causing mutations. J. Biol. Chem. 2000;275:36415–36422. doi: 10.1074/jbc.M006693200. [DOI] [PubMed] [Google Scholar]
- 3.Purdue P. E., Takada Y., Danpure C. J. Identification of mutations associated with peroxisome-to-mitochondrion mistargeting of alanine/glyoxylate aminotransferase in primary hyperoxaluria type 1. J. Cell Biol. 1990;111:2341–2351. doi: 10.1083/jcb.111.6.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Purdue P. E., Lumb M. J., Allsop J., Minatogawa Y., Danpure C. J. A glycine-to-glutamate substitution abolishes alanine:glyoxylate aminotransferase catalytic activity in a subset of patients with primary hyperoxaluria type 1. Genomics. 1992;13:215–218. doi: 10.1016/0888-7543(92)90225-h. [DOI] [PubMed] [Google Scholar]
- 5.Zhang X., Roe S. M., Hou Y., Bartlam M., Rao Z., Pearl L. H., Danpure C. J. Crystal structure of alanine:glyoxylate aminotransferase and the relationship between genotype and enzymatic phenotype in primary hyperoxaluria type 1. J. Mol. Biol. 2003;331:643–652. doi: 10.1016/s0022-2836(03)00791-5. [DOI] [PubMed] [Google Scholar]
- 6.Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danpure C. J., Jennings P. R. Further studies on the activity and subcellular distribution of alanine:glyoxylate aminotransferase in the livers of patients with primary hyperoxaluria type 1. Clin. Sci. 1988;75:315–322. doi: 10.1042/cs0750315. [DOI] [PubMed] [Google Scholar]
- 8.Cellini B., Bertoldi M., Borri Voltattorni C. Treponema denticola cystalysin catalyzes β-desulfination of L-cysteine sulfinic acid and β-decarboxylation of L-aspartate and oxalacetate. FEBS Lett. 2003;554:306–310. doi: 10.1016/s0014-5793(03)01178-5. [DOI] [PubMed] [Google Scholar]
- 9.Bertoldi M., Borri Voltattorni C. Reaction of dopa decarboxylase with L-aromatic amino acids under aerobic and anaerobic conditions. Biochem. J. 2000;352:533–538. [PMC free article] [PubMed] [Google Scholar]
- 10.Popovic B., Tang X., Chirgadze D. Y., Huang F., Blundell T. L., Spencer J. B. Crystal structures of the PLP- and PMP-bound forms of BtrR, a dual functional aminotransferase involved in butirosin biosynthesis. Proteins. 2006;65:220–230. doi: 10.1002/prot.21076. [DOI] [PubMed] [Google Scholar]
- 11.Gibrat J. F., Madej T., Bryant S. H. Surprising similarities in structure comparison. Curr. Opin. Struct. Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- 12.Shindyalov I. N., Bourne P. E. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 13.Yang I. Y., Harris C. M., Metzler D. E., Korytnyk W., Lachmann B., Potti P. P. Properties of 4-ethenyl and 4-ethynyl analogs of pyridoxal phosphate and their reactions with the apo form of asparatate aminotransferase. J. Biol. Chem. 1975;250:2947–2955. [PubMed] [Google Scholar]
- 14.Han Q., Robinson H., Gao Y. G., Vogelaar N., Wilson S. R., Rizzi M., Li J. Crystal structures of Aedes aegypti alanine glyoxylate aminotransferase. J. Biol. Chem. 2006;281:37175–37182. doi: 10.1074/jbc.M607032200. [DOI] [PubMed] [Google Scholar]
- 15.Han G. W., Schwarzenbacher R., Page R., Jaroszewski L., Abdubek P., Ambing E., Biorac T., Canaves J. M., Chiu H. J., Dai X., et al. Crystal structure of an alanine-glyoxylate aminotransferase from Anabaena sp. at 1.70 Å resolution reveals a noncovalently linked PLP cofactor. Proteins. 2005;58:971–975. doi: 10.1002/prot.20360. [DOI] [PubMed] [Google Scholar]
- 16.Gosling J. P., Fottrell P. F. Purification and characterisation of D-amino acid aminotransferase from Rhizobium japonicum. Biochim. Biophys. Acta. 1978;522:84–89. doi: 10.1016/0005-2744(78)90324-8. [DOI] [PubMed] [Google Scholar]
- 17.Hacking A. J., Hassall H. The purification and properties of L-histidine-2-oxoglutarate aminotransferase from Pseudomonas testosteroni. Biochem. J. 1975;147:327–334. doi: 10.1042/bj1470327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuramitsu S., Hiromi K., Hayashi H., Morino Y., Kagamiyama H. Pre-steady-state kinetics of Escherichia coli aspartate aminotransferase catalyzed reactions and thermodynamic aspects of its substrate specificity. Biochemistry. 1990;29:5469–5476. doi: 10.1021/bi00475a010. [DOI] [PubMed] [Google Scholar]
- 19.Yorifuji T., Ishihara T., Naka T., Kondo S., Shimizu E. Purification and characterization of polyamine aminotransferase of Arthrobacter sp. TMP-1. J. Biochem. (Tokyo) 1997;122:537–543. doi: 10.1093/oxfordjournals.jbchem.a021786. [DOI] [PubMed] [Google Scholar]
- 20.Noland B. W., Newman J. M., Hendle J., Badger J., Christopher J. A., Tresser J., Buchanan M. D., Wright T. A., Rutter M. E., Sanderson W. E., et al. Structural studies of Salmonella typhimurium ArnB (PmrH) aminotransferase: a 4-amino-4-deoxy-L-arabinose lipopolysaccharide-modifying enzyme. Structure. 2002;10:1569–1580. doi: 10.1016/s0969-2126(02)00879-1. [DOI] [PubMed] [Google Scholar]
- 21.Sivaraman J., Li Y., Larocque R., Schrag J. D., Cygler M., Matte A. Crystal structure of histidinol phosphate aminotransferase (HisC) from Escherichia coli, and its covalent complex with pyridoxal-5′-phosphate and L-histidinol phosphate. J. Mol. Biol. 2001;311:761–776. doi: 10.1006/jmbi.2001.4882. [DOI] [PubMed] [Google Scholar]
- 22.Danpure C. J. Primary hyperoxaluria type 1: AGT mistargeting highlights the fundamental differences between the peroxisomal and mitochondrial protein import pathways. Biochim. Biophys. Acta. 2006;1763:1776–1784. doi: 10.1016/j.bbamcr.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 23.Grishin N. V., Phillips M. A., Goldsmith E. J. Modeling of the spatial structure of eukaryotic ornithine decarboxylases. Protein Sci. 1995;4:1291–1304. doi: 10.1002/pro.5560040705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marceau M., Lewis S. D., Shafer J. A. The glycine-rich region of Escherichia coli D-serine dehydratase. Altered interactions with pyridoxal 5′-phosphate produced by substitution of aspartic acid for glycine. J. Biol. Chem. 1988;263:16934–16941. [PubMed] [Google Scholar]
- 25.Gong J., Kay C. J., Barber M. J., Ferreira G. C. Mutations at a glycine loop in aminolevulinate synthase affect pyridoxal phosphate cofactor binding and catalysis. Biochemistry. 1996;35:14109–14117. doi: 10.1021/bi961296h. [DOI] [PubMed] [Google Scholar]
- 26.Danpure C. J. Molecular etiology of primary hyperoxaluria type 1: new directions for treatment. Am. J. Nephrol. 2005;25:303–310. doi: 10.1159/000086362. [DOI] [PubMed] [Google Scholar]
- 27.Pirulli D., Marangella M., Amoroso A. Primary hyperoxaluria: genotype-phenotype correlation. J. Nephrol. 2003;16:297–309. [PubMed] [Google Scholar]
- 28.De Lano W. San Carlos, CA: DeLano Scientifics; 2002. The pyMol Molecular Graphics system. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.