

Abstract
β-Actin, once thought to be an exclusively cytoplasmic protein, is now known to have important functions within the nucleus. Nuclear β-actin associates with and functions in chromatin remodeling complexes, ribonucleic acid polymerase complexes, and at least some ribonucleoproteins. Proteins involved in regulating actin polymerization are also found in the interphase nucleus. We define the dynamic properties of nuclear actin molecules using fluorescence recovery after photobleaching. Our results indicate that actin and actin-containing complexes are reduced in their mobility through the nucleoplasm diffusing at ∼0.5 μm2 s−1. We also observed that ∼20% of the total nuclear actin pool has properties of polymeric actin that turns over rapidly. This pool could be detected in endogenous nuclear actin by using fluorescent polymeric actin binding proteins and was sensitive to drugs that alter actin polymerization. Our results validate previous reports of polymeric forms of nuclear actin observed in fixed specimens and reveal that these polymeric forms are very dynamic.
Introduction
Several recent studies have demonstrated that the cytoskeletal protein actin has important functions within the nucleoplasm (Olave et al., 2002; Pederson and Aebi, 2002; Goldman, 2003). The first identified function that unambiguously required a nuclear pool of nonmuscle actin was chromatin remodeling. β-Actin and a series of recently discovered actin-related proteins have been characterized both biochemically and genetically as components of chromatin remodeling complexes (Olave et al., 2002). The role of actin in the nucleus is not limited to chromatin remodeling. β-Actin was recently found to bind specifically to hrp65-2; blocking this interaction in vivo resulted in a dramatic reduction in RNA polymerase II transcription (Percipalle et al., 2003). β-Actin also resides as a component of the Balbiani ring gene messenger RNP (mRNP) throughout its nuclear maturation and export to the cytoplasm (Percipalle et al., 2001). These complexes appear to require β-actin in its monomeric form. More recently, studies have shown that actin is associated with and required for transcription by RNA polymerase I (Fomproix and Percipalle, 2004; Philimonenko et al., 2004), II (Hofmann et al., 2004; Kukalev et al., 2005), and III (Hu et al., 2004).
The cellular concentration of actin is typically at least 100–1,000 times higher than the concentration required for actin to spontaneously polymerize in vitro (Pollard et al., 2000). The concentration of nuclear actin is therefore sufficient to spontaneously polymerize and will require active processes to prevent polymerization. Indeed, several critical regulators of actin polymerization can be found in the nucleoplasm. For example, phosphoinositide signaling occurs within the nucleus and is initiated by a nucleus-specific isoform of phospholipase C (Irvine, 2003). The machinery for phosphoinositide signaling is enriched in splicing factor compartments (Boronenkov et al., 1998; Didichenko and Thelen, 2001; Tabellini et al., 2003). Much of the nuclear monomeric actin pool has recently been reported to be bound in a complex with actin depolymerizing factor/cofilin (Chhabra and dos Remedios, 2005). Other proteins that sequester monomers of actin (Huff et al., 2004), cap filaments (Van Impe et al., 2003; De Corte et al., 2004), or are directly involved in regulating the polymeric state of actin (Vetterkind et al., 2002; Mizutani et al., 2004; Archer et al., 2005) are present in the nuclei. The regulation of nuclear actin is therefore likely to include the controlled polymerization and depolymerization of actin and involve both mechanisms that stimulate polymerization, such as phosphoinositides, and molecules that inhibit actin polymerization or regulate the size of actin polymers.
Specific affinity reagents for globular species of actin and filamentous species of actin have been used to detect nuclear actin and nuclear actin filaments (Nakayasu and Ueda, 1983; Lachapelle and Aldrich, 1988; Kushnaryov et al., 1990; Milankov and De Boni, 1993; Amankwah and De Boni, 1994; Sauman and Berry, 1994; Gonsior et al., 1999; Percipalle et al., 2001; Okorokov et al., 2002; Zhang et al., 2002; Kiseleva et al., 2004). Unfortunately, the conclusions of these studies are complicated by the fact that the different reagents used in these studies lead to different results and different conclusions on localization and polymerization state of the nuclear actin pool. Consequently, current models of nuclear actin function are speculative, with most discussions on the subject proposing that nuclear actin exists and functions as a monomer (Boyer and Peterson, 2000) or as short oligomers (Olave et al., 2002; Goldman, 2003; Percipalle et al., 2003).
In this study, we take the first step toward quantifying the properties of actin within the living nucleus. Using fluorescently tagged actins and actin binding proteins, we measured the steady-state distribution and dynamics of nuclear actin in living HeLa cells. In fluorescence recovery after photobleaching (FRAP) experiments, nuclear actin, like cytoplasmic actin, presented as both rapidly and slowly moving kinetic populations. Based on the loss of this slow moving population when cells are treated with latrunculin and the absence of this population when a mutant actin incapable of polymerization is used in place of wild-type β-actin, we conclude that this slow recovering population is comprised of polymeric (F) actin. Thus, rather than polymerization, the only significant difference between the pools of cytoplasmic and nuclear actin is the bundling of cytoplasmic actin that generates the stress fibers that are prominent in cells stained with fluorescent phalloidin. Under normal physiological conditions, actin bundles are exclusively found in the cytoplasm.
Results
HeLa cells maintain a dynamic actin cytoskeleton in the presence of GFP-actin
The kinetics of actin polymerization and cytoskeletal dynamics can be quantified by microinjection of purified and fluorescently tagged β-actin (Miller et al., 1988; McGrath et al., 1998a,b; Schafer et al., 1998; Albuquerque and Flozak, 2001) or by the expression of a GFP-fusion protein (Doyle and Botstein, 1996; Fischer et al., 1998; Verkhusha et al., 1999; Shelden et al., 2002). Both fluorescent probes have been used successfully to estimate the dynamics of actin and F-actin in cell types ranging from yeast to mammals (Doyle and Botstein, 1996; Westphal et al., 1997; Ballestrem et al., 1998; Verkhusha et al., 1999; Herget-Rosenthal et al., 2001). Stable expression of GFP–β-actin (or spectral variants of GFP fused to actin) has been achieved in a wide variety of cell lines with different motility properties, cytoskeletal organizations, and dynamics of cytoskeleton remodeling (Ballestrem et al., 1998; Choidas et al., 1998; Herget-Rosenthal et al., 2001). Thus, fluorescent actins are well-established probes for investigating actin dynamics in living cells.
Because actin is normally found in the nucleoplasm, it stands to reason that fluorescent actins can also be used to measure the dynamic properties of nuclear actin. We first determined whether GFP-actin could be detected in sufficient quantity to allow kinetic measurements in living cells. Fig. 1 shows a field of HeLa cells that are stably transfected with GFP–β-actin (Fig. 1 A; and Fig. 1, B and C, green). We were unable to simultaneously detect GFP–β-actin with endogenous β-actin by immunoblotting despite using several independent antibody preparations recognizing different epitopes on β-actin. Because the protein was readily detected using an anti-GFP antibody, we believe that this is likely due to the low relative abundance of the EGFP-tagged actin relative to the endogenous actin pool. It was necessary to use a separate approach to estimate the nuclear actin concentration based on the concentration of diffusing EGFP-actin molecules. Using fluorescence correlation spectroscopy (FCS), we measured the diffusing pool of EGFP–β-actin to be present at 0.14 ± 0.11 μM. Measurements of actin concentrations in human cell lines (Pollard et al., 2000) indicate that the endogenous concentration of actin is at least 1,000 times higher than that of EGFP-actin.
Figure 1.

Dynamics of GFP-actin in living HeLa cells. HeLa cells that stably expressed the EGFP–β-actin construct were incubated with 1 μg/ml of Hoechst 33258 to fluorescently stain the nuclear chromatin and then imaged live by fluorescence microscopy. (A) The GFP image obtained at the onset of the time-lapse experiment. (B) A composite image where the DNA image (red) is superimposed on the GFP-actin image (green). The arrows indicate the positions of F-actin–dependent extensions from the cell surface that rapidly remodel throughout the time course (C).
The drug resistance–based selection of cells for stable expression reduced the heterogeneity in the expression level of the transfected proteins; therefore, stable transfectants were used in most experiments. The nuclei (Fig. 1, B and C, red) are visibly depleted in GFP-actin, but actin is nonetheless easily detected within the nucleus. Consistent with previous studies, GFP-actin was incorporated into a dynamically organized cytoskeleton. This was confirmed by counterstaining cells with fluorescently labeled phalloidin (Fig. S1, available at www.jcb.org/cgi/content/full/jcb.200507101/DC1). Fig. 1 C shows a subregion of this field over time. By examining the cell surface, the dynamics of the bundles of filaments at the cell surface are easily illustrated. This time series demonstrates that the GFP-actin is incorporated into the expected F-actin–containing structures and that these GFP-actin–containing structures can both assemble and disassemble filaments that incorporate the GFP-tagged actin hybrid protein.
Distribution of GFP–β-actin between cellular monomeric and polymeric pools
We employed a commonly used and simple extraction procedure to estimate the efficiency of GFP-actin incorporation into actin filaments and compare it to the efficiency of incorporation of the endogenous actin within the same cell population. The results of this experiment are shown in Fig. 2. The top three images show the distribution of GFP-actin among soluble (S) and filamentous (P) pools isolated from HeLa cells, HeLa cells transfected with EGFP, or HeLa cells transfected with EGFP–β-actin. The bottom two images show the distribution of EGFP–β-actin and EGFP in the soluble and cytoskeletal fractions. When the distribution between soluble and cytoskeletal fractions was quantified from these immunoblots, the distribution for the actin and EGFP actin ranged from 52% of the total pool found in the F-actin fraction for EGFP actin to 56% of the total pool found in the F-actin fraction for endogenous actin in HeLa cells. As expected, EGFP was found entirely within the soluble fraction. Stable transfection of EGFP or EGFP–β-actin did not significantly alter the distribution of the endogenous β-actin. Collectively, these results demonstrate that EGFP–β-actin is an effective probe for β-actin dynamics in vivo. This is consistent with the conclusions of previous studies of EGFP–β-actin as a probe for actin dynamics in living cells (Pang et al., 1998; Verkhusha et al., 1999; Hodgson et al., 2000).
Figure 2.

Immunoblotting of cytosolic and cytoskeletal cellular fractions. HeLa cells were lysed in the presence of Triton X-100 to solubilize the cytosol (S). The polymerized actin remains in the cell pellet (P). HeLa cells, HeLa cells stably expressing GFP–β-actin, and HeLa cells stably expressing GFP were analyzed using this procedure. The SDS-soluble proteins were then electrophoresed on an 8% polyacrylamide–SDS gel, transferred, and developed using a fluorescently tagged antibody. Quantitative immunoblots were then collected using an Odyssey system (Li-Cor). The labels on the left indicate the types of cells used for the experiment, and the labels on the right indicate the antigen recognized by the antibody (Ab) used to develop the blot.
Nuclear extracts support actin polymerization
Proteins that bind or sequester actin have been reported to be constituents of the nucleoplasm (Pederson and Aebi, 2005). Hence, it might be anticipated that the nucleoplasm is rich in proteins that regulate actin polymerization. To address this possibility, we generated nuclear extracts from cells treated for 60 min with 20 μM latrunculin B and tested the ability of added purified actin to polymerize in vitro. Under these conditions, nuclei are released more easily and, as judged by brightfield microscopy, appear free of cytoplasmic contaminants (unpublished data). To control for effects solely due to increased protein concentration, purified actin was incubated with an identical amount of BSA. After incubation with either nuclear extract or BSA, polymerized actin was separated from soluble actin by centrifugation. Fig. 3 shows the results of an assay with duplicate reactions for each sample. In the presence of BSA, very little actin polymerizes under the assay conditions. In contrast, when nuclear extract is mixed with purified actin, most of the actin polymerizes and consequently is found in the pellet fraction.
Figure 3.

In vitro polymerization of actin in the presence of nuclear extract. Partially polymerized actin was incubated alone, in the presence of nuclear extract, or in the presence of BSA. After centrifugation, supernatant and pellet fractions were resolved by SDS-PAGE and visualized with Coomassie blue. Each reaction was performed in duplicate, and both samples were loaded onto the gel. S indicates the monomer population, whereas P indicates the polymerized population.
GFP-actin is homogeneously distributed throughout the nucleoplasm
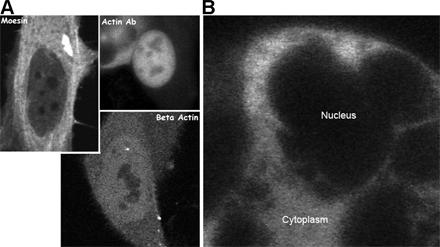
The involvement of the nuclear actin pool in chromatin remodeling or transcription/processing of RNAs may result in the compartmentalization of the nuclear actin population. Consistent with this hypothesis, there are several reports of the specific enrichment of actin within transcription or splicing-associated compartments (Milankov and De Boni, 1993; Sahlas et al., 1993; Nguyen et al., 1998; Gonsior et al., 1999). Because of the relative ease in identifying subnuclear domains within the mouse embryonic fibroblast nucleus, we have used this cell line to illustrate the subnuclear distribution of β-actin (Fig. 4 A). All cell lines examined, including HeLa, showed an identical subnuclear organization. Fig. 4 A illustrates that GFP-actin is homogeneously distributed throughout the nucleoplasm. Line scans were used to quantify actin distribution within the nucleus (Fig. 4 A). The nuclear regions of lowest GFP-actin concentration, the nucleoli, maintain concentrations of actin that are ∼10% of the cytoplasmic concentration (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200507101/DC1). As a control, we demonstrated that an exclusively cytoplasmic fluorescently tagged dextran does not show any evidence of nuclear fluorescence (Fig. S5 B). Because of the small size of the nucleolus, however, it is not possible to rule out the possibility that the source of the nucleolar signal originates in the nucleoplasm.
Figure 4.

The distribution of GFP-actin in the living nucleus. (A) Mouse cells (left) or HeLa cells (right) stably expressing GFP–β-actin were examined by laser-scanning confocal microscopy. The lines drawn across the cells indicate the path of a 3-pixel-wide linescan, and the arrowhead indicates the direction of the linescan. The linescan is shown as pixel number versus intensity as a percentage of the maximum value obtained in the linescan. (B) Rhodamine-conjugated β-actin was microinjected into living HeLa cells. After equilibration of the microinjected fluorescent actin throughout the cell volume, laser-scanning confocal images were collected. (left) An image obtained from the uppermost surface of a group of microinjected HeLa cells. (right) An image obtained near the midplane of most of the cell nuclei. The asterisks show the position of the nucleoli. Bar, 10 μm.
We confirmed that the failure to enrich in specific subnuclear domains was not solely a property of the GFP-tagged version of β-actin. Fig. 4 B shows the distribution of rhodamine actin in living HeLa cells. Like GFP-actin, rhodamine actin is present in significant quantities in the nucleus and is homogeneously distributed outside of the nucleoli (Fig. 4 B; examples of nucleoli are illustrated with asterisks).
FRAP measurements of cytoplasmic and nuclear F-actin pools
The generation of subnuclear patterns of molecular distribution reflects both molecular exclusion events (Gorisch et al., 2003) and binding interactions (Phair and Misteli, 2001). Binding interactions, if they occur with molecules that are much more massive than the free protein, will reduce the mobility of a protein through the local environment. If these binding sites are of sufficient affinity and spatial density, the concentration of the protein within the local environments that contain binding sites will be higher at steady state than the concentration that is unbound and diffusing through the nucleoplasm. When the spatial information obtained from live cell imaging is combined with kinetic information on the underlying molecular flux, it becomes possible to investigate the relationships between organization and function in a quantitative manner. Therefore, we examined the mobility properties of actin within the nucleoplasm of living cells to determine whether kinetic populations of actin could be resolved that may differ functionally.
The molecular flux of actin has been studied in the living cytoplasm. These experiments, which used microinjection of fluorescently labeled β-actin, easily resolved monomeric and polymeric pools of actin (Amato and Taylor, 1986; McGrath et al., 1998a; Star et al., 2002). This is expected when the cytoplasm is measured because the actin becomes incorporated into filaments that are sufficiently large that the rate of filament turnover, rather than diffusion, limits the rate of fluorescence recovery in these studies. We confirmed that the cytoplasmic pool resolved into two distinct populations during recovery from photobleaching of GFP–β-actin in living HeLa cells. When the recovery profiles from many individual cells are pooled and the mean kinetic properties of the cytoplasmic actin pool are compared with the nuclear pool, the recovery profiles are very similar (Fig. 5). The x axis (time) is plotted on a log scale to better illustrate both the fast and slow phases of the recovery profile. Like the cytoplasm, the nucleoplasm clearly resolves into at least two distinct kinetic pools. The principal difference between the recovery kinetics of β-actin in the cytoplasm and the recovery kinetics in the nucleus is a small difference in the distribution between the fast and slow recovering species. This is most evident near the 10-s mark, at which time the rapid recovery phase has reached equilibrium. At this point, 84% of the nucleoplasmic signal has equilibrated, whereas only 76% of the cytoplasmic pool has equilibrated. This difference was statistically significant (P < 0.02).
Figure 5.

Recovery of GFP-actin in the cytoplasmic and nuclear compartments. HeLa cells stably expressing GFP–β-actin were analyzed by FRAP. FRAP data was collected from cells where either the cytoplasm or the nucleoplasm was photobleached. The mean recovery profile is plotted versus recovery time. Time is plotted on a log scale to better illustrate both the rapid and the slow populations of molecules.
FRAP of EGFP nuclear actin under conditions that alter cellular polymeric actin pools
In the cytoplasm, the slow phase of the recovery profile is generated by the relatively slow turnover of fluorescence within the F-actin pool relative to the diffusion rate of the much larger diffusing pool (McGrath et al., 1998a,b).To determine whether the slow recovering phase of nucleoplasmic actin is rate limited by the turnover of actin filaments, we pretreated cells with latrunculin A, which depletes the cellular F-actin pool, and then analyzed the FRAP recovery kinetics. Fig. 6 shows a comparison between the recovery of GFP-actin in control cells and in cells treated for 1 h with latrunculin A. Latrunculin A completely eliminates the slow recovery phase that is otherwise found in the nucleoplasmic actin FRAP profile (Fig. 6 A; Table I). A second drug, swinholide, which can fragment existing polymeric actin, also resulted in the loss of the slow recovering fraction of nuclear GFP-actin (unpublished data). In contrast, a third drug, jasplakinolide, stabilizes actin polymers, and treatment with this drug resulted in a time-dependent decrease in the recovering fraction of nuclear actin (Fig. 6 B).
Figure 6.

Recovery of nuclear actin in the presence and absence of an inhibitor of actin polymerization. The recovery kinetics of GFP-actin was quantified in stably transfected HeLa cells, HeLa cells that were treated with latrunculin A, and HeLa cells that were treated with EDTA. The relative intensity is plotted versus time, with time plotted on a log scale.
Table I.
FRAP recovery of fluorescent actins and fluorescent actin binding proteins
| Protein | Latrunculin | Percentage recovery | Standard deviation | P-value |
|---|---|---|---|---|
| Moesin-GFP | − | 91.3 | 4.7 | |
| + | 97.2 | 3.5 | ≤0.001 | |
| Anti-actin antibody | − | 78.5 | 4.8 | |
| + | 85.9 | 4.9 | ≤0.001 | |
| Anti–rabbit IgG | − | 94.2 | 1.8 | |
| + | 93.5 | 1.3 | NS | |
| GFP-actin (transient) | − | 84.7 | 4.5 | |
| + | 97.8 | 2.5 | ≤0.001 | |
| R62D actin–GFP | − | 99.3 | 1.4 | |
| GFP-actin (stable) | − | 84.0 | 9.4 | |
| FITC actin | − | 90.3 | 4.9 |
Percentage of recovery was measured at 11.5 s after photobleaching for each protein examined. In some instances, cells were preincubated for 60 min with latrunculin A. The P-value was obtained using a t test.
Latrunculin, by destabilizing stress fibers, causes the treated cells to detach from the coverslip and adopt a more spherical morphology. To rule out the possibility that changes in cell shape either directly or indirectly alter the mobility of nucleoplasmic actin independent of any direct effects of nuclear actin, we also examined the FRAP recovery profile of nuclear actin in cells that have been pretreated with EDTA. EDTA treatment caused the cells to adopt a rounded shape similar to that of the latrunculin-treated cells. However, unlike the latrunculin-treated cells, the recovery profile of nuclear actin in cells detached by EDTA treatment is indistinguishable from that of the adherent control cells (Fig. 6 A).
We devised several approaches to confirm the presence of a nuclear pool of polymerized actin. Fluorescently labeled phalloidin was initially used but was found to be unsuitable because the binding was reversible when it was studied by FRAP after fixation with paraformaldehyde. Table I shows the different probes used and the proportion of fluorescence signal that has recovered by 11.5 s into the recovery curve. This time point represents a period sufficient to allow recovery of freely diffusing molecules but insufficient time for significant amounts of the slow recovering molecules to equilibrate. As such, where fluorescent actins are used, this point represents a good approximation of the size of the pool of unpolymerized actin. The probes were chosen to either incorporate into (FITC- and EGFP-actin) or bind to (FITC–anti-actin antibody and moesin-GFP; Edwards et al., 1997) F-actin. The subcellular distribution of these probes is illustrated in Fig. S5 A. In each case, a slow recovering population of nuclear actin was detected and, where tested, was lost or reduced upon treatment with latrunculin. The diffusion of the fluorescent anti–rabbit IgG antibody serves as a useful control for latrunculin-dependent changes that may indirectly alter diffusion in the nucleoplasm. There was no significant difference in the diffusion of microinjected anti–rabbit IgG antibody in the presence or absence of latrunculin treatment (Table I).
GFP-actin must be polymerization competent to incorporate into the slowly recovering nuclear fraction
To directly test the requirement for polymerization in defining the low-mobility pool of nuclear actin, we made use of a previously characterized R62D mutant of actin (Posern et al., 2002). This mutant actin does not incorporate into actin filaments. We prepared an EGFP fusion of the R62D mutant of actin and transfected cells with either the R62D variant or wild-type fusion proteins. This EGFP fusion protein was not evident in any of the more prominent components of the actin cytoskeleton (stress fibers and cell cortex). Because the expression of this protein altered cellular morphology, we chose to examine the kinetics of the mutant protein in transiently transfected cells. Fig. 7 shows time-lapse images of transiently transfected GFP–β-actin (top) and GFP–R62D–β-actin (bottom) collected during the first few seconds of the recovery phase. The differences between the two proteins are most evident at these earliest stages of the recovery period. The first image of each time lapse shows the cell before photobleaching a 2-μm-wide line across the cell nucleus. The differences in the mobility of the two proteins are obvious in the first image collected after photobleaching. In the wild-type GFP-actin images, the photobleached region is obvious, whereas in the R62D mutant GFP-actin, inhomogeneity is evident but there is no clearly defined photobleached region. The absence of a clear photobleached region reflects the rapid mobility of the protein being examined. The graph in Fig. 6 shows the mean recovery profiles of HeLa cells transiently transfected with GFP-actin or R62D mutant GFP-actin. For comparison, the recovery profile of GFP-actin obtained from cells treated with latrunculin is also plotted.
Figure 7.

Recovery of an actin mutant unable to incorporate into filaments. Photobleaching experiment performed using cells transiently transfected with wild type GFP-actin (WT) or actin containing a point mutation at amino acid 62 (R62D). From left to right, the images represent prebleach, 1.5 s postbleach, 5 s postbleach, and 10 s postbleach. Dark colors represent lower concentrations of GFP, whereas bright colors represent higher concentrations, with white being the most concentrated. The bottom panel shows the FRAP recovery profiles of GFP-actin, GFP-actin + latrunculin, and the R62D actin mutant.
Dynamic properties of nuclear polymeric actin
We analyzed the data using mathematical models to simulate two possible mechanisms for explaining the recovery profile of nuclear β-actin. The development of the mathematical simulations of nuclear GFP-actin and the fitting of these models to the experimental data has been published separately (Carrero et al., 2004a,b). In simple terms, we assumed that the recovery curve reflected a mixture of rapidly diffusing complexes containing one or a few actin molecules and much slower moving polymeric actin species. In this model, both species recover through diffusion of new fluorescently labeled macromolecules or polymers into the region that has undergone photobleaching. The second model assumes that although polymeric actin may diffuse, its rate of diffusion is slower than the rate of remodeling of the polymer with new fluorescent monomers through treadmilling of actin polymers. In this case, the first phase of the recovery curve reflects diffusion of actin monomers and complexes, whereas the second phase of the recovery curve represents the turnover time of monomers within larger polymeric actin structures. Both mathematical simulations produced a very good approximation of the experimental data (Carrero et al., 2004a), demonstrating that either a turnover process or a diffusion-dependent process can explain the observed recovery rates. The effective diffusion coefficients for the fast phase and the slow phase were estimated at 0.47 μm2 s−1 and 0.009 μm2 s−1, respectively. The measured rate of diffusion obtained by FCS, ∼30 μm2 s−1 is more consistent with the expected diffusion rate of a protein of this size. In vitro, the measured rate of actin monomer diffusion is ∼70 μm2 s−1 (Lanni and Ware, 1984). Thus, even this fast phase migrates at least 50 times slower than expected for a molecule of monomeric size diffusing through the nucleoplasm.
If we simulate the recovery of the slow phase as a reflection of the underlying kinetics of actin polymerization and depolymerization, the data can be fit by a population consisting of 84% of the nuclear β-actin molecules residing in the G-actin pool, whereas the remaining 16% are incorporated into actin filaments for a mean duration of 76 s (Carrero et al., 2004a). Polymeric actin found within the cytoplasm is known to be dynamic and show different turnover rates in different regions of the cell. For example, actin cables, which represent bundles of actin microfilaments, require several minutes to turn over their constituent actin monomers and represent relatively stable polymeric actin structures (Kreis et al., 1982; Wang, 1987). In contrast, the polymeric actin assembled at the leading edge of the cell turnover with half-lives of <30 s (Amato and Taylor, 1986; Star et al., 2002). The rate of actin turnover in the nucleus is relatively high but similar to what is observed for polymeric actin outside of actin cables (Amato and Taylor, 1986; McGrath et al., 1998b; Theriot and Mitchison, 1991; Sund and Axelrod, 2000).
Identification of multiple diffusing species of GFP-actin in the nucleus of living cells
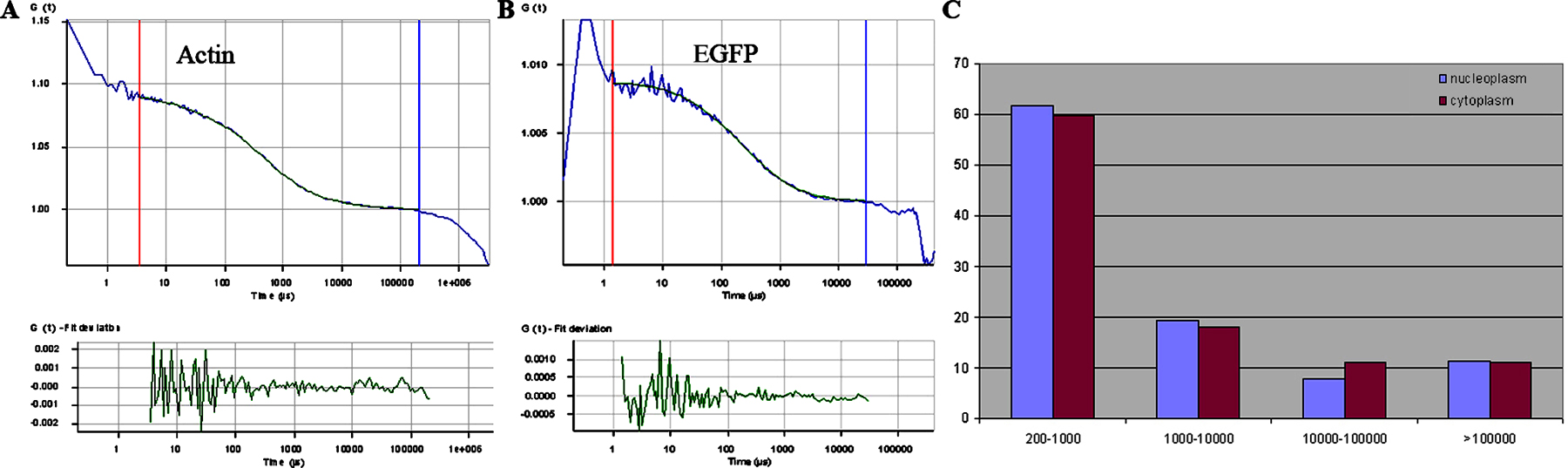
To further assess the properties of the diffusing forms of GFP-actin within the nucleoplasm, we used FCS. FCS is a technique that measures stochastic fluctuations in the fluorescence emission that corresponds to molecules moving into and out of the focal point of the objective lens. Because the focal plane has a defined volume, this information can be directly used to quantify the diffusion rate of the fluorescent species through the focal volume. In principal, current mathematical modeling of FCS data enables no more than three diffusing components to be estimated. When the nucleoplasm and the cytoplasm were simultaneously measured by FCS, the diffusing species resolve into roughly four separate effective diffusion rates (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200507101/DC1). Approximately 70% of both the nuclear and the cytoplasmic actin pools diffuse at a rate of ∼30 μm2 s−1, consistent with the diffusion of monomeric actin or small complexes containing actin. The remainder of the detected GFP-actin–containing molecules diffuse at rates that are between 10 and 500 times slower than the monomeric pool.
The relationship between actin polymerization and RNA polymerase II transcription
There is now evidence that actin is a required component of the RNA polymerase holoenzymes (Hofmann et al., 2004; Hu et al., 2004; Philimonenko et al., 2004). Based on this association, it is possible that the regulation of actin polymerization and nuclear transcription are interdependent. To address this possibility, we first determined whether inhibiting transcription altered the kinetic properties of the nuclear actin pools. Inhibition of RNA polymerase I transcription using actinomycin D or RNA polymerase II using 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) yielded similar results. Fig. 8 shows the FRAP recovery curves of nuclear actin after 4 h of pretreatment with the RNA polymerase II transcriptional inhibitor DRB relative to the FRAP recovery curves of nuclear actin under normal growth conditions or when cells are pretreated with latrunculin B. These results demonstrate that there is a similar equilibrium between polymeric and monomeric actin, regardless of whether transcription is occurring. We next examined whether depolymerization of cellular polymeric actin altered transcription rates. In this experiment, a significant effect was seen. Mouse 10T1/2 cells were the most sensitive of the cell lines tested. Upon treatment with latrunculin, the incorporation of [3H]uridine into nuclear RNA was inhibited by >90%. This was also evident in experiments incorporating 5-fluorouridine into nascent RNA, but analysis by indirect immunofluorescence is complicated by rounding and detachment of cells (unpublished data). A B cell line (Raji) was also examined for 5-fluorouridine incorporation in nascent RNA. These cells grow in suspension and do not contain the extensive cytoskeleton present in adherent cell lines. When Raji cells were analyzed by flow cytometry for the incorporation of 5-fluorouridine into nascent RNA, although less sensitive than the mouse embryonic fibroblasts, we also observed significant inhibition after treatment with latrunculin (Fig. 9).
Figure 8.

FRAP recovery curve of EGFP–β-actin in the presence of transcriptional inhibitors. The recovery kinetics of GFP-actin was quantified in stably transfected HeLa cells and HeLa cells that were treated with the RNA polymerase II transcription inhibitor, DRB, for 4 h before performing the FRAP experiment. The results obtained with latrunculin B are shown for comparison.
Figure 9.

Transcriptional activity in the presence of latrunculin. (A) The immunofluorescence images show 5-fluorouridine incorporation (left and right, green) in mouse 10T1/2 cells without (top) or with (bottom) pretreatment for 60 min with latrunculin. The corresponding color composite images show 5-fluorouridine (green), DAPI (red), and Alexa 546 phalloidin (blue). (B) Flow cytometry profiles of cells treated with DRB and actinomycin D (red line), 0.2 mM latrunculin B (blue line), or 0.05 mM latrunculin B (green line) or left untreated (black line).
The inhibition of transcription by the actin binding drug latrunculin could be due to the drug interfering with the association of actin monomers with the RNA polymerase complexes. To determine whether this was the mechanism responsible for the inhibition of transcription that we observed, we performed coimmunoprecipitation experiments. We observed that an antibody directed against β-actin coimmunoprecipitates RNA polymerase II independent of whether the cell extracts were obtained from latrunculin-treated or control cells (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200507101/DC1).
Discussion
In this study, we sought to define the kinetic properties of the nuclear actin pool in living cells. Our results demonstrate that nuclear actin exists in several different kinetic populations, including a significant pool of polymeric actin. The effective diffusion coefficient of the rapidly recovering pool of nuclear actin was ∼0.5 μm2 s−1. This mobility is ∼50–100 times slower than expected of actin monomers or multimolecular complexes incorporating actin and could contain some oligomeric complexes of actin. When translated into mass equivalents, this represents up to a 1,000,000-fold increase in mass that is required to explain the reduced diffusion of the rapidly recovering population of actin molecules. It is more reasonable to explain differences of this magnitude based on binding interactions (Phair and Misteli, 2001; Carrero et al., 2004b; Phair et al., 2004). In this respect, it is notable that the rate of effective diffusion is similar to what is observed for many chromatin binding proteins, including those that modify chromatin structure (Phair et al., 2004). Thus, the diffusion of monomers or oligomers that are incorporated into protein complexes as well as the reversible binding interactions that these complexes undergo in the nucleoplasm are likely to all contribute to the fast phase of actin recovery observed during FRAP.
The more slowly recovering population of nuclear actin was directly correlated with the ability of actin to polymerize and was also detected among endogenous actin pools when photobleaching experiments were performed on fluorescent proteins that specifically bind actin in the F-actin state. We found that the equilibrium between the monomer and polymer state differs only slightly between the nucleus and the cytoplasm. The nuclear pool of polymeric actin turns over slightly faster than what is observed in the cytoplasm. In addition, the nucleoplasm contained a slightly lower proportion of polymeric actin than the cytoplasm.
Based on the geometry of the interchromatin space, Pederson and Aebi (2002) have suggested that it is unlikely that long filaments of actin exist in the nucleoplasm. Others have speculated that oligomeric actin may associate with the nuclear lamina and/or RNA polymerase II transcription sites (Goldman, 2003; Fomproix and Percipalle, 2004). The slow recovering phase containing the polymeric fraction of nuclear actin could be explained by more than one potential mechanism. First, oligomeric actin or relatively small actin polymers could be complexed with larger structures within the nucleoplasm. In this case, polymers could be relatively small and yet essentially immobile within the nucleoplasm. Alternatively, the mass of the polymer, either as a filament or as a network of filaments, may be too large to diffuse through the nucleoplasm whether or not it binds to other nuclear structures. Given that the concentration of cellular actin is hundreds to thousands of times greater than the critical concentration for spontaneous polymerization of actin, even if there were a 10-fold reduction in the concentration of actin within the nucleoplasm, it would still far exceed what could be maintained in a monomeric form without regulation.
Although the FRAP recovery curve of nuclear actin revealed that ∼20% of the nuclear actin pool recovered with the kinetics of a polymeric actin pool, we wanted to verify that this fraction contained polymeric actin. The conclusion that this pool is predominantly or entirely polymeric actin is supported by the following observations: (a) the detection of the slow migrating population using fluorescently tagged actin, regardless of the fluorescent tag used or the method of introduction into the cell; (b) the detection of this same pool of slow migrating actin when fluorescent probes that bind specifically to endogenous actin or F-actin are used; (c) the loss of this population, independent of whether actin is fluorescently tagged or a fluorescent tag is present on an actin binding protein, when cells are incubated with latrunculin; (d) the failure to detect this slow phase of nuclear actin when an actin mutant that does not incorporate into filaments is measured using FRAP; and (e) the stabilization of this fraction in cells treated with jasplakinolide.
One of the principal mechanisms for initiating actin polymerization in the cytoplasm is through phosphoinositide release and the generation of phosphatidylinositol-4,5-bisphosphate; thus, the cell membrane is where much of the cytoplasmic actin polymerization is initiated (Yin and Janmey, 2003). Although the nuclear membrane may similarly function as a site where actin polymerization is initiated (Shumaker et al., 2003; Gruenbaum et al., 2005; Wilson et al., 2005), the nuclear phosphoinositide signaling machinery and nuclear phosphoinositides colocalize with splicing factor compartments (Boronenkov et al., 1998; Didichenko and Thelen, 2001) and thereby provide a potential site to nucleate actin polymerization distant from the nuclear membrane. Because these sites are centrally located within the euchromatic domains of the nucleus, these sites are well positioned to regulate the polymerization of actin throughout the nucleoplasm. With the exception of depletion in the nucleolus, we did not find evidence of significant inhomogeneities in the presence of either diffusing or polymeric and immobilized nuclear actin analogous to what are observed for proteins that enrich in splicing factor compartments or other nuclear bodies. Although this result is consistent with an essentially homogeneous distribution of actin and polymeric actin outside of the nucleolus, the large size of the monomer pool may require large differences in nucleoplasmic concentrations of polymeric actin to be evident using this approach.
When actin was depolymerized with latrunculin, we observed a significant, although not complete, inhibition of transcription. β-Actin has recently been found to directly associate with both RNA polymerase I and II holoenzymes and is required for transcription in vitro (Percipalle et al., 2003; Fomproix and Percipalle, 2004; Philimonenko et al., 2004; Kukalev et al., 2005). Although our result is consistent with a function of F-actin in the transcription process, until better tools are developed for specifically targeting nuclear polymeric actin without altering the cytoplasmic fraction, the experiment cannot be considered conclusive.
In summary, our results demonstrate that actin polymerization occurs within the living nucleoplasm and highlight the necessity to consider monomeric, oligomeric, or polymeric forms of actin as studies continue to reveal new functions and nuclear complexes that contain actin. To make significant progress in discriminating between these possibilities, it is essential that methods be developed that selectively target the state of polymerization within the nucleus without directly altering polymerization within the cytoplasm.
Materials and methods
Generation of cells stably expressing GFP-actin
Human β-actin containing an NH2-terminal EGFP was purchased from CLONTECH Laboratories, Inc. Indian muntjac fibroblast, mouse 10T1/2, HeLa, and SK-N-SH human neuroblastoma cells were each transfected with the GFP-actin construct, and stable expression of the plasmid was selected for using G418. In some instances, drug-resistant cultures were further selected by fluorescence-activated cell sorting for GFP expression. R62D actin was provided by R. Treisman (Cancer Research UK, London, UK), and moesin-GFP was provided by D. Kiehart (Duke University, Durham, NC).
FRAP
Our protocol for FRAP has been described in detail previously (Lever et al., 2000). In brief, living cells were photobleached in 2-μm-diameter circles or 1.5-μm strips (for mathematical modeling) across the width of the nucleus using a laser-scanning confocal microscope (LSM 510; Carl Zeiss MicroImaging, Inc.) and a 488-nm laser line at 100% intensity for 20 iterations. Photobleaching was completed in <250 ms. The recovery of the fluorescence signal over time was then monitored and used to measure the mobility of the actin. Because the stress fibers are the brightest GFP-actin–containing structures within the cells, it was necessary to close the pinhole aperture on the LSM 510 to the extent that light was collected from sections no thicker than 2 μm. Quantification of data was not performed on mouse 10T1/2 or Indian muntjac fibroblasts because the flat profile of these cells made it difficult to exclude the possibility that some of the fluorescence was contributed by stress filaments located above or below the nucleus. It was also necessary, in some instances, to perform image summing or frame averaging to improve the signal-to-noise ratio and maximize the dynamic range during data collection. Finally, the number of scans during collection was optimized by increasing the time interval between scans from <1 to 5 s after the first 5–10 s of recovery. Recovery curves were generated from between 20 and 30 individual cells recorded from at least three separate experiments. P-values were determined using the t test.
For quantification, each image was normalized for total fluorescence intensity relative to the first image collected after photobleaching to correct for any photobleaching that occurred during the collection of the postbleach time series. This was accomplished by measuring the total cellular fluorescence at each time point. Mathematical modeling of mobility using the compartment model (Carrero et al., 2004a,b) used a 2-μm-wide line photobleached across the width of the cell nucleus and extended the duration of the experiment to ∼2 min.
Fluorescence microscopy
Images were collected using MetaMorph (Universal Imaging Corp.) to control an Axiovert 200 M (Carl Zeiss MicroImaging, Inc.) equipped and acquired with a 12-bit charge-coupled device camera (Sensicam; Cooke Corp.) or a 14-bit charge-coupled device camera (Cascade; Photometrics). In some cases, confocal sections were acquired using a laser-scanning confocal microscope and a pinhole aperture setting of 1 Airy unit. GFP was excited using a Xenon lamp or a 488-nm laser line. Rhodamine was excited using a 514-nm laser line. The spatial sampling ranged from 0.07 to 0.15 μm per pixel in the xy plane and 0.2 to 0.4 μm in the z plane. For images acquired on fields of cells, a 1.3 NA Plan Fluor 40× objective (Carl Zeiss MicroImaging, Inc.) was used. For higher magnification images, a 1.4 NA Plan Apo 63× objective (Carl Zeiss MicroImaging, Inc.) was used. Time-lapse experiments involving living cells were typically acquired at 23°C in standard DME with added fetal calf serum.
Image processing and figure construction
Images from the LSM 510 were imported into MetaMorph software if it was necessary to apply a 3 × 3 median filter to reduce pixel noise in the figure images. Otherwise, images were directly exported as 16-bit TIFF files and rescaled over an 8-bit data range. In most cases, the background fluorescence of the medium and the base signal from the detector are minimized to better represent the dynamic range of the data content in the image. In some instances, three-dimensional image sets were imported into Imaris 4.12 (BitPlane) and three-dimensional image sets were generated. In this instance, the image was scaled to map the data over the range of the display and the screen capture function in Imaris 4.12 was used to capture the image used in the figure. Figures were prepared in Photoshop CS (Adobe) for Windows. In general, images were scaled to span the 8-bit data depth, reducing background in the process, and then pasted into a composite canvas that was either 8-bit grayscale or 24-bit RGB color. If necessary, images were interpolated to 300 dpi using Photoshop.
Drug treatments and cell staining
Cells growing in culture, either in culture flasks or plated onto glass coverslips, were treated with latrunculin A or B (Calbiochem) at the concentrations indicated. No difference was observed between cells treated with latrunculin A or B, with the exception that treatment with latrunculin B required a 10-fold increase in concentration. This was expected based on the known properties of the two latrunculins. Jasplakinolide was used at a final concentration of 1 μM. Alexa 546–conjugated phalloidin was used as recommended by the manufacturer (Invitrogen). Fluorouridine was used to determine the synthesis of RNA as described previously (Bleoo et al., 2001; Boisvert et al., 2001). For these studies, incubations of between 10 and 20 min were used for RNA labeling, depending on the rate of incorporation into the cells (which varies between cell types). For flow cytometry analysis, cells were fixed with 100% methanol. Unless otherwise indicated, all other fixations were performed with 4% paraformaldehyde.
Microinjection of actins and antibodies into HeLa cells
HeLa cells were plated on glass coverslips embedded in 35-mm dishes. Microcapillaries were drawn from 1.0-mm outer diameter aluminosilicate filaments on a flaming/brown micropipette puller (model P-87; Sutter Instrument Co.). They were loaded from the rear with fluorescent actin at 0.5 μg/ml in 2 mM Tris HCl, pH 8, 0.1 mM ATP, 0.1 mM DTT, and 0.1 mM CaCl2. Injection was attempted in the nucleus, but occasionally the actin appeared to go in the cytoplasm. However, when looking at the injected cells under fluorescent light, the two were indistinguishable. The cells were then incubated in fresh media for 1–2 h before FRAP experiments.
FCS
FCS data was collected with an LSM 510 NLO using a 488-nm excitation source and collected with a 40× C-Apochromat 1.2 NA water immersion objective (Carl Zeiss MicroImaging, Inc.) and a ConfoCor 2 detector (Carl Zeiss MicroImaging, Inc.). Autocorrelation analysis was performed using the software provided with the ConfoCor 2 module. Curve fitting was performed by comparing the fit (r2 value) for single, two, and three parameter fits. The data was summarized and tabulated as described previously (Politz et al., 1998).
Actin polymerization assays
The distribution of actin in soluble and insoluble fractions was determined as described previously (Posern et al., 2002). Nuclear extracts were prepared as described previously (Andrin and Hendzel, 2004). An Ultrafree-MC centrifugal filter (10-kD cutoff; Millipore) was used to concentrate the nuclear extract approximately fivefold and perform a buffer exchange back to buffer A. The concentrated extract was precleared by centrifuging at 125,000 g for 1 h at 22°C.
Lyophilized nonmuscle actin was resuspended in 5 mM Tris, pH 8, 0.2 mM CaCl2, 0.2 mM MgCl2, and 0.2 mM ATP at 2 mg/ml and incubated on ice for 30 min. Nonmuscle actin was partially polymerized on ice for 60 min by diluting the actin to 1 mg/ml and adding 1 mM ATP, 2 mM MgCl2, and 50 mM KCl (all final concentrations). This solution was then used directly in the spin assay.
Nuclear extract from ∼8 × 105 − 1.0 × 106 cells was gently mixed with 40 μg F-actin and allowed to incubate at room temperature for 30 min. As controls, buffer A alone or 8 μg BSA solubilized in buffer A was incubated with 40 μg F-actin under the same conditions. The samples were then centrifuged at 125,000 g for 90 min at 22°C. The supernatant was removed, and an appropriate amount of 3× concentrated SDS sample buffer was added. The pellets were solubilized in 3× concentrated SDS sample buffer and heated at 60°C for 15 min. Approximately one third of the supernatant and pellet from each sample condition was then separated by 10% SDS-PAGE and visualized by Coomassie blue staining.
Coimmunoprecipitation experiments
HeLa S3 cells were either treated with 20 μM latrunculin B for 60 min before extraction or left untreated and used for control extracts. Whole cell extracts were prepared using modified RIPA buffer (50 mM Tris, pH 7.4, 1% NP-40, 150 mM NaCl, and 1.0 mM EDTA) for 60 min on ice. Insoluble material was removed by centrifugation at 21,000 g for 10 min. The supernatant was used as the extract for the immunoprecipitation experiments.
Immunoprecipitations were performed as described by Hoffman et al. (2004) with minor alterations. In brief, 10 μg anti–β-actin (AC-15) antibody were added to precleared, untreated, or latrunculin B–treated whole cell extracts (300 μg diluted 10-fold in 10 mM Tris-HCl, pH 7.9, 20 mM Hepes, pH 7.9, 8% glycerol, 45 mM KCl, 8 mM MgCl2, 5 mM (NH4)2SO4, 2% polyethylene glycol, 4.5 mM β-mercaptoethanol, 0.05 mM EDTA, and 0.025% sodium lauryl sarcrosine) and incubated for 2 h at 4°C. Protein G–Sepharose (150 μl of a 50% solution; GE Healthcare) was added, and the mixture was incubated for an additional 2 h at 4°C. The beads were washed five times with five volumes of IP dilution buffer. Bound proteins were recovered by boiling the washed beads in SDS sample buffer. The eluted proteins were separated by 10% SDS-PAGE and analyzed by protein immunoblotting using antibodies to RNA polymerase II or β-actin.
Mathematical modeling of FRAP recovery curves
For mathematical modeling of FRAP recovery curves, see Carrero et al. (2004a,b).
Online supplemental material
Fig. S1 shows costaining of cells expressing GFP-tagged β-actin with phalloidin. Fig. S2 shows an FCS analysis of GFP-tagged β-actin. Fig. S3 shows that RNA polymerase II coimmunoprecipitates with β-actin in the presence and absence of latrunculin. Fig. S4 shows a series of laser-scanning confocal optical sections through cells expressing EGFP–β-actin. Fig. S5 shows fluorescence microscopy of indirect actin probes and fluorescent dextrans. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200507101/DC1.
Supplementary Material
Acknowledgments
We thank Ellen Crawford and Carmen Tomkinson for excellent technical assistance. We thank Dr. Richard Treisman and Dr. Daniel Kiehart for gifts of R62D and moesin-GFP, respectively. We thank Dr. X. Sun for assistance in the application of FCS.
This work was supported by research grants awarded by the Alberta Cancer Foundation (to M.J. Hendzel) and the Natural Sciences and Engineering Research Council (to G. de Vries). M.J. Hendzel is a scholar of the Alberta Heritage Foundation for Medical Research and a Canadian Institutes of Health Research new investigator.
Abbreviations used in this paper: DRB, 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole; FCS, fluorescence correlation spectroscopy; FRAP, fluorescence recovery after photobleaching; mRNP, messenger RNP.
References
- Albuquerque, M.L., and A.S. Flozak. 2001. Patterns of living beta-actin movement in wounded human coronary artery endothelial cells exposed to shear stress. Exp. Cell Res. 270:223–234. [DOI] [PubMed] [Google Scholar]
- Amankwah, K.S., and U. De Boni. 1994. Ultrastructural localization of filamentous actin within neuronal interphase nuclei in situ. Exp. Cell Res. 210:315–325. [DOI] [PubMed] [Google Scholar]
- Amato, P.A., and D.L. Taylor. 1986. Probing the mechanism of incorporation of fluorescently labeled actin into stress fibers. J. Cell Biol. 102:1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrin, C., and M.J. Hendzel. 2004. F-actin-dependent insolubility of chromatin-modifying components. J. Biol. Chem. 279:25017–25023. [DOI] [PubMed] [Google Scholar]
- Archer, S.K., C. Claudianos, and H.D. Campbell. 2005. Evolution of the gelsolin family of actin-binding proteins as novel transcriptional coactivators. Bioessays. 27:388–396. [DOI] [PubMed] [Google Scholar]
- Ballestrem, C., B. Wehrle-Haller, and B.A. Imhof. 1998. Actin dynamics in living mammalian cells. J. Cell Sci. 111:1649–1658. [DOI] [PubMed] [Google Scholar]
- Bleoo, S., X. Sun, M.J. Hendzel, J.M. Rowe, M. Packer, and R. Godbout. 2001. Association of human DEAD box protein DDX1 with a cleavage stimulation factor involved in 3′-end processing of pre-mRNA. Mol. Biol. Cell. 12:3046–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert, F.M., M.J. Kruhlak, A.K. Box, M.J. Hendzel, and D.P. Bazett-Jones. 2001. The transcription coactivator CBP is a dynamic component of the promyelocytic leukemia nuclear body. J. Cell Biol. 152:1099–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boronenkov, I.V., J.C. Loijens, M. Umeda, and R.A. Anderson. 1998. Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing pre-mRNA processing factors. Mol. Biol. Cell. 9:3547–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer, L.A., and C.L. Peterson. 2000. Actin-related proteins (Arps): conformational switches for chromatin-remodeling machines? Bioessays. 22:666–672. [DOI] [PubMed] [Google Scholar]
- Carrero, G., E. Crawford, M.J. Hendzel, and G. de Vries. 2004. a. Characterizing fluorescence recovery curves for nuclear proteins undergoing binding events. Bull. Math. Biol. 66:1515–1545. [DOI] [PubMed] [Google Scholar]
- Carrero, G., E. Crawford, J. Th'ng, G. de Vries, and M.J. Hendzel. 2004. b. Quantification of protein-protein and protein-DNA interactions in vivo, using fluorescence recovery after photobleaching. Methods Enzymol. 375:415–442. [DOI] [PubMed] [Google Scholar]
- Chhabra, D., and C.G. dos Remedios. 2005. Cofilin, actin and their complex observed in vivo using fluorescence resonance energy transfer. Biophys. J. 89:1902–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choidas, A., A. Jungbluth, A. Sechi, J. Murphy, A. Ullrich, and G. Marriott. 1998. The suitability and application of a GFP-actin fusion protein for long-term imaging of the organization and dynamics of the cytoskeleton in mammalian cells. Eur. J. Cell Biol. 77:81–90. [DOI] [PubMed] [Google Scholar]
- De Corte, V., K. Van Impe, E. Bruyneel, C. Boucherie, M. Mareel, J. Vandekerckhove, and J. Gettemans. 2004. Increased importin-β-dependent nuclear import of the actin modulating protein CapG promotes cell invasion. J. Cell Sci. 117:5283–5292. [DOI] [PubMed] [Google Scholar]
- Didichenko, S.A., and M. Thelen. 2001. Phosphatidylinositol 3-kinase c2alpha contains a nuclear localization sequence and associates with nuclear speckles. J. Biol. Chem. 276:48135–48142. [DOI] [PubMed] [Google Scholar]
- Doyle, T., and D. Botstein. 1996. Movement of yeast cortical actin cytoskeleton visualized in vivo. Proc. Natl. Acad. Sci. USA. 93:3886–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, K.A., M. Demsky, R.A. Montague, N. Weymouth, and D.P. Kiehart. 1997. GFP-moesin illuminates actin cytoskeleton dynamics in living tissue and demonstrates cell shape changes during morphogenesis in Drosophila. Dev. Biol. 191:103–117. [DOI] [PubMed] [Google Scholar]
- Fischer, M., S. Kaech, D. Knutti, and A. Matus. 1998. Rapid actin-based plasticity in dendritic spines. Neuron. 20:847–854. [DOI] [PubMed] [Google Scholar]
- Fomproix, N., and P. Percipalle. 2004. An actin-myosin complex on actively transcribing genes. Exp. Cell Res. 294:140–148. [DOI] [PubMed] [Google Scholar]
- Goldman, M.A. 2003. The epigenetics of the cell. Genome Biol. 4:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsior, S.M., S. Platz, S. Buchmeier, U. Scheer, B.M. Jockusch, and H. Hinssen. 1999. Conformational difference between nuclear and cytoplasmic actin as detected by a monoclonal antibody. J. Cell Sci. 112:797–809. [DOI] [PubMed] [Google Scholar]
- Gorisch, S.M., K. Richter, M.O. Scheuermann, H. Herrmann, and P. Lichter. 2003. Diffusion-limited compartmentalization of mammalian cell nuclei assessed by microinjected macromolecules. Exp. Cell Res. 289:282–294. [DOI] [PubMed] [Google Scholar]
- Gruenbaum, Y., A. Margalit, R.D. Goldman, D.K. Shumaker, and K.L. Wilson. 2005. The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 6:21–31. [DOI] [PubMed] [Google Scholar]
- Herget-Rosenthal, S., M. Hosford, A. Kribben, S.J. Atkinson, R.M. Sandoval, and B.A. Molitoris. 2001. Characteristics of EYFP-actin and visualization of actin dynamics during ATP depletion and repletion. Am. J. Physiol. Cell Physiol. 281:C1858–C1870. [DOI] [PubMed] [Google Scholar]
- Hodgson, L., W. Qiu, C. Dong, and A.J. Henderson. 2000. Use of green fluorescent protein-conjugated beta-actin as a novel molecular marker for in vitro tumor cell chemotaxis assay. Biotechnol. Prog. 16:1106–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann, W.A., L. Stojiljkovic, B. Fuchsova, G.M. Vargas, E. Mavrommatis, V. Philimonenko, K. Kysela, J.A. Goodrich, J.L. Lessard, T.J. Hope, et al. 2004. Actin is part of pre-initiation complexes and is necessary for transcription by RNA polymerase II. Nat. Cell Biol. 6:1094–1101. [DOI] [PubMed] [Google Scholar]
- Hu, P., S. Wu, and N. Hernandez. 2004. A role for beta-actin in RNA polymerase III transcription. Genes Dev. 18:3010–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff, T., O. Rosorius, A.M. Otto, C.S. Muller, E. Ballweber, E. Hannappel, and H.G. Mannherz. 2004. Nuclear localisation of the G-actin sequestering peptide thymosin beta4. J. Cell Sci. 117:5333–5341. [DOI] [PubMed] [Google Scholar]
- Irvine, R.F. 2003. Nuclear lipid signalling. Nat. Rev. Mol. Cell Biol. 4:349–360. [DOI] [PubMed] [Google Scholar]
- Kiseleva, E., S.P. Drummond, M.W. Goldberg, S.A. Rutherford, T.D. Allen, and K.L. Wilson. 2004. Actin- and protein-4.1-containing filaments link nuclear pore complexes to subnuclear organelles in Xenopus oocyte nuclei. J. Cell Sci. 117:2481–2490. [DOI] [PubMed] [Google Scholar]
- Kreis, T.E., B. Geiger, and J. Schlessinger. 1982. Mobility of microinjected rhodamine actin within living chicken gizzard cells determined by fluorescence photobleaching recovery. Cell. 29:835–845. [DOI] [PubMed] [Google Scholar]
- Kukalev, A., Y. Nord, C. Palmberg, T. Bergman, and P. Percipalle. 2005. Actin and hnRNP U cooperate for productive transcription by RNA polymerase II. Nat. Struct. Mol. Biol. 12:238–244. [DOI] [PubMed] [Google Scholar]
- Kushnaryov, V.M., J.J. Sedmak, R.R. Markwald, M.L. Faculjak, and P.M. Loo. 1990. Actin and lamin comprised filaments in the nuclei of Chinese hamster ovary cells affected with Clostridium difficile enterotoxin A. Cytobios. 64:181–196. [PubMed] [Google Scholar]
- Lachapelle, M., and H.C. Aldrich. 1988. Phalloidin-gold complexes: a new tool for ultrastructural localization of F-actin. J. Histochem. Cytochem. 36:1197–1202. [DOI] [PubMed] [Google Scholar]
- Lanni, F., and B.R. Ware. 1984. Detection and characterization of actin monomers, oligomers, and filaments in solution by measurement of fluorescence photobleaching recovery. Biophys. J. 46:97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever, M.A., J.P. Th'ng, X. Sun, and M.J. Hendzel. 2000. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature. 408:873–876. [DOI] [PubMed] [Google Scholar]
- McGrath, J.L., J.H. Hartwig, Y. Tardy, and C.F. Dewey Jr. 1998. a. Measuring actin dynamics in endothelial cells. Microsc. Res. Tech. 43:385–394. [DOI] [PubMed] [Google Scholar]
- McGrath, J.L., Y. Tardy, C.F. Dewey Jr., J.J. Meister, and J.H. Hartwig. 1998. b. Simultaneous measurements of actin filament turnover, filament fraction, and monomer diffusion in endothelial cells. Biophys. J. 75:2070–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milankov, K., and U. De Boni. 1993. Cytochemical localization of actin and myosin aggregates in interphase nuclei in situ. Exp. Cell Res. 209:189–199. [DOI] [PubMed] [Google Scholar]
- Miller, L., M. Phillips, and E. Reisler. 1988. Polymerization of actin modified with fluorescein isothiocyanate. Eur. J. Biochem. 174:23–29. [DOI] [PubMed] [Google Scholar]
- Mizutani, K., S. Suetsugu, and T. Takenawa. 2004. FBP11 regulates nuclear localization of N-WASP and inhibits N-WASP-dependent microspike formation. Biochem. Biophys. Res. Commun. 313:468–474. [DOI] [PubMed] [Google Scholar]
- Nakayasu, H., and K. Ueda. 1983. Association of actin with the nuclear matrix from bovine lymphocytes. Exp. Cell Res. 143:55–62. [DOI] [PubMed] [Google Scholar]
- Nguyen, E., D. Besombes, and P. Debey. 1998. Immunofluorescent localization of actin in relation to transcription sites in mouse pronuclei. Mol. Reprod. Dev. 50:263–272. [DOI] [PubMed] [Google Scholar]
- Okorokov, A.L., C.P. Rubbi, S. Metcalfe, and J. Milner. 2002. The interaction of p53 with the nuclear matrix is mediated by F-actin and modulated by DNA damage. Oncogene. 21:356–367. [DOI] [PubMed] [Google Scholar]
- Olave, I.A., S.L. Reck-Peterson, and G.R. Crabtree. 2002. Nuclear actin and actin-related proteins in chromatin remodeling. Annu. Rev. Biochem. 71:755–781. [DOI] [PubMed] [Google Scholar]
- Pang, K.M., E. Lee, and D.A. Knecht. 1998. Use of a fusion protein between GFP and an actin-binding domain to visualize transient filamentous-actin structures. Curr. Biol. 8:405–408. [DOI] [PubMed] [Google Scholar]
- Pederson, T., and U. Aebi. 2002. Actin in the nucleus: what form and what for? J. Struct. Biol. 140:3–9. [DOI] [PubMed] [Google Scholar]
- Pederson, T., and U. Aebi. 2005. Nuclear actin extends, with no contraction in sight. Mol. Biol. Cell. 16:5055–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percipalle, P., J. Zhao, B. Pope, A. Weeds, U. Lindberg, and B. Daneholt. 2001. Actin bound to the heterogeneous nuclear ribonucleoprotein hrp36 is associated with Balbiani ring mRNA from the gene to polysomes. J. Cell Biol. 153:229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percipalle, P., N. Fomproix, K. Kylberg, F. Miralles, B. Bjorkroth, B. Daneholt, and N. Visa. 2003. An actin-ribonucleoprotein interaction is involved in transcription by RNA polymerase II. Proc. Natl. Acad. Sci. USA. 100:6475–6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phair, R.D., and T. Misteli. 2001. Kinetic modelling approaches to in vivo imaging. Nat. Rev. Mol. Cell Biol. 2:898–907. [DOI] [PubMed] [Google Scholar]
- Phair, R.D., P. Scaffidi, C. Elbi, J. Vecerova, A. Dey, K. Ozato, D.T. Brown, G. Hager, M. Bustin, and T. Misteli. 2004. Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell. Biol. 24:6393–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philimonenko, V.V., J. Zhao, S. Iben, H. Dingova, K. Kysela, M. Kahle, H. Zentgraf, W.A. Hofmann, P. de Lanerolle, P. Hozak, and I. Grummt. 2004. Nuclear actin and myosin I are required for RNA polymerase I transcription. Nat. Cell Biol. 6:1165–1172. [DOI] [PubMed] [Google Scholar]
- Politz, J.C., E.S. Browne, D.E. Wolf, and T. Pederson. 1998. Intranuclear diffusion and hybridization state of oligonucleotides measured by fluorescence correlation spectroscopy in living cells. Proc. Natl. Acad. Sci. USA. 95:6043–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard, T.D., L. Blanchoin, and R.D. Mullins. 2000. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 29:545–576. [DOI] [PubMed] [Google Scholar]
- Posern, G., A. Sotiropoulos, and R. Treisman. 2002. Mutant actins demonstrate a role for unpolymerized actin in control of transcription by serum response factor. Mol. Biol. Cell. 13:4167–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlas, D.J., K. Milankov, P.C. Park, and U. De Boni. 1993. Distribution of snRNPs, splicing factor SC-35 and actin in interphase nuclei: immunocytochemical evidence for differential distribution during changes in functional states. J. Cell Sci. 105:347–357. [DOI] [PubMed] [Google Scholar]
- Sauman, I., and S.J. Berry. 1994. An actin infrastructure is associated with eukaryotic chromosomes: structural and functional significance. Eur. J. Cell Biol. 64:348–356. [PubMed] [Google Scholar]
- Schafer, D.A., M.D. Welch, L.M. Machesky, P.C. Bridgman, S.M. Meyer, and J.A. Cooper. 1998. Visualization and molecular analysis of actin assembly in living cells. J. Cell Biol. 143:1919–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelden, E.A., J.M. Weinberg, D.R. Sorenson, C.A. Edwards, and F.M. Pollock. 2002. Site-specific alteration of actin assembly visualized in living renal epithelial cells during ATP depletion. J. Am. Soc. Nephrol. 13:2667–2680. [DOI] [PubMed] [Google Scholar]
- Shumaker, D.K., E.R. Kuczmarski, and R.D. Goldman. 2003. The nucleoskeleton: lamins and actin are major players in essential nuclear functions. Curr. Opin. Cell Biol. 15:358–366. [DOI] [PubMed] [Google Scholar]
- Star, E.N., D.J. Kwiatkowski, and V.N. Murthy. 2002. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat. Neurosci. 5:239–246. [DOI] [PubMed] [Google Scholar]
- Sund, S.E., and D. Axelrod. 2000. Actin dynamics at the living cell submembrane imaged by total internal reflection fluorescence photobleaching. Biophys. J. 79:1655–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabellini, G., R. Bortul, S. Santi, M. Riccio, G. Baldini, A. Cappellini, A.M. Billi, R. Berezney, A. Ruggeri, L. Cocco, and A.M. Martelli. 2003. Diacylglycerol kinase-theta is localized in the speckle domains of the nucleus. Exp. Cell Res. 287:143–154. [DOI] [PubMed] [Google Scholar]
- Theriot, J.A., and T.J. Mitchison. 1991. Actin microfilament dynamics in locomoting cells. Nature. 352:126–131. [DOI] [PubMed] [Google Scholar]
- Van Impe, K., V. De Corte, L. Eichinger, E. Bruyneel, M. Mareel, J. Vandekerckhove, and J. Gettemans. 2003. The Nucleo-cytoplasmic actin-binding protein CapG lacks a nuclear export sequence present in structurally related proteins. J. Biol. Chem. 278:17945–17952. [DOI] [PubMed] [Google Scholar]
- Verkhusha, V.V., S. Tsukita, and H. Oda. 1999. Actin dynamics in lamellipodia of migrating border cells in the Drosophila ovary revealed by a GFP-actin fusion protein. FEBS Lett. 445:395–401. [DOI] [PubMed] [Google Scholar]
- Vetterkind, S., H. Miki, T. Takenawa, I. Klawitz, K.H. Scheidtmann, and U. Preuss. 2002. The rat homologue of Wiskott-Aldrich syndrome protein (WASP)-interacting protein (WIP) associates with actin filaments, recruits N-WASP from the nucleus, and mediates mobilization of actin from stress fibers in favor of filopodia formation. J. Biol. Chem. 277:87–95. [DOI] [PubMed] [Google Scholar]
- Wang, Y.L. 1987. Mobility of filamentous actin in living cytoplasm. J. Cell Biol. 105:2811–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal, M., A. Jungbluth, M. Heidecker, B. Muhlbauer, C. Heizer, J.M. Schwartz, G. Marriott, and G. Gerisch. 1997. Microfilament dynamics during cell movement and chemotaxis monitored using a GFP-actin fusion protein. Curr. Biol. 7:176–183. [DOI] [PubMed] [Google Scholar]
- Wilson, K.L., J.M. Holaska, R.M. de Oca, K. Tifft, M. Zastrow, M. Segura-Totten, M. Mansharamani, and L. Bengtsson. 2005. Nuclear membrane protein emerin: roles in gene regulation, actin dynamics and human disease. Novartis Found. Symp. 264:51–58. [PubMed] [Google Scholar]
- Yin, H.L., and P.A. Janmey. 2003. Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol. 65:761–789. [DOI] [PubMed] [Google Scholar]
- Zhang, S., K. Buder, C. Burkhardt, B. Schlott, M. Gorlach, and F. Grosse. 2002. Nuclear DNA helicase II/RNA helicase A binds to filamentous actin. J. Biol. Chem. 277:843–853. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}