

Abstract
Titin, the largest protein known to date, has been linked to sarcomere assembly and function through its elastic adaptor and signaling domains. Titin's M-line region contains a unique kinase domain that has been proposed to regulate sarcomere assembly via its substrate titin cap (T-cap). In this study, we use a titin M line–deficient mouse to show that the initial assembly of the sarcomere does not depend on titin's M-line region or the phosphorylation of T-cap by the titin kinase. Rather, titin's M-line region is required to form a continuous titin filament and to provide mechanical stability of the embryonic sarcomere. Even without titin integrating into the M band, sarcomeres show proper spacing and alignment of Z discs and M bands but fail to grow laterally and ultimately disassemble.
The comparison of disassembly in the developing and mature knockout sarcomere suggests diverse functions for titin's M line in embryonic development and the adult heart that not only involve the differential expression of titin isoforms but also of titin-binding proteins.
Introduction
Sarcomere assembly is a process orchestrated by the sequential expression of structural and signaling proteins, which ultimately leads to the formation of mature myofibrils. It involves the exchange of nonmuscle myosin IIB for muscle myosin II, the incorporation of titin and titin-binding proteins into the nascent myofibril, the lateral alignment of sarcomeric proteins, and the fusion of α-actinin–rich Z bodies into Z bands (Dabiri et al., 1997). During maturation of the sarcomere, titin's NH2 terminus is localized in Z bodies, and muscle myosin II is aligned along the developing myofibril, presumably in a titin-dependent process (Rhee et al., 1994). Mature myofibrils are characterized by the alignment of muscle myosin II filaments to form A bands and the fusion of Z bodies to form Z bands (Tokuyasu and Maher, 1987; Ehler et al., 1999; Sanger et al., 2000). They contain a continuous elastic filament system along the myofibril with titin molecules overlapping at the Z disc and M band, which has been regarded as a molecular ruler or blueprint for sarcomere assembly (Labeit and Kolmerer, 1995; Trinick, 1996; van der Loop et al., 1996; Obermann et al., 1997; Gregorio et al., 1998).
In addition to its structural role in myofibrillogenesis, titin's M-line region has been implicated in sarcomere assembly through the titin kinase domain and its in vitro substrate titin cap (T-cap), which is also known as telethonin. Activation of the titin kinase and phosphorylation of T-cap in differentiating myocytes has been hypothesized to be involved in reorganization of the cytoskeleton during myofibrillogenesis (Mayans et al., 1998). So far, no suitable animal or tissue culture model was available to test this hypothesis.
We have successfully used the Cre-lox recombination system to excise titin's M-line exons (MExs) 1 and 2 in striated muscle and demonstrated their importance in both skeletal and cardiac muscle (Gotthardt et al., 2003; Peng et al., 2006). Loss of titin's M line leads to impaired stability of the muscle fiber with the disassembly of existing sarcomeres. This results in reduced cardiac output followed by a failure to thrive and lethality dependent on the onset and level of Cre expression. The conditional knockout approach enabled the generation of adult animals to study titin's function in the mature heart and skeletal muscle, but expression kinetics of the Cre recombinase transgene preclude the analysis of titin's role in sarcomere assembly during early embryonic development. To distinguish a role in sarcomere assembly from a role in stabilizing preexisting sarcomeres and to address potential nonmuscle functions, we have converted our conditional M-line titin knockout into a complete knockout using germline recombination. In this study, we show that titin's M-line region is dispensable for initial sarcomere assembly, including the correct localization of M-band proteins, but that it is required to fortify the sarcomere structure and for lateral growth. Although the titin M line–deficient hearts start to contract and loop properly, wall thickness and trabeculation are reduced from embryonic day (E) 9.5 followed by apoptosis secondary to the reduced cardiac output. Monitoring the localization and embryonic expression of M-band proteins and proposed substrates of the titin kinase, we were able to attribute the sarcomere disassembly to titin's structural functions. Unlike in the adult knockout sarcomere, kinase-deficient titin does not integrate into the A band and, thus, fails to form a continuous filament system. The failure to cross-link myomesin and titin results in increased mobility of titin's COOH terminus. These structural changes lead to reduced stability and ultimately to disassembly of the sarcomere.
Results
Titin deficiency leads to early embryonic lethality
The titin M-line region is critical for the maintenance of sarcomere structure and function in adult muscle (Gotthardt et al., 2003). To address its role in early cardiac development and possible nonmuscle functions, we have used germline expression of the Cre recombinase under control of the protamine promoter to convert our conditional knockout model into a constitutive titin M line–deficient animal (Fig. 1 A). After recombination, the mutant titin allele is transmitted through the male germline and leads to the expression of a titin protein that is deficient in the titin kinase region encoded by titin's MEx1 and 2 (Fig. 1, B–D). In addition to the kinase domain, this region contains binding sites for the ubiquitin ligase MuRF-1 (muscle-specific RING finger protein 1), signaling proteins such as calmodulin and FHL2 (four and a half LIM-only protein 2), and the M-band protein myomesin (a detailed M-band protein map indicating the deletion is provided in Fig. 7). Although heterozygous knockout animals are fertile and do not display any phenotypic abnormalities, homozygous knockouts die in midgestation (Fig. 1 B and Table I). Knockout, heterozygous, and wild-type animals are present at the appropriate Mendelian ratios and express titin isoforms of the expected sizes. Unlike in embryonic development, the adult M line–deficient titin is expressed at lower levels than the wild-type isoform (compare adult with embryonic heterozygous animals; Fig. 1 D).
Figure 1.

Conversion of the inducible into a constitutive titin kinase region knockout. (A) Outline of the exon/intron structure of titin's M-line region and location of the genotyping primers (PL1, 2, and 4) and in situ probes (prekin and kinase). Cre-mediated recombination leads to the deletion of exons 358 and 359 (MEx1 and 2). (B) PCR-based genotyping of the protamine-Cre transgenic mouse (lane 1) and Recf mouse with loxP sites flanking MEx1 and 2 (lane 2) that were mated to obtain double heterozygotes (lane 3). After germline recombination, offspring contain the deleted Rec allele (lane 4). PCR analysis of embryos at E9.5 confirms early embryonic survival of homozygous knockouts (lanes 9, 11, and 14). (C) Pedigree for animals analyzed in B. (D) SDS-agarose gels of wild-type (lane 1), heterozygous (lane 2), and knockout hearts (lane 3) of E9.5 animals and adult heterozygous (lane 4) and wild-type animals (lane 5). Titin proteins of the predicted sizes are expressed in knockout animals. Because embryonic titin is expressed as a larger isoform, differences in migration are more prevalent in T2 titin. Truncated titin (Rec) is more stable in the embryo compared with adult heterozygotes (ratio of wild-type/knockout protein in lanes 2 and 4). Homozygous adults could not be obtained. (E) In situ hybridization of whole-mount E9.5 embryos using an antisense probe directed against the kinase region (a and b) and a probe that recognizes a region upstream of the kinase domain (prekin in c and d). Both heart and somites are stained in all controls, whereas the kinase probe in knockout animals does not produce a signal (b). Note the smaller body size of titin M-line knockout animals (KO). WT, wild type. Bar, 500 μm.
Figure 7.

Signaling and structural functions of titin's M-line region. The titin M-line region (gray) mainly consists of Ig domains labeled M1–M10, a fibronectin (FN3), and a kinase domain (CD + RD). The 214-kD region deleted in our titin M line–deficient animals (bracket) contains binding sites for MuRF-1, Nbr1, calmodulin, FHL2 (signaling proteins in yellow), and myomesin (green). Myomesin integrates into the M band through interaction with myosin (My1), titin (My4), and dimerization (My13). In vitro substrates for the truncated soluble kinase are T-cap, p62, and Nbr1 (blue). The titin M-line region has been proposed to cover structural (sarcomere assembly) as well as signaling functions (hypertrophy/atrophy) through its multiple binding partners. Proteins expressed at low levels or below the detection limit of our Western blot in the embryonic heart are semitransparent. Protein–protein interactions are indicated by double-pointed arrows. The protein domains depicted are PB1 (Phox and Bem1p domain), Z (zinc-binding domain), U (ubiquitin-associated domain), IG (Ig-like domain), FN3 (fibronectin type 3 domain), CD (titin kinase catalytic domain), RD (titin kinase regulatory domain), R (RING finger), B (B box–type zinc finger), E (EF hand calcium-binding motif), and L (LIM domain).
Table I.
Embryonic lethality of titin M-line knockout mice
| Stage | +/+ | +/− | −/− |
|---|---|---|---|
| % | % | % | |
| E8.5 | 26 (12) | 57 (26) | 17 (8) |
| E9.0 | 25 (8) | 52 (16) | 23 (7) |
| E9.5 | 23 (44) | 55 (106) | 22 (43) |
| E10.0 | 24 (7) | 55 (16) | 21 (6) |
| E10.5 | 18 (19) | 62 (64) | 20 (21) |
| E11.0 | 13 (1) | 62 (5) | 25 (2) |
| E11.5 | 23 (7) | 58 (18) | 19 (6) |
| P1a | 29 (21) | 71 (51) | 0 (0) |
Stage is indicated as the embryonic day after coitum. Absolute numbers of wild-type (+/+), heterozygous (+/−), and knockout animals (−/−) are provided in parentheses.
Postnatal day 1.
Expression of the wild-type and truncated titin is restricted to the heart and somites in the developing embryo (Fig. 1 E), as shown by in situ hybridization. At E9.5, there is no titin MEx1 expression detectable in knockout hearts. Compared with the wild-type and heterozygous animals, head and body size are reduced. The symmetrical body shape is unlike the shrunken head phenotype observed in the Shru mutant, which maps to the titin locus (May et al., 2004).
A beating heart can develop in the absence of titin's kinase region
We followed the embryonic phenotype from E9, when knockout embryos are of similar size as their littermates and display proper development. This is not only reflected in identical cardiac morphology but also in appropriate initiation and maintenance of the heartbeat in both knockout and wild-type embryos (Fig. 2, A and B; and Figs. S1 and S2, available at http://www.jcb.org/cgi/content/full/jcb.200601014/DC1). Because embryonic development at the organ, cellular, and ultrastructural level did not differ between wild-type and heterozygous animals, we restricted all further comparisons to wild-type and homozygous knockout animals.
Figure 2.

Knockout embryos develop normally up to E9.0, followed by a failure to thrive. (A) The wild-type (WT) and knockout (KO) embryos at E9.0 (a and b) develop normally with appropriate cardiac size and function. The mutant embryo at E10.0 (d) appears normal, including normal cardiac morphology, but is small for its age. At E11.0, embryonic and cardiac development of the mutant is delayed with reduced body size and pericardial hemorrhage (f). Arrows point to the heart. (B) Histological analysis demonstrates normal development of the left ventricle and myocardium until E9.0. At E10.0, proper looping takes place, but ventricular wall thickness and trabeculation are reduced compared with wild-type animals (f vs. e; and Fig S2, available at http://www.jcb.org/cgi/content/full/jcb.200601014/DC1). At E11.0, the difference in wall thickness becomes more prominent and is accompanied by cellular infiltration of the knockout pericardium (f, arrowheads). A, atrium; LA, left atrium; RA, right atrium; V, ventricle; M, myocardium; T, trabeculation. Bars, 500 μm.
At E10, the mutant embryo appears normal (including the ratio of heart to body size) but small for its age. The heart undergoes proper looping, but trabeculation and wall thickness are reduced (Fig. S2). By day 11, atrophy results in instability of the ventricular wall with pericardial effusion (Fig. 2 B). At this stage, the developmental delay is reflected in the decreased number of somites in M-line knockout embryos (Fig. 1 E, c vs. d).
Failure to thrive is secondary to impaired cardiac function
The reduced cardiac wall thickness and asystole (loss of cardiac activity) from E9.5, together with the failure to thrive, led us to investigate apoptosis in muscle and nonmuscle tissues as a potential mechanism. At E9.5, when we first see a genotype-dependent size difference, the level of apoptosis is comparable between knockout and wild-type animals (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200601014/DC1). At E10.5, apoptosis is significantly increased in knockout animals, particularly around the peritoneal cavity in the abdominal part of the trunk (Fig. S3, bottom). Because nonmuscle knockout cells divide and differentiate normally until cardiac function is affected, there is no primary defect in nonmuscle cells that would interfere with the cell cycle or cause apoptosis. The increased apoptosis from E9.5 in knockout animals affects all tissues. It is not increased in cardiac versus extracardiac tissue (abdominal cavity, limbs, and brain), which would also indicate a secondary change and does not imply a nonmuscle function of titin's M-line region such as an anti-apoptotic effect.
Titin's kinase region is dispensable for initial sarcomere assembly but is required for lateral growth and stabilization of the sarcomere
Titin's kinase domain has been proposed to be required for sarcomere assembly. As a mechanism, it has been suggested that phosphorylation of the titin kinase substrate T-cap, which transiently localizes to the M band during development, coordinates the assembly of Z-line and M-band lattices during myofibrillogenesis (Mayans et al., 1998). To investigate titin's structural functions, we have used electron microscopy to follow sarcomere assembly, growth, and disassembly. At E9, assembled sarcomeres were detected in the knockout, and no structural alteration compared with wild-type sarcomeres was apparent (Fig. 3 A). SDS-PAGE revealed the exclusive expression of kinase-deficient titin in our knockout animals (Fig. 1 D, lane 3). Therefore, our ultrastructural data suggest that the titin kinase domain is not required for sarcomere assembly. From E9.5, knockout myofibrils fail to grow laterally (quantification in Fig. 3 B), but Z-disc and M-band structure are maintained through E10. Thereafter, knockout sarcomeres disassemble, and, at E11, only a few filaments in disarray remain.
Figure 3.

Ultrastructural analysis of cardiac sarcomere maturation. (A) Sarcomeres assemble in both wild-type (WT) and knockout animals (KO), but lateral growth is impaired from day 10 in knockout animals followed by sarcomere disassembly (h and j). At E11, nearly all sarcomeric structures are dissolved. (B) Myofibril diameter was quantified for >50 fibers in duplicate with two animals per group. The lateral growth of knockout myofibrils lags behind the increase in wild-type myofibril diameter. The loss of sarcomere structure precludes the analysis of E10.5 and 11.0 sarcomeres in knockout animals. **, P < 0.01. (C) Sarcomere length was not changed in wild-type versus knockout animals before disassembly at E10.5. Error bars represent SD. (D) M-band and Z-disc structures at E10.0 do not differ significantly between knockouts and wild-type animals, and there is no change in the A/I alignment. Slight variation in M-band alignment is present in both knockout and wild-type animals (arrowheads in a and b). Embryonic sarcomeres do not display an electron-dense M band. L, lipid; Mi, mitochondrium; N, nucleus; Z, Z disc; M, M band. Bars, 0.5 μm.
Thus, the titin M-line region has multiple effects on the molecular development of the sarcomere: it is involved in regulating lateral growth and fortification of the sarcomere. The underlying mechanism could involve a role in signal transduction or as a scaffold for structural proteins.
Proper localization of Z-disc and M-band proteins proceeds even without M-line–deficient titin forming a continuous filament
The embryonic heart does not reveal M bands as an electron-dense area (Smolich, 1995), so electron microscopy is blind to early changes in the developing cardiac M band. Thus, although titin M line–deficient and wild-type sarcomeres are indistinguishable at the ultrastructural level early in development, their structure and molecular composition could already be affected.
Accordingly, we followed the integration of titin into the sarcomere using antibodies directed against titin's I-band region proximal to the Z disc (N2B) and its COOH terminus (M8/M9) as well as an anti–α-actinin antibody as a marker for the Z disc (Fig. 4; also see the localization of titin epitopes in Fig. S2).
Figure 4.

Integration of titin into the sarcomere. Coimmunostaining with antibodies directed against α-actinin and titin's N2B region (A), the A/I region (B), and the M-line region (C) reveals that titin is expressed and incorporated into the I band of the sarcomere in both wild-type (WT) and knockout (KO) animals (A). The N2B region is located proximal to the Z disc, and, thus, N2B staining partially overlaps with α-actinin staining at E9.5 (yellow). In knockouts at day 10.5, sarcomeres disassemble with few areas of striation remaining (open arrowheads). In areas of disassembly where α-actinin localizes in spotted aggregates (closed arrowheads), the N2B region is distributed diffusely. (B) The T3 antibody stains titin at the beginning of the A band. Correct localization in wild-type and knockouts (doublets marked with arrowheads) indicates that titin transitions properly into the A band. (C) Unlike the N2B region and the T3 epitope, titin's M line (M8/M9) is not integrated into the sarcomere in the absence of the kinase region. Although wild-type sarcomeres at different stages of embryonic development and in the adult animal show the expected striated pattern with alternating Z-disc and M-band epitopes, knockout animals fail to incorporate titin into the M band. Already at E9.5, titin's COOH terminus localizes diffusely. (D) To confirm the presence of titin's M8/M9 epitope in the kinase region–deficient protein, we used Western blot analysis of the wild-type and truncated titin in heterozygous embryos. Bars, 5 μm.
In wild-type animals, titin molecules overlap at the Z disc and M band to form a continuous filament system (Fig. 4, compare A with B; Obermann et al., 1996; Gregorio et al., 1998; Young et al., 1998). In knockout animals, titin's NH2 terminus is incorporated into the Z disc, which results in close proximity of α-actinin and the titin N2B region at E9.5. Upon disassembly, Z bodies distribute from regular striation (Fig. 4 A, open arrowheads) to random patches of variable size (Fig. 4 A, closed arrowheads). This sign of disintegration of the Z bodies is complemented by the diffuse staining for titin's N2B region, which is now separate from α-actinin.
Unlike the NH2 terminus of kinase-deficient titin, its M-line region is not integrated into the developing sarcomere at all (Fig. 4 B). Even at E9.5, there is no accumulation of titin's M8/M9 epitope between Z discs. The absence of a distinct M8/M9 staining could result either from the mislocalization of M8/M9 or from the expression of a truncated protein, which does not only lack the kinase region but fails to include the M8/M9 domains. We used Western blot analysis of embryonic hearts derived from wild-type and heterozygous animals to confirm proper expression of the truncated M-line region (Fig. 4 C). Because the M8/M9 epitope is included in the wild-type and knockout protein of heterozygous animals, the loss of M-line staining is indeed the result of the partial integration of kinase region–deficient titin into the sarcomere at the Z disc only.
Next, we used immunofluorescence staining to follow M-band assembly and the fate of myomesin in the absence of titin's myomesin-binding site (see Fig. 7). The periodic myomesin staining indicates that even in the absence of titin's M line, myomesin is incorporated into the sarcomere (Fig. 5 A). It localizes properly between Z discs, as demonstrated by costaining with the Z-disc protein α-actinin. Nevertheless, in knockout animals, myomesin staining is more diffuse. This might reflect the reduced number of binding sites in the absence of titin's kinase region because myomesin levels are not up-regulated in knockout versus wild-type animals (Fig. 5 B).
Figure 5.

M-band assembly in the absence of titin's M-line region. (A) Although titin is not incorporated into the M band, myomesin (Myom1 EH) localizes between Z discs (α-actinin staining). (B) The more diffuse myomesin staining in titin kinase region knockout (KO) animals is not related to the overexpression of myomesin, as demonstrated by Western blotting at both E9.5 and 10.5. Expression was normalized to actin. WT, wild type. Bar, 5 μm.
Differential expression of titin-binding proteins is linked to diverse functions of M-line titin in the adult and embryonic sarcomere
In both our conditional titin knockout animals (Gotthardt et al., 2003; Peng et al., 2006) and the conventional knockout described in this study, titin kinase-deficient sarcomeres disassemble. Nevertheless, structure is preserved better in the adult than in the developing sarcomere with titin's M line integrated properly, even in the absence of titin's kinase region. Titin's sarcomeric functions are in part inherent in the protein (spacer/spring) and in part relayed through protein–protein interactions. Thus, we used expression analysis of both titin and its binding proteins at various stages of development to help discriminate their roles in the embryonic and adult heart. Overall mRNA levels of titin and its binding proteins are reduced in embryonic development and increase by up to two orders of magnitude from E9.5 to adulthood.
To monitor titin expression, we used TaqMan probes that distinguish the Z disc, kinase region, M line, and an internal region of titin (Fig. 6 A). Adult titin isoforms lack various internal exons of the Ig and PEVK regions (Lahmers et al., 2004; Opitz et al., 2004). The novex-3 isoform, which encodes a truncated 700-kD protein, does not even include the M-line region (Bang et al., 2001).
Figure 6.
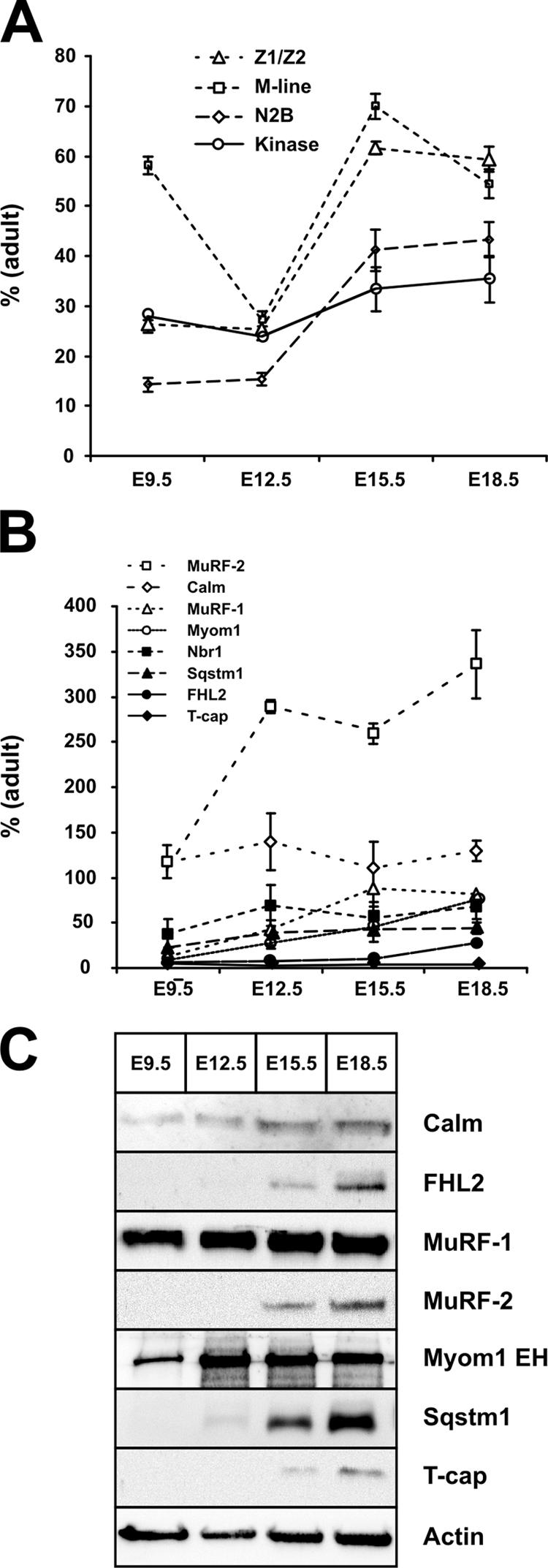
Sarcomere metabolism in embryonic development versus adulthood. Real-time RT-PCR analysis of titin (A) and its M line–binding proteins (B) demonstrates that transcript levels change by up to two orders of magnitude from early cardiac development to adulthood. (A) The cardiac-specific N2B region is barely expressed in the embryonic heart. Differences in Z-disc and M-line levels indicate differences in the ratio of full-length titin to the NH2-terminal novex isoforms, with more full-length titin expressed in the developing embryo, as depicted in Fig. S2 A (available at http://www.jcb.org/cgi/content/full/jcb.200601014/DC1). (B) Except for calmodulin, MuRF-2, and Nbr1, transcript levels of most binding proteins are <20% of adult levels at E9.5. The proposed kinase substrate T-cap shows the lowest embryonic expression of all transcripts tested. (C) Most M-line titin–binding proteins can be detected by Western blotting only after E9.5, when cardiac pathology in knockout animals is already present. Notable exceptions are MuRF-1, which is expressed at similar levels throughout embryonic development, calmodulin, and myomesin. Error bars represent SD. Calm, calmodulin; FHL2, four and a half LIM-only protein 2; Nbr1, neighbor of BRCA1 gene 1; Sqstm1, sequestosome 1; MuRF, muscle-specific RING finger protein; T-cap, titin cap.
Correlating titin's Z-disc and M-line expression allows us to determine the amount of full-length versus novex-3 titin. Increased embryonic M-line RNA levels indicate a higher ratio of full-length titin compared with the truncated novex-3 isoform in the developing embryo from E9.5 (Fig. 6 A). In early development, we see a reduced expression of the heart-specific titin N2B region, which would imply altered elastic properties of the titin filament system.
Various titin-binding proteins have been proposed to act as a substrate for the titin kinase and regulate embryonic sarcomere assembly. Of these, T-cap, Sqstm1 (sequestosome 1), and Nbr1 (neighbor of BRCA1 gene 1) are expressed at <20% of adult levels in the embryonic heart (Fig. 6 B). Combining protein and RNA data, MuRF-1, myomesin, and calmodulin are the only proteins expressed in significant amounts at the time when the knockout phenotype develops (Fig. 6, B and C; and Fig. S2). This would argue against a role of the proposed kinase substrates T-cap, Sqstm1, and Nbr1 in the development of the phenotype.
Sarcomere disassembly in embryonic knockout hearts is not compensated for by increased expression of sarcomeric proteins or hypertrophic signal transduction
Although the sarcomeric phenotype can be explained by structural features of M-line titin alone, additional changes in signal transduction or secondary effects could precipitate or modify the phenotype. Therefore, we compared the expression of proteins involved in sarcomere assembly and hypertrophic signal transduction in wild-type and knockout embryos. Unexpectedly, neither titin nor its binding proteins are up-regulated in response to the elimination of titin's kinase region (Fig. S2). Furthermore, the hypertrophic gene response accompanying the adult kinase region knockout phenotype (unpublished data) is not elicited in the embryonic heart.
We conclude that the initial sarcomere assembly and maturation can proceed normally, even in the absence of titin's kinase region, up until titin is incorporated into the M band. The lack of both changes in signal transduction and the compensatory up-regulation of titin and its binding proteins implies that titin's M-line region, including the kinase domain, covers structural more than signaling functions in the developing sarcomere.
Discussion
Because of its size, it has been notoriously difficult to study titin's signaling and structural functions in vivo. Although multiple titin-deficient animals and cell lines have been generated (Person et al., 2000; Garvey et al., 2002; Xu et al., 2002; Gotthardt et al., 2003; Miller et al., 2003; Peng et al., 2006), it has not been possible so far to address titin's role in early sarcomere assembly.
In this study, we present a novel animal model to investigate titin in the developing sarcomere. We have generated an internal homozygous deletion in the titin gene, which excises titin's kinase region (MEx1 and 2), allowing us to study its role in sarcomere assembly and address its potential functions in nonmuscle cells.
Nonmuscle functions of M-line titin
Titin was originally discovered in striated muscle, but nonmuscle titins have been described in various tissues such as the duodenal epithelium and smooth muscle (Eilertsen and Keller, 1992; Eilertsen et al., 1994; Keller et al., 2000). Titin's role in nonmuscle cells is poorly understood, but it has been implicated in cytokinesis through localization to the cleavage furrows (Keller et al., 2000) and in chromosome condensation and mitosis through localization to mitotic chromosomes and the spindle machinery (Machado et al., 1998; Wernyj et al., 2001). So far, functional data are only available for the Drosophila homologue D-titin. Drosophila deficient in D-titin show chromosome undercondensation, premature sister chromatid separation, and aneuploidy (Machado and Andrew, 2000). Our titin-deficient animals as well as the published zebrafish and mouse titin mutants do not display any nonmuscle phenotypes apart from defects that are secondary to impaired cardiac and skeletal muscle function (Garvey et al., 2002; Xu et al., 2002; May et al., 2004). The differences between Drosophila and mouse/zebrafish might be attributed to the different titin domains involved; in particular, the titin kinase region is not present in D-titin.
Because our titin M line–deficient animals develop normally until midgestation, we can exclude an essential cellular function of the kinase region in cell division and filament assembly. Knockout muscle and nonmuscle cells undergo apoptosis only after E9.5, which can be attributed to impaired cardiac function. The severity of the cardiac phenotype precludes the analysis of smooth muscle and intestinal functions, which would have to be addressed in the respective tissue-specific knockout animals.
The titin kinase in sarcomere assembly
Various models of sarcomerogenesis have been proposed using data derived from cultured chick cardiomyocytes (Schultheiss et al., 1990; Rhee et al., 1994). Although they differ with respect to early thin and thick filament assembly, they ultimately agree on the joint assembly of actin and myosin filaments along a common filament plane late in sarcomere development and on the necessity of adaptor proteins to cross-link Z bodies and M-band proteins into periodic A and I bands (Dabiri et al., 1997; van der Ven et al., 1999; Rudy et al., 2001).
Titin has been proposed to act as a template for sarcomere assembly through its unique domain structure with multiple protein interaction domains and its ability to multimerize into filaments extending throughout the muscle fiber (Labeit and Kolmerer, 1995; Trinick, 1996; van der Loop et al., 1996; Obermann et al., 1997; Gregorio et al., 1998). The titin kinase domain in particular and its substrate T-cap have been implicated in the early stages of sarcomere assembly based on structural and in vitro data (Mayans et al., 1998).
The compartmentalization of titin's mechanical, structural, and signaling functions facilitates the analysis of individual subdomains. Nevertheless, a truncated, overexpressed protein fragment does not necessarily reflect the activity of a domain integrated into the native protein in the context of the sarcomere. This does not only apply to titin's elastic domains but also to titin's kinase region. To circumvent this problem, we used site-specific recombination to excise titin's kinase region and compared sarcomere assembly in our titin M line–deficient and wild-type embryos. This approach combines the advantage of eliminating tissue culture artifacts and providing a clean genetic model to address the role of titin's kinase domain in sarcomere assembly. For the first time, we show that phosphorylating T-cap by the titin kinase is not important for initial sarcomere assembly. Not only do sarcomeres form in the absence of titin's kinase domain, but, furthermore, the T-cap protein is not detectable in early sarcomere development (E9.5) by Western blotting (Fig. 6) or by immunoprecipitation (not depicted). Although T-cap overexpression in tissue culture interferes with sarcomere assembly, the physiological role is debatable because the earliest published detection in mice is E10.5 (transcript level only) when fully assembled sarcomeres already exist (Gregorio et al., 1998).
Patients deficient in T-cap develop limb girdle muscular dystrophy type 2G (Moreira et al., 2000). Because even homozygous patients do not show symptoms before 2 yr of age, sarcomeres can assemble even in the absence of T-cap. The truncated T-cap protein expressed in these patients (premature stop codon in exon 2) is not detected in the sarcomere; nevertheless, ultrastructure is maintained (Vainzof et al., 2002).
Integration of titin into the developing sarcomere
Sarcomere assembly differs between skeletal muscle and cardiomyocytes. In skeletal muscle, titin's Z-disc and M-band regions integrate into the sarcomere almost at the same stage of differentiation, before internal A- and I-band epitopes are localized (Fürst et al., 1989; Kontrogianni-Konstantopoulos et al., 2006), whereas in cardiomyocytes, the integration of titin into the M band is delayed (Ehler et al., 1999). In our titin kinase knockout mice, titin is not integrated into the embryonic M band and, thus, fails to form a continuous elastic titin filament. Nevertheless, cardiac sarcomeres in titin M line–deficient animals assemble and start to contract. Moreover, the M-band protein myomesin targets between Z discs even in the absence of its binding site in M-line titin, although the localization is more diffuse.
The ruler hypothesis states that the titin filament covers an essential role as a blueprint in sarcomerogenesis (Labeit and Kolmerer, 1995; Trinick, 1996; van der Loop et al., 1996; Obermann et al., 1997; Gregorio et al., 1998). Our data show that sarcomere length is not affected after the removal of M-line titin, which might indicate that at least some of titin's scaffold function can be taken over by other proteins in the myofilament. This is facilitated by most sarcomeric proteins interacting with multiple partners, such as myomesin, which not only dimerizes (Lange et al., 2005a) but also binds to both myosin and titin (Auerbach et al., 1999). The proper localization of the T3 epitope at the A→I transition would argue for the proper transition of titin into the A band, where it is cross-linked with the thick filament. Sarcomere spacing would thus be maintained through the titin–myosin filament.
Structural, mechanical, and signaling functions of titin in the embryonic sarcomere
Titin has been proposed to keep thick filaments centered in relaxed as well as in activated myofibrils, generating restoring forces to counteract the unequal contraction of neighboring sarcomeres and to keep multiple sarcomeres in register (Horowits and Podolsky, 1987; Horowits et al., 1989). In fact, dislocation of the A band has been demonstrated in continuously activated muscle (Page and Huxley, 1963), but in our contracting knockout hearts, neither the mislocalization of A bands nor the misalignment of the M band could be detected.
Nevertheless, sarcomere disassembly in our titin-deficient animals clearly demonstrates that a discontinuous titin filament is incompatible with the maintenance of sarcomere structure under mechanical load. This could be a primary defect of titin or be caused by the altered composition of titin-deficient M bands with the mislocalization of myomesin (Agarkova et al., 2003; Agarkova and Perriard, 2005). It is tempting to speculate that titin's integration into the M band allows it to monitor shearing forces between neighboring thick filaments (Agarkova and Perriard, 2005). How this might be affected in our M line–deficient animals will be the focus of future mechanical studies.
The integration of titin into the M band allows it to stabilize the sarcomere (Horowits and Podolsky, 1987). This alone would explain the sarcomeric phenotype in our M-line titin–deficient animals in the absence of changes in signal transduction. We were not able to confirm an additional signaling function for the titin kinase domain, which has been proposed to act as a stretch sensor based on in silico data (Grater et al., 2005). Indeed, patients suffering from hereditary myopathy with early respiratory failure carry a mutant regulatory kinase domain that is supposedly involved in mechanosignaling, which only causes pathology in the adult (Lange et al., 2005b). In our adult titin kinase knockout mice, changes in signal transduction involve increased MAPK signal transduction (unpublished data). This is part of the hypertrophic signaling response secondary to the cardiomyopathy phenotype and could not be detected in the kinase region–deficient embryonic heart using hypertrophy markers, including genes in the MAPK pathway.
The MAPK pathway has been implicated in regulating the serial versus parallel assembly of sarcomeres (Nicol et al., 2001). Because we do not see changes in MAPK signaling in the titin knockout, the mechanism underlying impaired growth of the myofibril is independent of leukemia inhibitory factor/MAPK signal transduction. Lateral growth with the assembly of α-actinin into broad Z lines is a sign of proper maturation of the nascent to mature myofibril (Dabiri et al., 1997). The immature dots we observe in immunostaining thus indicate a problem of sarcomere maturation, not of signal transduction.
Adult versus embryonic sarcomere dynamics and structure
Differentiated embryonic cardiomyocytes can undergo mitosis, which requires the disassembly of preexisting sarcomeres (Ahuja et al., 2004). Based on this finding, the inability of adult cardiomyocytes to divide has been attributed to differences in sarcomere structure and dynamics in the embryonic versus adult animal. These differences could also be the basis of phenotypic differences in our adult and embryonic kinase region–deficient animals. In kinase-deficient adult hearts, we have demonstrated by immunoelectron microscopy that in the absence of titin's kinase region (loss of MuRF-1 localization), titin is still integrated into the sarcomere (Gotthardt et al., 2003). This includes titin's M8/M9 epitope, which remains at the widening M band despite progressive deletion of the kinase region. In the embryonic sarcomere, mutant titin is not localized at the M band despite proper integration into the Z disc and expression of the M8/M9 epitope. This difference might be attributable to changes in titin isoform or titin-binding protein expression during cardiac development. Increased expression of titin's novex isoform, T-cap, as well as all titin M line–binding proteins in the adult could account for increased stability of the sarcomere. Conversely, the lack of titin-binding protein expression in the embryonic M line–deficient animals makes it unlikely that titin signaling through T-cap or FHL2 is the basis for impaired lateral growth and disassembly of the embryonic sarcomere (compare translucent proteins in Fig. 7).
Apart from titin, extrasarcomeric filament systems can promote sarcomere stability. This includes the spectrin, ankyrin, obscurin, and possibly the intermediate filament system (Flick and Konieczny, 2000; Bagnato et al., 2003; Kontrogianni-Konstantopoulos et al., 2004, 2006). These structural proteins could provide the basis for early sarcomerogenesis and make the titin filament system dispensable in the initial assembly of the sarcomere and localization of M-band proteins. The subsequent disassembly is modulated by sarcomere stability, which depends on the proteome of embryonic versus adult cardiomyocytes and on the connection between the sarcomere and additional filament systems.
Differences in the expression and assembly of cardiac proteins determine both structure and function of the adult and embryonic sarcomere. The ability to regenerate sarcomeres and allow for mitosis in the embryo comes at the expense of impaired stability and contractile function.
Understanding how titin and its binding proteins shape and regulate sarcomere assembly and dynamics could help develop novel therapeutic strategies to improve sarcomere stability in patients with muscle disease.
Materials and methods
Generation of titin M-line knockout mice
The transgenic mice with loxP sites flanking titin MEx1 and 2 have been described previously (Gotthardt et al., 2003). They were converted from a conditional to a complete knockout using protamine-Cre transgenic mice (The Jackson Laboratory; O'Gorman et al., 1997). Male double heterozygotes (protamine-Cre/wild type; titin MEx1/2lox/wt) were backcrossed to 129/SvEms-+Ter?/J to obtain a clean colony of heterozygous knockouts devoid of the Cre transgene (TiMEx1/2rec/wt).
Genotyping
Template DNA was prepared from yolk sac or tail according to standard procedures, and recombination of the titin locus was monitored by PCR (primers PL1 and PL4). Lox and wild-type loci were typed using primers PL1 and PL2. All primers used have been described previously (Gotthardt et al., 2003).
Animal procedures (breeding and maintenance)
Timed matings were set up between heterozygotes (MEx1/2rec/wt). The morning of vaginal plug detection was regarded as day 0.5 after conception. Embryos were harvested at E8.5, 9.0, 9.5, 10.0, 10.5, 11.0, and 11.5. All experiments involving animals were performed according to institutional and National Institutes of Health (NIH) Using Animals in Intramural Research guidelines.
Gel electrophoresis and Western blot analysis
SDS lysates for titin gels were prepared by homogenization of pooled dissected hearts from at least three different litters in 0.5 M Tris-HCl, pH 6.8, followed by DNase digestion for 30 min at 37°C and lysis in a buffer containing 8 M urea, 2 M thiourea, 3% SDS, 75 mM DTT, 0.05 M Tris-HCl, pH 8.6, and 0.03% bromphenolblue. Titin isoforms were separated using an SDS-agarose gel electrophoresis system followed by staining with Coomassie (Warren et al., 2003) or Sypro Ruby (Invitrogen).
For Western blot analysis, heart extracts were prepared as described above except using lysis buffer containing 6 M urea, 2% Chaps, 1 mM DTT, 1 mM PMSF, and 1 μg/ml leupeptin. Samples were equally loaded on an 8 or 12% SDS-PAGE gel or SDS-agarose gels for titin and transferred onto nitrocellulose membranes (GE Healthcare). Membranes were blocked with 5% skim milk in PBS–Tween 20 followed by incubation with the following antibodies: 1:500 affinity-purified polyclonal anti–T-cap antibody (for the generation of antibody; see below), 1:750 monoclonal anti-FHL2 (MBL International Corporation), 1:10,000 polyclonal antimyomesin EH (gift from J.-C. Perriard, Eidgenössische Technische Hochschule [ETH] Zürich, Zurich, Switzerland), 1:200 monoclonal anti–MuRF-2 (gift from S. Labeit, Universitätsklinikum Mannheim, Mannheim, Germany), 1:500 affinity-purified polyclonal anti–MuRF-1 (gift from D. Glass, Regeneron Pharmaceuticals, Inc., Tarrytown, NY), and 1:2,000 polyclonal anti-actin (Sigma-Aldrich). All antibodies were diluted in 5% skim milk in PBS and incubated for 2 h at RT or at 4°C overnight. HRP-conjugated goat anti–rabbit IgG (Sigma-Aldrich) and goat anti–mouse IgG (Calbiochem) were used as the secondary antibodies. The blots were washed in PBS–Tween 20 and developed by chemiluminescence staining using ECL (Supersignal West Pico Chemiluminescent Substrate; Pierce Chemical Co.).
Morphological and histological analysis
Embryos were dissected in RNase-free PBS and fixed overnight in freshly prepared 4% PFA in PBS, pH 7.4, at 4°C. The developmental stage of embryos was determined by the days after coitum and the number of somites.
For paraffin embedding, embryos were hydrated followed by dehydration through graded ethanol solutions. Transverse sections were cut at 5 μm, deparaffinized, and stained with 0.5% hematoxylin and 0.1% eosin. Multiple serial sections were stained and analyzed for each developmental stage and genotype.
In situ hybridization
Whole-mount RNA in situ hybridization for the exon preceding MEx1 and for MEx1 was performed using digoxigenin-labeled antisense riboprobes. RNA labeling of linearized plasmid templates was accomplished with digoxigenin-UTP by in vitro transcription with T7 polymerase according to the manufacturer's specifications (Roche Diagnostics). After fixation, embryos were washed in PBS plus 0.15% Tween 20, dehydrated, bleached with 25% H2O2 in MeOH, and rehydrated in an ascending/descending series of methanol (25, 50, and 75% in PBS–Tween 20 and 100% methanol). The tissue was permeabilized with 20 μg/ml proteinase K and refixed with 4% PFA and 0.2% glutaraldehyde for 20 min at room temperature. Embryos were prehybridized in 50% formamide, 5× SSC (0.75 M NaCl and 0.75 M sodium citrate, pH 7.0), 0.1 mg/ml single-stranded DNA (Sigma-Aldrich), 40 μg/ml heparin, 50 μg/ml tRNA, 0.15% Tween 20, and 60 mM citric acid. Hybridization of the digoxigenin-labeled probe was performed overnight at 65°C. Embryos were washed in solution 1 (50% formamide, 5× SSC, and 0.15% Tween 20) at 65°C twice for 40 min, washed in solution 2 (10 mM Tris, pH 7.5, 50 mM NaCl, and 0.15% Tween 20) three times for 15 min, and washed in solution 3 (50% formamide, 2× SSC, and 0.15% Tween 20) three times for 60 min. Embryos were incubated with 0.75 U antidigoxigenin antibody, Fab fragments (Roche Diagnostics) in 5% goat serum in TBS–Tween 20 overnight at 4°C, and washed with TBS–Tween 20 for at least 8 h at room temperature. Embryos were stained with NBT/BCIP solution (350 and 175 μg/ml, respectively; Roche Diagnostics). Images were taken after refixation with 4% PFA and clearance with 80% glycerol.
Immunofluorescence staining
Embryos were dissected followed by overnight fixation in 4% PFA, equilibration with 30% sucrose in PBS, and embedding in Tissue Tek (optimal cutting temperature compound; Vogel). 5-μm sagittal sections were air dried, fixed with 4% PFA, blocked, and permeabilized in 0.3% Triton X-100, 0.2% BSA, and 10% normal goat serum for 60 min. Cryosections were incubated overnight with primary antibodies at 4°C followed by fluorescent-conjugated secondary antibodies (AlexaFluor488 goat anti–rabbit [Invitrogen] and Cy3 goat anti–mouse [Jackson ImmunoResearch Laboratories]). Primary antibodies were used at the following dilutions: 1:500 monoclonal antisarcomeric α-actinin (Sigma-Aldrich), 1:100 monoclonal anti-titin T3 (gift from D.O. Fürst, Universität Bonn, Bonn, Germany; Fürst et al., 1988), 1:10,000 polyclonal antimyomesin EH (gift from E. Ehler and J.-C. Perriard, ETH Zürich; Auerbach et al., 1997), and rabbit polyclonal antibodies 1:1,000 anti–titin-Z1/Z2, 1:200 titin-N2B, and 1:500 anti–titin-M8/M9 (both were gifts from S. Labeit, Universitätsklinikum Mannheim; Trombitas et al., 2000). Stained tissue was mounted with fluorescent mounting medium (DakoCytomation) and analyzed at room temperature using Immersol 518N (Carl Zeiss MicroImaging, Inc.) on a confocal scanning laser microscope (LSM5 Pascal with software version 3.0 SP2; Carl Zeiss MicroImaging, Inc.) with a plan-Neofluar 100× 1.3 NA lens (Carl Zeiss MicroImaging, Inc.). Images were assembled using Photoshop 9.0 and Corel Draw 12.0.
Apoptosis assay
For TUNEL assay (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling), cryosections of mouse embryos were processed using the in situ cell death detection kit (Roche Diagnostics) according to the protocol supplied by the manufacturer. In brief, sections were fixed in 4% PFA and permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2 min on ice. After washing, slides were incubated with TdT terminal transferase and fluorescein-dUTP for nick end labeling. Sections were counterstained with 1:500 antisarcomeric α-actinin (Sigma-Aldrich). Stained tissue was analyzed using a microscope (BX51; Olympus), a CCD camera (Visitron Systems 7.4 Slider; Diagnostic Instruments), and MetaMorph software version 6.2r2 (Universal Imaging Corp.).
Electron microscopy
Embryos for ultrastructural analysis of the sarcomere assembly were dissected and fixed with 3% formaldehyde in 0.2 M Hepes, pH 7.4, for 30 min followed by immersion in 8% formaldehyde/0.1% glutaraldehyde in 0.2 M Hepes, pH 7.4, overnight. Embryos were postfixed with 1% OsO4 for 2 h, dehydrated in a graded ethanol series and propylene oxide, and embedded in Poly/Bed 812 (Polysciences, Inc.). Ultrathin sections (70 nm) were contrasted with uranyl acetate and lead citrate and were examined with an electron microscope (model 910; Carl Zeiss MicroImaging, Inc.). Digital images were taken with a 1k × 1k high speed slow scan CCD camera (Proscan). The diameter of ∼25 sarcomeres per embryo was measured on a range of ultrathin sections taken from two embryos per genotype and day of gestation using the analySIS 3.2 software (Soft Imaging System).
Real-time PCR
For isolation of total RNA, the hearts of 5–10 embryos from at least three litters were pooled (depending on the size and age of the embryo). RNA was isolated by using the RNeasy Mini Kit (QIAGEN) followed by the digestion of contaminating genomic DNA (RNase-Free DNase Set; QIAGEN). RNA concentration and purity were determined by spectrophotometry. 1 μg of total RNA was used for cDNA synthesis (Thermoscript First-Strand Synthesis System; Invitrogen). Quantitative real-time RT-PCR was performed using the TaqMan probe-based chemistry (Applied Biosystems). Primers and probes were designed using the Primer Express 1.5 software (Applied Biosystems) and were ordered from BioTez GmbH for the following titin amplicons: Z1/2 forward, CGATGGCCGCGCTAGA; reverse, CTCAGGGAGTATCGTCCACTGTT; probe, 6-FAM-TGATGATCCCCGCCGTGACTAAAGC-TAMRA; N2B forward, ACAGTGGGAAAGCAAAGACATC; reverse, AGGTGGCCCAGAGCTACTTC; probe, 6-FAM-GAAAGAGCTGCCCCTGTGATCA-TAMRA; kinase forward, CCGATGGACTCAAGTACAGGATT; reverse, CCCATGCCTTCGAGAGTCTT; probe, 6-FAM-TCCTTGGAAGTGGAAGTTCCAGCTAAGATACAC-TAMRA; M-line forward, GCCTTGTGTGGTAGTTCTAAATTCAA; reverse, TTTGCTGTGGCTCATTGCTT; probe, 6-FAM-TTTCACCGGGAACTGGGCAA-TAMRA; MuRF-1 forward (titin-binding proteins), CCGAGTGCAGACGATCATCTC; reverse, CCTTCACCTGGTGGCTATTCTC; probe, 6-FAM-AGCTGGAGGACTCGTGCAGAGTGACC-TAMRA; MuRF-2 forward, TGGAGAACGTATCCAAGTTGGT; reverse, CCTTTGATGCTTCCACGATCT; probe, 6-FAM-CATGGATGAGCCCGAAATGGCA-TAMRA; ANP forward (hypertrophy markers), TTCTAGGCGCAGCCCCT; reverse, GCAGAGCCCTCAGTTTGCTT; probe, 6-FAM-ACCCCTCCGATAGATCTGCCCTCTTGAA-TAMRA; MAPKAP2 forward, GTGTGGGTATCCCCCCTTCT; reverse, TACGAGTCTTCATGCCCGG; probe, 6-FAM-TCCAATCACGGCCTTGCCATCTC-TAMRA; Mef2C forward, GGCTCTGTAACTGGCTGGCA; reverse, TCCCAACTGACTGAGGGCAG; probe, 6-FAM-CAGCAGCACCTACATAACATGCCGCC-TAMRA; TGF-β2 forward, CCCCTGCTACTGCAAGTCAG; reverse, GTCCTCAGGTCCTGCCTCCT; probe, 6-FAM-CTTCTTGGCACTGCGCTGTCTCGC-TAMRA; and endogenous control 18S RNA forward, CGCCGCTAGAGGTGAAATTC; reverse, TGGGCAAATGCTTTCGCTC; and probe, 6-FAM-TGGACCGGCGCAAGACGGAC-TAMRA. All remaining primer probe sets were ordered as TaqMan Gene Expression Assays from Applied Biosystems (calmodulin, FHL2, myomesin, Nbr1, Sqstm1, and T-cap). Real-time PCR amplification reaction was performed on a Sequence Detection System (7900 HT; Applied Biosystems) using the qPCR MasterMix Plus (Eurogentec) according to the manufacturer's instructions with 2× TaqMan universal PCR master mix, 900 nM of primers, and 250 nM of probe.
Thermal cycling conditions were as follows: 50°C for 2 min, 95°C for 10 min followed by 59 cycles of 95°C for 10 s, and 60°C for 1 min. Data were collected and analyzed with the Sequence Detection System 2.1 software (Applied Biosystems). The comparative CT Method (ΔΔCT Method) was used as described in the User Bulletin 2: ABI PRISM 7700 Sequence Detection System.
Statistical analysis
All results are expressed as means ± SD. An unpaired two-tailed t test was performed to assess differences between two groups. A P value of <0.05 was considered significant.
Online supplemental material
Fig. S1 shows the morphology and contractile function of titin-deficient hearts. Fig. S2 provides the location of antibody epitopes and primer-binding sites and shows expression changes in titin-deficient and wild-type animals. Fig. S3 shows increased apoptosis in knockout embryos that are secondary to the cardiac phenotype. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200601014/DC1.
Supplementary Material
Acknowledgments
The authors thank Beate Goldbrich, Katrin Räbel, Cathrin Rudolph, and Marianne Vannauer for expert technical assistance and Siegfried Labeit, Jean-Claude Perriard, and Dieter Fürst for antibodies against titin and myomesin.
This work was supported by grants from the NIH (RO-1 69008) and the Sofja-Kovalevskaya program of the Alexander von Humboldt Foundation.
Abbreviations used in this paper: FHL2, four and a half LIM-only protein 2; MEx, M-line exon; MuRF, muscle-specific RING finger protein; Nbr1, neighbor of BRCA1 gene 1; Sqstm1, sequestosome 1; T-cap, titin cap.
References
- Agarkova, I., and J.C. Perriard. 2005. The M-band: an elastic web that crosslinks thick filaments in the center of the sarcomere. Trends Cell Biol. 15:477–485. [DOI] [PubMed] [Google Scholar]
- Agarkova, I., E. Ehler, S. Lange, R. Schoenauer, and J.C. Perriard. 2003. M-band: a safeguard for sarcomere stability? J. Muscle Res. Cell Motil. 24:191–203. [DOI] [PubMed] [Google Scholar]
- Ahuja, P., E. Perriard, J.C. Perriard, and E. Ehler. 2004. Sequential myofibrillar breakdown accompanies mitotic division of mammalian cardiomyocytes. J. Cell Sci. 117:3295–3306. [DOI] [PubMed] [Google Scholar]
- Auerbach, D., B. Rothen-Ruthishauser, S. Bantle, M. Leu, E. Ehler, D. Helfman, and J.C. Perriard. 1997. Molecular mechanisms of myofibril assembly in heart. Cell Struct. Funct. 22:139–146. [DOI] [PubMed] [Google Scholar]
- Auerbach, D., S. Bantle, S. Keller, V. Hinderling, M. Leu, E. Ehler, and J.C. Perriard. 1999. Different domains of the M-band protein myomesin are involved in myosin binding and M-band targeting. Mol. Biol. Cell. 10:1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnato, P., V. Barone, E. Giacomello, D. Rossi, and V. Sorrentino. 2003. Binding of an ankyrin-1 isoform to obscurin suggests a molecular link between the sarcoplasmic reticulum and myofibrils in striated muscles. J. Cell Biol. 160:245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang, M.L., T. Centner, F. Fornoff, A.J. Geach, M. Gotthardt, M. McNabb, C.C. Witt, D. Labeit, C.C. Gregorio, H. Granzier, and S. Labeit. 2001. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 89:1065–1072. [DOI] [PubMed] [Google Scholar]
- Dabiri, G.A., K.K. Turnacioglu, J.M. Sanger, and J.W. Sanger. 1997. Myofibrillogenesis visualized in living embryonic cardiomyocytes. Proc. Natl. Acad. Sci. USA. 94:9493–9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehler, E., B.M. Rothen, S.P. Hammerle, M. Komiyama, and J.C. Perriard. 1999. Myofibrillogenesis in the developing chicken heart: assembly of Z-disk, M-line and the thick filaments. J. Cell Sci. 112:1529–1539. [DOI] [PubMed] [Google Scholar]
- Eilertsen, K.J., and T.C. Keller. 1992. Identification and characterization of two huge protein components of the brush border cytoskeleton: evidence for a cellular isoform of titin. J. Cell Biol. 119:549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilertsen, K.J., S.T. Kazmierski, and T.C. Keller. 1994. Cellular titin localization in stress fibers and interaction with myosin II filaments in vitro. J. Cell Biol. 126:1201–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flick, M.J., and S.F. Konieczny. 2000. The muscle regulatory and structural protein MLP is a cytoskeletal binding partner of betaI-spectrin. J. Cell Sci. 113:1553–1564. [DOI] [PubMed] [Google Scholar]
- Fürst, D.O., M. Osborn, R. Nave, and K. Weber. 1988. The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: a map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. J. Cell Biol. 106:1563–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürst, D.O., M. Osborn, and K. Weber. 1989. Myogenesis in the mouse embryo: differential onset of expression of myogenic proteins and the involvement of titin in myofibril assembly. J. Cell Biol. 109:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvey, S.M., C. Rajan, A.P. Lerner, W.N. Frankel, and G.A. Cox. 2002. The muscular dystrophy with myositis (mdm) mouse mutation disrupts a skeletal muscle-specific domain of titin. Genomics. 79:146–149. [DOI] [PubMed] [Google Scholar]
- Gotthardt, M., R.E. Hammer, N. Hubner, J. Monti, C.C. Witt, M. McNabb, J.A. Richardson, H. Granzier, S. Labeit, and J. Herz. 2003. Conditional expression of mutant M-line titins results in cardiomyopathy with altered sarcomere structure. J. Biol. Chem. 278:6059–6065. [DOI] [PubMed] [Google Scholar]
- Grater, F., J. Shen, H. Jiang, M. Gautel, and H. Grubmuller. 2005. Mechanically induced titin kinase activation studied by force-probe molecular dynamics simulations. Biophys. J. 88:790–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio, C.C., K. Trombitas, T. Centner, B. Kolmerer, G. Stier, K. Kunke, K. Suzuki, F. Obermayr, B. Herrmann, H. Granzier, et al. 1998. The NH2 terminus of titin spans the Z-disc: its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. J. Cell Biol. 143:1013–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowits, R., and R.J. Podolsky. 1987. The positional stability of thick filaments in activated skeletal muscle depends on sarcomere length: evidence for the role of titin filaments. J. Cell Biol. 105:2217–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowits, R., K. Maruyama, and R.J. Podolsky. 1989. Elastic behavior of connectin filaments during thick filament movement in activated skeletal muscle. J. Cell Biol. 109:2169–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, T.C., K. Eilertsen, M. Higginbotham, S. Kazmierski, K.T. Kim, and M. Velichkova. 2000. Role of titin in nonmuscle and smooth muscle cells. Adv. Exp. Med. Biol. 481:265–277. [DOI] [PubMed] [Google Scholar]
- Kontrogianni-Konstantopoulos, A., D.H. Catino, J.C. Strong, W.R. Randall, and R.J. Bloch. 2004. Obscurin regulates the organization of myosin into A bands. Am. J. Physiol. Cell Physiol. 287:C209–C217. [DOI] [PubMed] [Google Scholar]
- Kontrogianni-Konstantopoulos, A., D.H. Catino, J.C. Strong, and R.J. Bloch. 2006. De novo myofibrillogenesis in C2C12 cells: evidence for the independent assembly of M bands and Z disks. Am. J. Physiol Cell Physiol. 290:C626–C637. [DOI] [PubMed] [Google Scholar]
- Labeit, S., and B. Kolmerer. 1995. Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science. 270:293–296. [DOI] [PubMed] [Google Scholar]
- Lahmers, S., Y. Wu, D.R. Call, S. Labeit, and H. Granzier. 2004. Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ. Res. 94:505–513. [DOI] [PubMed] [Google Scholar]
- Lange, S., M. Himmel, D. Auerbach, I. Agarkova, K. Hayess, D.O. Fürst, J.C. Perriard, and E. Ehler. 2005. a. Dimerisation of myomesin: implications for the structure of the sarcomeric M-band. J. Mol. Biol. 345:289–298. [DOI] [PubMed] [Google Scholar]
- Lange, S., F. Xiang, A. Yakovenko, A. Vihola, P. Hackman, E. Rostkova, J. Kristensen, B. Brandmeier, G. Franzen, B. Hedberg, et al. 2005. b. The kinase domain of titin controls muscle gene expression and protein turnover. Science. 308:1599–1603. [DOI] [PubMed] [Google Scholar]
- Machado, C., and D.J. Andrew. 2000. D-Titin. A giant protein with dual roles in chromosomes and muscles. J. Cell Biol. 151:639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado, C., C.E. Sunkel, and D.J. Andrew. 1998. Human autoantibodies reveal titin as a chromosomal protein. J. Cell Biol. 141:321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May, S.R., N.J. Stewart, W. Chang, and A.S. Peterson. 2004. A Titin mutation defines roles for circulation in endothelial morphogenesis. Dev. Biol. 270:31–46. [DOI] [PubMed] [Google Scholar]
- Mayans, O., P.F. van der Ven, M. Wilm, A. Mues, P. Young, D.O. Fnrst, M. Wilmanns, and M. Gautel. 1998. Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature. 395:863–869. [DOI] [PubMed] [Google Scholar]
- Miller, G., H. Musa, M. Gautel, and M. Peckham. 2003. A targeted deletion of the C-terminal end of titin, including the titin kinase domain, impairs myofibrillogenesis. J. Cell Sci. 116:4811–4819. [DOI] [PubMed] [Google Scholar]
- Moreira, E.S., T.J. Wiltshire, G. Faulkner, A. Nilforoushan, M. Vainzof, O.T. Suzuki, G. Valle, R. Reeves, M. Zatz, M.R. Passos-Bueno, and D.E. Jenne. 2000. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat. Genet. 24:163–166. [DOI] [PubMed] [Google Scholar]
- Nicol, R.L., N. Frey, G. Pearson, M. Cobb, J. Richardson, and E.N. Olson. 2001. Activated MEK5 induces serial assembly of sarcomeres and eccentric cardiac hypertrophy. EMBO J. 20:2757–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Gorman, S., N.A. Dagenais, M. Qian, and Y. Marchuk. 1997. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc. Natl. Acad. Sci. USA. 94:14602–14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermann, W.M., M. Gautel, F. Steiner, P.F. van der Ven, K. Weber, and D.O. Fürst. 1996. The structure of the sarcomeric M band: localization of defined domains of myomesin, M-protein, and the 250-kD carboxy-terminal region of titin by immunoelectron microscopy. J. Cell Biol. 134:1441–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermann, W.M., M. Gautel, K. Weber, and D.O. Fürst. 1997. Molecular structure of the sarcomeric M band: mapping of titin and myosin binding domains in myomesin and the identification of a potential regulatory phosphorylation site in myomesin. EMBO J. 16:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz, C.A., M.C. Leake, I. Makarenko, V. Benes, and W.A. Linke. 2004. Developmentally regulated switching of titin size alters myofibrillar stiffness in the perinatal heart. Circ. Res. 94:967–975. [DOI] [PubMed] [Google Scholar]
- Page, S., and H.E. Huxley. 1963. Filament length in striated muscle. J. Cell Biol. 19:369–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, J., K. Raddatz, S. Labeit, H. Granzier, and M. Gotthardt. 2006. Muscle atrophy in Titin M-line deficient mice. J. Muscle Res. Cell Motil. 10:1–8. [DOI] [PubMed] [Google Scholar]
- Person, V., S. Kostin, K. Suzuki, S. Labeit, and J. Schaper. 2000. Antisense oligonucleotide experiments elucidate the essential role of titin in sarcomerogenesis in adult rat cardiomyocytes in long-term culture. J. Cell Sci. 113:3851–3859. [DOI] [PubMed] [Google Scholar]
- Rhee, D., J.M. Sanger, and J.W. Sanger. 1994. The premyofibril: evidence for its role in myofibrillogenesis. Cell Motil. Cytoskeleton. 28:1–24. [DOI] [PubMed] [Google Scholar]
- Rudy, D.E., T.A. Yatskievych, P.B. Antin, and C.C. Gregorio. 2001. Assembly of thick, thin, and titin filaments in chick precardiac explants. Dev. Dyn. 221:61–71. [DOI] [PubMed] [Google Scholar]
- Sanger, J.W., J.C. Ayoob, P. Chowrashi, D. Zurawski, and J.M. Sanger. 2000. Assembly of myofibrils in cardiac muscle cells. Adv. Exp. Med. Biol. 481:89–102. [DOI] [PubMed] [Google Scholar]
- Schultheiss, T., Z.X. Lin, M.H. Lu, J. Murray, D.A. Fischman, K. Weber, T. Masaki, M. Imamura, and H. Holtzer. 1990. Differential distribution of subsets of myofibrillar proteins in cardiac nonstriated and striated myofibrils. J. Cell Biol. 110:1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolich, J.J. 1995. Ultrastructural and functional features of the developing mammalian heart: a brief overview. Reprod. Fertil. Dev. 7:451–461. [DOI] [PubMed] [Google Scholar]
- Tokuyasu, K.T., and P.A. Maher. 1987. Immunocytochemical studies of cardiac myofibrillogenesis in early chick embryos. II. Generation of α-actinin dots within titin spots at the time of the first myofibril formation. J. Cell Biol. 105:2795–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinick, J. 1996. Titin as a scaffold and spring. Cytoskeleton. Curr. Biol. 6:258–260. [DOI] [PubMed] [Google Scholar]
- Trombitas, K., A. Freiburg, M. Greaser, S. Labeit, and H. Granzier. 2000. From connecting filaments to co-expression of titin isoforms. Adv. Exp. Med. Biol. 481:405–418. [DOI] [PubMed] [Google Scholar]
- Vainzof, M., E.S. Moreira, O.T. Suzuki, G. Faulkner, G. Valle, A.H. Beggs, O. Carpen, A.F. Ribeiro, E. Zanoteli, J. Gurgel-Gianneti, et al. 2002. Telethonin protein expression in neuromuscular disorders. Biochim. Biophys. Acta. 1588:33–40. [DOI] [PubMed] [Google Scholar]
- van der Loop, F.T., P.F. van der Ven, D.O. Fürst, M. Gautel, G.J. van Eys, and F.C. Ramaekers. 1996. Integration of titin into the sarcomeres of cultured differentiating human skeletal muscle cells. Eur. J. Cell Biol. 69:301–307. [PubMed] [Google Scholar]
- van der Ven, P.F., E. Ehler, J.C. Perriard, and D.O. Fürst. 1999. Thick filament assembly occurs after the formation of a cytoskeletal scaffold. J. Muscle Res. Cell Motil. 20:569–579. [DOI] [PubMed] [Google Scholar]
- Warren, C.M., P.R. Krzesinski, and M.L. Greaser. 2003. Vertical agarose gel electrophoresis and electroblotting of high-molecular-weight proteins. Electrophoresis. 24:1695–1702. [DOI] [PubMed] [Google Scholar]
- Wernyj, R.P., C.M. Ewing, and W.B. Isaacs. 2001. Multiple antibodies to titin immunoreact with AHNAK and localize to the mitotic spindle machinery. Cell Motil. Cytoskeleton. 50:101–113. [DOI] [PubMed] [Google Scholar]
- Xu, X., S.E. Meiler, T.P. Zhong, M. Mohideen, D.A. Crossley, W.W. Burggren, and M.C. Fishman. 2002. Cardiomyopathy in zebrafish due to mutation in an alternatively spliced exon of titin. Nat. Genet. 30:205–209. [DOI] [PubMed] [Google Scholar]
- Young, P., C. Ferguson, S. Banuelos, and M. Gautel. 1998. Molecular structure of the sarcomeric Z-disk: two types of titin interactions lead to an asymmetrical sorting of alpha-actinin. EMBO J. 17:1614–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}