

Abstract
The first steps of transcription initiation include binding of RNA polymerase to a promoter to form an inactive, unstable, closed complex (described by an equilibrium constant, KB) and isomerization of the closed complex to an active, stable, open complex (described by a forward rate constant, kf). λ cI protein activates the PRM promoter by specifically increasing kf. A positive control mutant, cI-pc2, is defective for activation because it fails to raise kf. An Arg to His change in the σ70 subunit of RNA polymerase was previously obtained as an allele-specific suppressor of cI-pc2. To elucidate how the mutant polymerase restores the activation function of the mutant activator, abortive initiation assays were performed, using purified cI proteins and RNA polymerase holoenzymes. The change in σ does not significantly alter KB or kf in the absence of cI protein. As expected, cI-pc2 activates the mutant polymerase in the same way that wild-type cI activates the wild-type polymerase, by increasing kf. An unexpected and novel finding is that the wild-type activator stimulates the mutant polymerase, but not wild-type polymerase, by increasing KB.
Keywords: Escherichia coli RNA polymerase, σ subunit, suppression, compensatory mutations, abortive initiation
Gene expression is frequently regulated by activator proteins that bind to specific DNA sites and increase the rate of transcription initiation by RNA polymerase at a nearby promoter. In most cases, activation is thought to involve a favorable protein–protein interaction between the activator and RNA polymerase on the template DNA. A classic example is phage λ cI protein (also called λ repressor), which activates transcription of its own gene from PRM, the promoter for synthesis of repressor during maintenance of lysogeny (1). PRM is a weak, leftward promoter that is oriented divergently from a strong, rightward promoter (PR) in the OR (right operator) region (Fig. 1). Both promoters are controlled by binding of cI protein to three related 17-bp sites. PR overlaps with OR1 and OR2, while PRM overlaps with OR2 and OR3. At the normal concentration of cI in a lysogen, cI dimers bind cooperatively to OR1 and OR2, leaving OR3 vacant most of the time. PR is severely repressed, while the cI dimer bound to OR2 activates PRM. At higher concentrations, cI also binds to OR3, thereby repressing PRM.
Figure 1.

Sequence of the λ OR region. The 5′ ends of RNAs made by natural transcription are shown, as well as trinucleotides made in abortive initiation reactions. Critical promoter elements are marked with asterisks. The OR3-r2 mutation was used in assays in vitro.
Activation of PRM involves an interaction between an acidic patch on the surface of the helix–turn–helix motif of cI protein (2–4) and a target region near the C terminus of the σ70 subunit of Escherichia coli RNA polymerase (5, 6). We previously showed that a single amino acid change in σ (Arg-596 to His; abbreviated RH596) restores activation by a mutant form of cI that has a single amino acid change in the activation patch (Asp-38 to Asn; called pc2 for its specific defect in positive control). The change in σ fully suppressed the cI-pc2 defect in vivo, allowing the mutant activator to activate a PRM lacZ fusion. Two other mutant activators, cI-pc1 (2) and cI-pc3 (3), were not suppressed by the mutant σ. In this paper, we address two questions that could not be resolved by genetic experiments. The first issue was the formal possibility that the mutant σ might exert its effect indirectly (e.g., by altering expression of other genes, since nearly all transcription is σ70-dependent). This idea is ruled out here by the demonstration that the mutant activator stimulates initiation by the mutant polymerase in a purified system in vitro. The second question was whether cI-pc2 stimulates the mutant polymerase by the same mechanism used by wild-type cI to stimulate the wild-type polymerase.
λ cI activates PRM by speeding up the rate at which RNA polymerase forms an “open,” transcriptionally active enzyme–DNA complex. According to a simplified two-step model for open complex formation, RNA polymerase (R) binds to the promoter (P) to form an inactive, unstable closed complex, RPC, that then undergoes a relatively slow isomerization to form the open complex, RPO:
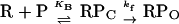 |
Initial binding is characterized by an apparent equilibrium constant, KB; isomerization is characterized by a forward rate constant, kf. When wild-type E. coli RNA polymerase was used, Hawley and McClure (7) found that wild-type cI activates PRM by enhancing kf about 10-fold, without having a significant effect on KB. The cI-pc2 mutant fails to stimulate kf without creating a severe defect in KB (8). In the experiments reported here, similar results were obtained using wild-type RNA polymerase. In addition, we show that (i) the Arg-596 to His change in σ does not significantly alter PRM promoter strength in the absence of cI protein; and (ii) with the mutant polymerase, cI-pc2 raises kf without changing KB significantly, which implies that cI-pc2 stimulates the mutant enzyme by the same mechanism that wild-type cI stimulates the wild-type enzyme. An interesting and unexpected discovery is that wild-type cI increases KB for the mutant polymerase, instead of activating the isomerization step.
MATERIALS AND METHODS
Reagents.
TE is 10 mM Tris·HCl, pH 8.0/0.1 mM EDTA. Lysis buffer is 100 mM Tris·HCl, pH 8.0/200 mM NaCl/10 mM MgCl2/2 mM CaCl2/1 mM EDTA/0.1 mM dithiothreitol/glycerol (5%, vol/vol)/hen egg white lysozyme (150 μg/ml). SB is 10 mM Tris·HCl, pH 8.0/2 mM CaCl2/0.1 mM EDTA/0.1 mM dithiothreitol/5% glycerol. Standard reaction buffer is 40 mM Hepes, pH 8.0/100 mM potassium glutamate/10 mM MgCl2/1 mM dithiothreitol/bovine serum albumin (30 μg/ml). Cytidylyl(3′-5′)adenosine (CpA) and uridylyl(3′-5′)adenosine (UpA) were purchased from ICN and Sigma, respectively.
Plasmids and Bacteria.
Plasmids are listed in Table 1. A pair of isogenic E. coli strains, rpoD+ and rpoD-RH596, were constructed by phage P1 transduction, using zgh::Tn10, an insertion that is linked to rpoD and confers resistance to tetracycline (20 μg/ml). Donors were CAG4018 (zgh::Tn10) and CAG4025 (zgh::Tn10 rpoD2, which is identical to rpoD-RH596) (15) carrying pMS1382, an rpoD+ plasmid that suppressed the temperature-sensitive phenotype of the rpoD2 strain and enhanced growth of P1. The recipient was E. coli RV308, which is Δ(lac)χ74 galPO-308::IS2 rpsL (16). Tetracycline-resistant transductants were checked for absence of pMS1382 and P1, and rpoD genotypes were determined by PCR amplification of chromosomal DNA and PCR sequencing, using the Promega fmol system. The resulting strains are MS4274 (RV308 zgh::Tn10) and MS4276 (RV308 zgh::Tn10 rpoD2). Although the rpoD2 (RH596) mutation does not confer a temperature-sensitive phenotype in the RV308 genetic background, both strains were maintained at 30°C.
Table 1.
Plasmids
| Plasmid | Description |
|---|---|
| pAED4 | pBR322-based Ampr T7 expression vector (D. S. Doering); derivative of pET-3a (9) |
| pCICK | pACYC177-based Camr plasmid carrying λ cI-ts857 (10) |
| pLysS | pACYC184-based Camr plasmid carrying T7 gene 3.5 (11) |
| pMRG8 | pBR322-based Ampr plasmid carrying E. coli rpoD+ fused to λ PLOL (12) |
| pMS802 | pSC101-based Spcr plasmid carrying E. coli lacIq (13) |
| pMS1382 | pBR322-based Ampr plasmid carrying E. coli rpoD+ fused to PlacUV5 |
| pMS1454 | NdeI–KpnI cI fragment* in the NdeI–KpnI backbone of pAED4 |
| pMS1456 | NdeI–KpnI cI-pc2 fragment* in the NdeI–KpnI backbone of pAED4 |
| pMS1488 | Replace BamHI–HindIII rpoD+ fragment of pMRG8 with the corresponding BamHI–BssHII rpoD-RH596 fragment from pMS1366 (5). HindIII and BssHII ends were filled in, using DNA polymerase I large fragment. |
| pPR1347 | pSC101-based Kanrcosλ cosmid carrying Salmonella typhimurium rfb and rfc operons (14) |
Antibiotic resistance: Amp, ampicillin; Cam, chloramphenicol; Spc, spectinomycin; and Kan, kanamycin.
Fragment extends from a site-directed NdeI site overlapping the start codon to a KpnI linker inserted at a filled-in MunI site downstream of cI. The gene contains four site-directed mutations that create restriction sites but do not alter the amino acid sequence.
These E. coli strains were made transiently sensitive to Salmonella phage P22, using cosmid pPR1347 (14), which directs synthesis of the O antigen of S. typhimurium. The cosmid is maintained by selecting resistance to kanamycin (10 μg/ml), but is spontaneously lost when kanamycin is removed. MS4274 and MS4276 were transduced to kanamycin resistance with λ particles containing pPR1347. The resulting strains were purified, checked for absence of λ, and infected in liquid culture with high multiplicities of one or both of the following: P22 PRM-lac8 ΔAB (5), which carries E. coli lacZ fused to λ PRMOR; and P22 Plac-cIA (5), which carries λ cI fused to a weak constitutive version of the E. coli lac promoter, and also carries the bla gene for resistance to ampicillin. Infected cultures were diluted, incubated overnight at 30°C, and plated on LB agar (17) containing 5-bromo-4-chloro-3-indolyl β-d-galactopyranoside (150 μg/ml; to detect presence of the lacZ prophage) with or without ampicillin (50 μg/ml; to select for the second prophage). Candidates for double lysogens were purified repeatedly on LB agar and screened for retention of both prophages by testing for retention of lacZ and bla; unstable double lysogens (which probably carried tandem prophages that were subsequently resolved by recombination) were discarded. Stable double lysogens presumably have nontandem prophages: PRM-lac8 ΔAB at the P22 attachment site and Plac-cIA at the λ attachment site (Fig. 2). [A recombinant prophage containing both fusions cannot arise, because PRM-lac8 ΔAB att22 is allelic to Plac-cIA attλ (5).]
Figure 2.

System used to measure activation in vivo. Strains have only one form of σ70, either wild-type or mutant, encoded by the rpoD gene at its normal locus in the E. coli chromosome (thick line). A prophage (thin line) at the λ attachment site produces a low level of cI protein encoded by various cI alleles fused to a weak, constitutive promoter. Effective combinations of σ and cI activate transcription of a lacZ reporter gene fused to wild-type λ PRMOR on a prophage at the P22 attachment site. λ cI protein does not control its own synthesis and does not control maintenance of lysogeny by either prophage.
Overexpression plasmids pMS1454 and pMS1456 (which carry λ cI fused to the T7 φ10 promoter and gene 10 ribosome binding site) were used in derivatives of E. coli BL21(λDE3)/pLysS (11) that also carry pMS802, a compatible lacIq plasmid that improved retention of pMS1454 and pMS1456. These strains, MS4456 and MS4458, were propagated in media containing chloramphenicol (20 μg/ml), spectinomycin (50 μg/ml), and ampicillin and were used to overproduce wild-type cI and cI-pc2 protein, respectively. [λDE3 does not carry the λ cI gene (9).]
Overexpression plasmids pMRG8 and pMS1488 carry E. coli rpoD+ and rpoD-RH596, respectively, fused to λ PL, which is controlled by temperature-sensitive λ repressor encoded by pCICK. E. coli MS4522 is MS4274/pCICK/pMRG8; MS4523 is MS4276/pCICK/pMS1488. Because the rpoD allele in the E. coli chromosome is the same as the rpoD allele in the overproducing plasmid, each strain produces only one type of σ70, either wild-type or mutant.
RNA Polymerase Holoenzymes.
σs were overproduced in parallel in MS4522 and MS4523 and were purified as described by Gribskov and Burgess (12) through the DEAE-Sephadex column step, with minor modifications. Partially purified σs were mixed with the same batch of purified wild-type E. coli RNA polymerase core enzyme (18) at a molar ratio of 1.5. The resulting holoenzymes were then purified (18). Each was estimated to be >90% pure as judged by Coomassie brilliant blue staining of samples analyzed by SDS/PAGE. Fractional activity of the holoenzymes, assayed on a poly[d(AT)] template (19), was about 40%. The active concentrations were used for the TAU plot analyses that yielded the reported values for KB.
Purification of cI.
MS4456 and MS4458 were grown at 37°C in LB plus antibiotics (see above) until OD550 reached 0.7, induced by adding isopropyl β-d-thiogalactopyranoside to 0.5 mM, incubated for 3 hr, and harvested by centrifugation. Methods for cell lysis and purification of cI were adapted from ref. 20. Cells were suspended in lysis buffer, sodium deoxycholate (0.05%) and phenylmethylsulfonyl fluoride (35 μg/ml) were added, and mixtures were incubated at 4°C for 20 min. To reduce viscosity, crude lysates were sonicated, diluted ≈5-fold with SB + 0.2 M NaCl, and treated with DNase I (50 μg/ml) at 4°C for 30 min. NaCl concentration was adjusted to 0.55 M before low-speed centrifugation to remove debris. Cleared lysates were mixed with polyethyleneimine (0.1%) at 4°C and the resulting precipitates were removed by centrifugation. Ammonium sulfate was added (40 g per 100 ml of supernatant), and the resulting precipitates were collected by centrifugation, dissolved in SB + 0.2 M NaCl, and dialyzed against SB + 0.1 M NaCl overnight at 4°C. Dialysates were clarified by centrifugation and loaded on Affi-Gel Blue (Bio-Rad) columns equilibrated with SB + 0.1 M KCl. Columns were washed with 2 bed vol of SB + 0.1 M KCl and eluted with SB containing a linear gradient of KCl (0.1 to 1 M). Fractions containing cI (identified by SDS/PAGE) were pooled, diluted 2-fold with SB, and loaded on HA-Ultrogel (Sigma) columns equilibrated with SB + 0.1 M KCl. Columns were washed with 1 bed vol of 0.1 M potassium phosphate, pH 7.4, and eluted with a linear gradient of potassium phosphate (0.1 to 1 M), pH 7.4. Peak fractions containing cI were pooled and concentrated by precipitation with ammonium sulfate. Pellets were dissolved in SB + 0.2 M NaCl (≈4 mg of protein per ml) and dialyzed against SB + 0.2 M NaCl overnight at 4°C. Dialysates were clarified by centrifugation and supernatants were stored frozen at −20°C (short-term) or −70°C (long-term). Both cI preparations were estimated to be about 90% pure. Activities were determined by repression assays, which correlate well with filter-binding assays of DNA-binding activity (8): 11 nM cI gives 50% repression of PR, using wild-type RNA polymerase (see Fig. 3A). The active fraction (measured by comparing activity with A280) was 15% for wild-type cI and 30% for cI-pc2.
Figure 3.

Effect of wild-type cI and cI-pc2 on repression of PR and activation of PRM. The activities of PR (A and C) and PRM (B and D) in the presence of various concentrations of wild-type cI (•) or cI-pc2 (○) were measured by monitoring the rate of synthesis of abortive products by wild-type RNA polymerase holoenzyme (A and B) or holoenzyme containing σ70-RH596 (C and D) as described in the text. The maximal rate for PR (set at 100) corresponds to 361 or 337 CpApU per promoter per min in A and C, respectively; the nonactivated rate for PRM (set at 1) corresponds to 71 or 81 UpApU per promoter per min in B and D, respectively.
DNA Template.
A fragment containing λ PRM and PR was obtained by PCR amplification, using a HindIII digest of λ112 cI-sus34 OR3-r2 (16) DNA as template (7.9 pM) and limiting amounts of primers (200 nM each). The product (343 bp; +140 to −203 with respect to PRM) was extracted with phenol/chloroform (1:1) and chloroform, and dialyzed against TE + 0.1 M NaCl and TE. DNA concentration was determined by measuring A260 and confirmed by gel electrophoresis.
Activation-Repression Curves.
Effects of cI on PRM and PR were measured by monitoring the rate of synthesis of abortive products as described (7, 8). DNA, UpA or CpA, [α-32P]UTP, and cI were mixed and prewarmed at 37°C for 8 min in 40 μl of standard reaction buffer. Reactions were initiated by adding 10 μl of RNA polymerase prewarmed in standard reaction buffer. Final concentrations were 1.1 nM DNA, 0.5 mM UpA or CpA, 50 μM [α-32P]UTP (≈400 cpm/pmol), and 40 nM RNA polymerase. The PRM reaction (UpA + UTP → UpApU) was assayed after 6 and 12 min. The PR reaction (CpA + UTP → CpApU) was assayed after 8 and 16 min. Product trinucleotides were separated from UTP by ascending chromatography (8), and radioactivity in the product peak was normalized to total radioactivity on the chromatogram (≈300,000 cpm). Reaction rates (product per promoter per min) were calculated, normalized to the controls (without added cI), and plotted against the concentration of active cI.
TAU Plot Analysis.
The average time required for open complex formation (τOBS) at PRM was measured by lag assays (7). DNA, nucleotides, and cI were mixed in 256 μl of standard reaction buffer and prewarmed at 37°C for 8 min before initiating the reaction by addition of 64 μl of RNA polymerase prewarmed in standard reaction buffer. Final concentrations were 1 nM DNA, 0.2 or 0.5 mM UpA, 50 μM [α-32P]UTP (≈400 cpm/pmol), and 0 or 50 nM cI protein; RNA polymerase was varied from 12 to 80 nM. UpA concentration was 0.2 mM in reactions without cI and 0.5 mM in reactions with cI; in both cases, substrate depletion was <12%. Portions (20 μl) were removed at various times and spotted onto the origin of a chromatogram prestreaked with 20 μl of 0.1 M EDTA to stop the reaction. The percentage of UTP incorporated (see above) was plotted versus time for each reaction, and τOBS was determined by a computer program (7) that performed a least-squares fit of the data to the equation
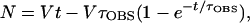 |
where N is the amount of UpApU synthesized per promoter, V is the final steady-state velocity, and t is time (min). Values of τOBS were measured at various RNA polymerase concentrations ([R]) and were plotted against 1/[R]. These TAU plots produce straight lines, as predicted by the equation
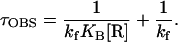 |
The reciprocal of the intercept yields kf and the ratio of intercept to slope yields KB. In this study, τOBS values were uniformly shorter and kf values were uniformly higher than those obtained previously (7, 8). Our standard reaction buffer contained potassium glutamate instead of KCl, to minimize experimental variability due to small differences in chloride concentrations (21); this change probably contributes to the increase in kf (unpublished data). Another difference is that the template used here has the OR3-r2 mutation, which was not present in any of the templates used previously in abortive initiation assays (7, 8). The OR3 mutation does not block activation of PRM by wild-type cI, but reduces binding of cI to OR3 and repression of PRM (16). Though OR3-r2 is at a nonconserved position (−18; Fig. 1), it may slightly improve PRM activity (16).
RESULTS
Effect of σ70-RH596 in E. coli.
In the experiments reported previously, the effect of the Arg-596 to His change in E. coli σ70 was examined in S. typhimurium strains carrying the E. coli rpoD gene on a high-copy plasmid and the S. typhimurium rpoD+ gene in the chromosome (5). To examine the effect of the mutant σ in E. coli strains that have only one copy of rpoD, we made use of the fact that the rpoD-RH596 mutation was previously isolated as a viable, haploid allele; the rpoD2 mutation characterized by Hu and Gross (15) is identical to rpoD-RH596. Isogenic E. coli rpoD+ and rpoD2 strains were constructed in a Δ(lac) genetic background as described above. These strains were then lysogenized with phages that provide a system for assaying activation of PRM by λ cI in vivo (Fig. 2).
The results obtained using E. coli haploids (Table 2) are strikingly similar to those obtained using rpoD plasmids in S. typhimurium (5). σ70-RH596 fully restores the activation function of cI-pc2. This effect is specific: the mutant σ does not respond to cI-pc1 or -pc3 and is slightly defective in responding to wild-type cI. The basal activity of PRM (activity with no cI) is not affected by the change in σ.
Table 2.
Effect of rpoD-RH596 in E. coli haploids
| Activator | Relative β-galactosidase activity
|
|
|---|---|---|
| rpoD+ | rpoD-RH596 | |
| None | 15.0 ± 1.1 | 15.6 ± 1.6 |
| cI wild type | (100) | 79.1 ± 4.3 |
| cI-pc1 (Gly-43 to Arg) | 4.7 ± 1.1 | 5.4 ± 1.2 |
| cI-pc2 (Asp-38 to Asn) | 15.5 ± 0.7 | 133 ± 10 |
| cI-pc3 (Glu-34 to Lys) | 0.6 ± 0.1 | 2.0 ± 0.5 |
Strains are derivatives of MS4274 and MS4276 carrying a PRM-lac8 ΔAB prophage plus either no second prophage or one of four Plac-cIA prophages. Four cultures of each strain were grown at 30°C in M9CAA (22), harvested in exponential phase, and assayed as described (23). Specific activities of the four control cultures (rpoD+ cI+) were averaged to yield the mean specific activity of the control (set at 100; corresponds to 131 Miller units). Specific activity of each of the other cultures was expressed as a percentage of the mean specific activity of the control. The four percentages for a particular genotype were used to calculate the mean and standard deviation given.
We note that the cI-pc1 and -pc3 mutants interfere with PRM activity, reducing expression below the basal level. The repression observed here, about 70% for cI-pc1 and 90% for -pc3, was comparable with both σs. This effect has been seen previously (3, 5, 24). The possibility that cI-pc1 and -pc3 inhibit PRM, rather than simply fail to activate PRM, led to the original choice of cI-pc2 for kinetic studies of the positive control defect (8). It has been suggested that cI-pc1 and -pc3 repress PRM by binding to OR3, but the evidence presented does not strongly support that model (24). An alternative model is that cI-pc1 and -pc3 bound to OR2 (and not to OR3) have an unfavorable interaction with RNA polymerase, because each mutation introduces a basic amino acid residue in the patch of cI that normally contacts a basic patch of RNA polymerase.
Effect of σ70-RH596 on Activation and Repression in Vitro.
To show that σ70-RH596 directly restores the activation function of cI-pc2, abortive initiation assays were performed, using purified cI proteins (wild-type or cI-pc2) and purified RNA polymerase holoenzymes containing σ70 (wild-type or σ70-RH596). On a template carrying both PR and PRM, open complexes at each promoter were monitored by their ability to repeatedly synthesize a trinucleotide corresponding to −1, +1, and +2: open complexes at PR convert CpA and UTP to CpApU, whereas open complexes at PRM convert UpA and UTP to UpApU (see Fig. 1). Fig. 3 shows the effects of the cI proteins on occupancy of both promoters. PR was repressed by increasing amounts of either wild-type cI or cI-pc2, and the repression curves were similar when wild-type RNA polymerase (Fig. 3A) and RNA polymerase containing σ70-RH596 (Fig. 3C) were used. With wild-type RNA polymerase, PRM was activated 3-fold by wild-type cI, but less than 2-fold by cI-pc2 (Fig. 3B). With the mutant RNA polymerase, PRM was activated about 5-fold by cI-pc2, but less than 2-fold by wild-type cI (Fig. 3D). These results are in excellent agreement with the activation assays in vivo (Table 2). The activation-repression experiments of Fig. 3 cannot be used to determine the full extent or identity of the activated step, but these results indicated the choice of cI concentration to be used for the kinetic experiments described next.
Kinetics of Open Complex Formation.
Lag assays were used to determine τOBS, the average time required for RNA polymerase to form an open complex at PRM in the presence or absence of cI protein. τOBS was measured at various RNA polymerase concentrations and plotted against the reciprocal of RNA polymerase concentration. These “TAU plots” were used to calculate the apparent binding constant, KB, and isomerization rate constant, kf (Table 3).
Table 3.
Kinetic parameters of open complex formation at PRM
| Activator | Wild-type RNA polymerase
|
Mutant RNA polymerase
|
||
|---|---|---|---|---|
| KB × 10−6, M−1 | kf × 103, s−1 | KB × 10−6, M−1 | kf × 103, s−1 | |
| None | 13 | 5.0 | 7.3 | 5.6 |
| cI wild type | 11 | 86 | 63 | 13 |
| cI-pc2 | 25 | 5.0 | 4.3 | 150 |
In the absence of cI, the values for KB and kf obtained with the mutant RNA polymerase and those obtained with wild-type polymerase are the same within experimental error (estimated as ±30–40% from the linear least-squares analyses). This finding agrees with the results of Table 2, which show that the mutant σ had no effect on the basal activity of PRM in vivo.
With wild-type RNA polymerase, activation of PRM occurred as described previously (7, 8). Wild-type cI increased kf more than 10-fold, but it had no significant effect on KB. The cI-pc2 mutant activator failed to raise kf, but it did not create a defect in initial binding.
The mutant RNA polymerase is strikingly different from wild-type polymerase in its response to wild-type cI and cI-pc2. The Arg-596 to His change in σ fully restores the activation function of cI-pc2. With the mutant polymerase, the mutant activator raised kf more than 20-fold and had no significant effect (within experimental error) on KB. Thus, cI-pc2 stimulates open complex formation at PRM by the mutant polymerase in the same way that wild-type cI stimulates the wild-type polymerase. Somewhat surprisingly, in the presence of the mutant RNA polymerase, wild-type cI raised KB about 9-fold and kf only about 2-fold.
DISCUSSION
Activation of PRM was investigated with all possible combinations of wild-type and mutant RNA polymerase holoenzymes and wild-type and mutant cI proteins. As shown previously, wild-type cI stimulates wild-type RNA polymerase to form open complexes on PRM by increasing kf (7), whereas the mutant cI-pc2 activator does not (8). With holoenzyme containing σ70-RH596, however, cI-pc2 stimulates formation of open complexes on PRM by increasing kf more than 20-fold, with little or no effect on KB (Table 3). This result shows that the change in σ directly suppresses the cI-pc2 defect, since the only proteins in the abortive initiation assays are RNA polymerase and cI. Because the same rate parameter is affected, cI-pc2 apparently stimulates the mutant polymerase in the same way that wild-type cI stimulates the wild-type polymerase. The cI-pc2 defect is fully suppressed, both in vivo and in vitro: the mutant activator works with the mutant polymerase slightly better than the wild-type activator works with the wild-type polymerase. The mutant σ has no significant effect on the basal activity of PRM, in vivo or in vitro. Thus, σ70-RH596 specifically, fully, and mechanistically suppresses the cI-pc2 defect.
Two general types of models can be proposed to explain the activation defect of cI-pc2 and the restoration of activation by the change in σ. The first model envisions that the pc2 mutation (Asp-38 to Asn) removes a favorable contact between the activator and RNA polymerase, and the Arg-596 to His change in σ provides an alternative, equally favorable contact. The second model is that Asp-38 of cI does not normally play a critical role in activation, but the Asp to Asn change (pc2) introduces an unfavorable interaction that prevents activation. Replacing Arg-596 of σ with His could simply relieve the clash without creating a new, favorable contact. Either model can explain the observation that activation of the mutant polymerase by the mutant activator resembles activation of the wild-type polymerase by the wild-type activator. Both models imply that the activation patch of cI is close to residue 596 of σ, an idea that is supported by molecular modeling (25). Other mutational studies (ref. 4; unpublished data) favor the second model, because activation works reasonably well with several other combinations of amino acids at position 596 of σ and position 38 of cI. Of course, elements of both models may apply here, just as they do in detailed studies of other protein–protein or protein–nucleic acid interactions.
A novel and surprising finding is that the mutant polymerase differs from the wild-type polymerase in its response to wild-type cI. With the wild-type enzyme, cI raises kf and has little effect on KB; with the mutant enzyme, cI raises KB about 9-fold and has a small effect on kf. The surprising aspect of this finding is that a single amino acid change in σ switches the activation of PRM from the isomerization step to the initial binding step. Although similar switches have not yet been reported for other combinations of polymerases and activators, such changes in simpler enzyme–substrate interactions are well documented (26). In those cases, a structural change that favorably affects the interaction between a substrate and an enzyme subsite can decrease Km (analogous to an increase in KB) or increase kcat (analogous to kf) or both. The partitioning between the two steps depends on whether the change in structure stabilizes the ground state or the activated enzyme–substrate complex. Applying the same reasoning to interactions between cI and RNA polymerase at PRM, cI wild-type stabilizes the transition state between RPC and RPO for the wild-type enzyme by about 1.6 kcal/mol; with the mutant enzyme, wild-type cI stabilizes the closed complex (RPC) by about 1.3 kcal/mol. The observation that single amino acid changes in σ or in cI can affect the partitioning between the two steps implies that favorable interactions affecting the first step are not profoundly different from those affecting the second step.
Initial binding of polymerase to the promoter is conceptually simpler than isomerization, since initial binding presumably resembles DNA binding by simpler proteins (e.g., cI binding to its operators). Enhancement of initial polymerase–promoter binding by an activator is also relatively easy to understand. For example, a better fit between complementary patches on the activator and polymerase is readily grasped as leading to an increase in a binding constant. The real mystery in promoter function and activation is the nature of the isomerization step, which involves separation of the DNA strands around the startpoint of transcription, uptake of ions, and possibly a conformational change in the enzyme (27). These reactions are not fully understood, and neither are the mechanisms by which activators speed them up. If additional amino acid changes in activators and polymerases can be found that have discrete effects on the steps leading to the open complex, it may be possible to interpret the partitioning between the steps in terms of structural changes that go beyond the current, still rather nebulous, descriptions.
Acknowledgments
We salute Carol Gross for generously providing E. coli strains and plasmids, even at the worst of times. We also thank E. Freund, R. Maurer, L. Mendelman, I. Molineux, P. Reeves, and C. Waldburger for providing strains and D. Burz and G. Ackers for advice on cI purification. This work was supported by grants from the National Institutes of Health: GM36811 to M.M.S. and GM30375 to W.R.M.
References
- 1.Ptashne M. A Genetic Switch. Oxford: Cell/Blackwell Scientific; 1992. [Google Scholar]
- 2.Guarente L, Nye J S, Hochschild A, Ptashne M. Proc Natl Acad Sci USA. 1982;79:2236–2239. doi: 10.1073/pnas.79.7.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hochschild A, Irwin N, Ptashne M. Cell. 1983;32:319–325. doi: 10.1016/0092-8674(83)90451-8. [DOI] [PubMed] [Google Scholar]
- 4.Bushman F D, Shang C, Ptashne M. Cell. 1989;58:1163–1171. doi: 10.1016/0092-8674(89)90514-x. [DOI] [PubMed] [Google Scholar]
- 5.Li M, Moyle H, Susskind M M. Science. 1994;263:75–77. doi: 10.1126/science.8272867. [DOI] [PubMed] [Google Scholar]
- 6.Kuldell N, Hochschild A. J Bacteriol. 1994;176:2991–2998. doi: 10.1128/jb.176.10.2991-2998.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hawley D K, McClure W R. J Mol Biol. 1982;157:493–525. doi: 10.1016/0022-2836(82)90473-9. [DOI] [PubMed] [Google Scholar]
- 8.Hawley D K, McClure W R. Cell. 1983;32:327–333. doi: 10.1016/0092-8674(83)90452-x. [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg A H, Lade B N, Chui D S, Lin S W, Dunn J J, Studier F W. Gene. 1987;56:125–135. doi: 10.1016/0378-1119(87)90165-x. [DOI] [PubMed] [Google Scholar]
- 10.Zerbib D, Polard P, Escoubas J M, Galas D, Chandler M. Mol Microbiol. 1990;4:471–477. doi: 10.1111/j.1365-2958.1990.tb00613.x. [DOI] [PubMed] [Google Scholar]
- 11.Studier F M. J Mol Biol. 1991;219:37–44. doi: 10.1016/0022-2836(91)90855-z. [DOI] [PubMed] [Google Scholar]
- 12.Gribskov M, Burgess R R. Gene. 1983;26:109–118. doi: 10.1016/0378-1119(83)90180-4. [DOI] [PubMed] [Google Scholar]
- 13.Waldburger C, Susskind M M. J Mol Biol. 1994;235:1489–1500. doi: 10.1006/jmbi.1994.1103. [DOI] [PubMed] [Google Scholar]
- 14.Neal B L, Brown P K, Reeves P R. J Bacteriol. 1993;175:7115–7118. doi: 10.1128/jb.175.21.7115-7118.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu J C, Gross C A. Mol Gen Genet. 1985;199:7–13. doi: 10.1007/BF00327502. [DOI] [PubMed] [Google Scholar]
- 16.Maurer R, Meyer B J, Ptashne M. J Mol Biol. 1980;139:147–161. doi: 10.1016/0022-2836(80)90302-2. [DOI] [PubMed] [Google Scholar]
- 17.Susskind M M, Wright A, Botstein D. Virology. 1971;45:638–652. doi: 10.1016/0042-6822(71)90178-4. [DOI] [PubMed] [Google Scholar]
- 18.Lowe P A, Hager D A, Burgess R R. Biochemistry. 1979;18:1344–1352. doi: 10.1021/bi00574a034. [DOI] [PubMed] [Google Scholar]
- 19.Hansen U M, McClure W R. J Biol Chem. 1980;255:9556–9563. [PubMed] [Google Scholar]
- 20.Beckett D, Koblan K S, Ackers G K. Anal Biochem. 1991;196:69–75. doi: 10.1016/0003-2697(91)90118-d. [DOI] [PubMed] [Google Scholar]
- 21.Liermo S, Harrison C, Kayley D S, Burgess R R, Record M T., Jr Biochemistry. 1987;26:2095–2101. doi: 10.1021/bi00382a006. [DOI] [PubMed] [Google Scholar]
- 22.Smith H O, Levine M. Proc Natl Acad Sci USA. 1964;52:356–363. doi: 10.1073/pnas.52.2.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller J. Experiments in Molecular Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1972. [Google Scholar]
- 24.Kolkhof P, Müller-Hill B. J Mol Biol. 1994;242:23–36. doi: 10.1006/jmbi.1994.1554. [DOI] [PubMed] [Google Scholar]
- 25.Busby S, Ebright R H. Cell. 1994;79:743–746. doi: 10.1016/0092-8674(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 26.Fersht A. Enzyme Structure and Mechanism. 2nd Ed. New York: Freeman; 1985. pp. 311–346. [Google Scholar]
- 27.Suh W-C, Ross W, Record M T., Jr Science. 1993;259:358–361. doi: 10.1126/science.8420002. [DOI] [PubMed] [Google Scholar]