

Abstract
The inner membrane of mitochondria is organized in two morphologically distinct domains, the inner boundary membrane (IBM) and the cristae membrane (CM), which are connected by narrow, tubular cristae junctions. The protein composition of these domains, their dynamics, and their biogenesis and maintenance are poorly understood at the molecular level. We have used quantitative immunoelectron microscopy to determine the distribution of a collection of representative proteins in yeast mitochondria belonging to seven major processes: oxidative phosphorylation, protein translocation, metabolite exchange, mitochondrial morphology, protein translation, iron–sulfur biogenesis, and protein degradation. We show that proteins are distributed in an uneven, yet not exclusive, manner between IBM and CM. The individual distributions reflect the physiological functions of proteins. Moreover, proteins can redistribute between the domains upon changes of the physiological state of the cell. Impairing assembly of complex III affects the distribution of partially assembled subunits. We propose a model for the generation of this dynamic subcompartmentalization of the mitochondrial inner membrane.
Introduction
Mitochondria are made up from two different types of noncontiguous membranes, the outer membrane (OM), and the inner membrane. The OM forms an envelope. It presents a barrier only for macromolecules, as it contains pore-forming proteins that allow the free passage of solutes up to a molecular mass of a few thousand Dalton. The inner membrane encloses the matrix space. It is a membrane in the strictest sense, as even small solutes like ions and metabolic substrates cannot pass through it without the help of carrier proteins. Further, it is one of the most protein-rich lipid bilayers in biological systems, with a protein/lipid mass ratio of ∼75:25 (Ardail et al., 1990; Simbeni et al., 1991). Various multisubunit protein complexes are located in this membrane, fulfilling several fundamental processes. The most abundant complexes are by far those involved in oxidative phosphorylation (OXPHOS). In addition, several other processes, such as protein translocation, metabolite exchange, protein assembly, iron–sulfur biogenesis, and protein degradation, take place in this membrane as well; proteins involved in inheritance of mitochondrial DNA, fusion and fission of mitochondria, and apoptosis are important for functionality of mitochondria.
The inner membrane can be subdivided in two morphologically and presumably functionally distinct subdomains. The first domain is the inner boundary membrane (IBM). It is closely apposed to the OM and can be considered as a second envelope structure. It interacts with the OM in many ways. In particular, it forms contact sites with the OM. These have been termed “morphological contact sites.” They appear to represent tight attachments, as they survive when mitochondria are subjected to procedures to separate outer and inner membranes by mechanical means (Reichert and Neupert, 2002). The OM and the IBMs also interact in functional terms. During import of proteins, the TOM and TIM complexes engage in close interaction (Schwaiger et al., 1987). ATP is exported from the matrix by the ADP/ATP carrier in conjunction with porin of the OM (Dolder et al., 2001). Outer and inner membrane are fusing and dividing in a coordinated manner (Okamoto and Shaw, 2005).
The second subdomain of the inner membrane is the cristae membrane (CM). In most mitochondria, it makes up the majority of the inner membrane surface, in particular in mitochondria of cells with a high energy demand, such as muscle cells. It forms invaginations of the IBM, in which two leaflets of inner membrane are juxtaposed to each other. In most cases, they form extended sheets. In some organisms or tissues, however, cristae have other shapes, such as tubules or fenestrated sheets (Munn, 1974; Mannella et al., 1994, 2001; Scheffler, 1999; Frey et al., 2002).
Other distinct substructures of the inner membrane are the cristae junctions. They connect the IBM with the cristae. In most types of mitochondria, these are narrow ring- or tubulelike structures, so small that they were proposed to form barriers between the intracristal space and the intermembrane space (Mannella et al., 1994, 2001; Frey et al., 2002). They were further proposed to undergo remodeling during apoptosis (Scorrano et al., 2002). Cristae junctions have been studied in mitochondria from various organisms by EM and electron tomography (Daems and Wisse, 1966; Perkins et al., 1997; Perkins et al., 1998; Frey and Mannella, 2000; Nicastro et al., 2000; Frey et al., 2002; Renken et al., 2002).
Despite the rather detailed insights into the composition, structure, function, and dynamics of mitochondria, the molecular basis of the structural and functional diversity of the inner membrane has remained elusive. Little is known about how proteins are distributed between the various subdomains of the inner membrane or the dynamics of their lateral movements. Even less is known about which proteins are responsible for the architecture of the various inner membrane substructures. Biogenesis and maintenance of subdomains are virtually a blank area on the landscape of mitochondrial biology. Attempts have been made to characterize the different parts of the inner membrane by biochemical means (Ohlendieck et al., 1986; Schwaiger et al., 1987; Pon et al., 1989; Rassow et al., 1989). Fractionation of submitochondrial membrane vesicles obtained by sonication and enzyme activity determinations were used, yet the information from such studies has remained rather limited, mostly because of the inability to correlate the fragments generated with their origin in the intact mitochondria.
In this study, we have used quantitative immuno-EM of intact yeast cells to characterize the various parts of the mitochondrial inner membrane. This appears to be the least distorting method to localize proteins in different subdomains of mitochondria. The high resolution of transmission EM allowed us to analyze wild-type yeast mitochondria that were reported to have a mean diameter of only 350 nm (Egner et al., 2002). We have combined this with the analysis of various processes in mitochondria, such as protein import, protein synthesis, and complex assembly. This was done not only to verify the morphological data but also to obtain insights into the mechanisms that determine mobility of proteins in the inner membrane. We show that each protein has a characteristic distribution between the various subcompartments of the inner membrane. We discuss these results in terms of the dynamic architecture of the subdomains, of mitochondrial functions in metabolism, and of mitochondrial biogenesis.
Results
Localization of proteins by quantitative immuno-EM
To determine the submitochondrial location of mitochondrial proteins, we used immunogold labeling of chemically fixed, cryosectioned yeast cells grown under respiratory conditions. The various steps of the procedure were optimized for obtaining sensitive low-background readout. The amount of antibodies added was kept very low to minimize unspecific labeling. For quantitative analysis, we plotted mitochondrially located gold particles found in hundreds of sections of mitochondria onto a single model representing part of the OM, the IBM, the CM, and the matrix (Fig. 1 A). For each protein, on average, ∼300 gold particles (174–1,254) were plotted, yielding a graphical representation of the mean distribution of the protein under investigation. We assigned each gold particle to one of the following three predefined zones: OM/IBM, CM, or background. The relative density was calculated after subtracting the background signal and taking into account that the length of the CM is, on average, 1.5-fold longer than that of the IBM under the growth conditions used in these experiments. Examples of raw data for immunogold labeling are presented for Core1, which is a subunit of complex III in the inner membrane, for the OM protein Tom20, and for matrix-targeted GFP (mtGFP; Fig. 1, B–F). Core1 is located predominantly in the CM (Fig. 1, B and D), consistent with its known role in OXPHOS, which is reported to take place predominantly in the CM in bovine heart mitochondria (Gilkerson et al., 2003). Tom20 shows a distribution that is consistent with its known location in the OM protein (Fig. 1, C and E). Also, the matrix location of the mtGFP is apparent (Fig. 1 F). Collectively, this type of representation of gold particles accumulated in silico allows the localization of mitochondrial proteins on a subcompartmental level.
Figure 1.

Localization of mitochondrial proteins by immuno-EM and in silico accumulation of gold particles onto an empiric model. S. cerevisiae wild-type (D273-10B) or matrix-targeted GFP-expressing cells were grown to early log phase in liquid complete media containing 2% lactate, chemically fixed, cryosectioned, and immunogold labeled. The location of gold particles found in mitochondria showing clearly resolvable CMs connected by cristae junctions to the IBM were plotted onto a single, empirically determined, drawn to scale model (see Materials and methods). (A) Model representing part of OM, IBM, CM, and matrix. The zones were defined as follows: OM/IBM, the center of a gold particle is ≤14 nm from the OM or IBM; CM, the center of a gold particle is ≤14 nm in distance from the CM and not in the OM/IBM zone; background, the center of a gold particle is neither in the OM/IBM nor the CM zone. Assignment to the matrix was done for gold particles that are in the background zone located on the matrix side of the inner membrane. (B and C) Exemplary alignment of the model with mitochondria that were immunogold labeled for Core1 or Tom20, respectively. Arrow points to gold particle plotted onto model shown. (D–F) Graphical representations of the distribution of Core1, Tom20, and mtGFP. Numbers at x and y axes represent distances in nanometers. Bars, 100 nm.
Because localization of proteins by immuno-EM is known to be fraught with unspecific effects, and because this method has to be validated for each organism in the first place, we performed a series of control experiments. First, when samples were analyzed by omitting primary antibodies and using only IgG-gold, virtually no gold particles were detected. Second, we decorated sections of cells in which the epitope to be detected was or was not present. Gold particles were detected only when the epitope was present. In a few cases, a background signal was found in the absence of the epitope, reaching up to 20% compared with the strain containing the epitope. Third, we tested several dilutions of individual antibodies. Within a certain range, the distribution of gold particles did not depend on the dilution used. Nevertheless, in all experiments, the lowest feasible antibody concentration was used to minimize background signals. Fourth, we tested the reproducibility of our method for several antibodies. In all cases, we observed very similar protein distributions with SDs of 7% or lower. Fifth, we analyzed proteins whose locations were limited to the OM and found a background of <15–20% in the matrix and the CM. Sixth, we expressed a protein in a manner that it was directed to different locations in mitochondria and found a background of ∼10–15%. Seventh, we analyzed the location of different proteins that are known to form a tight complex and observed a very similar distribution for them. Eighth, we analyzed the dynamic behavior of proteins that change their submitochondrial location during specific processes and found a consistent kinetic behavior. Data for these specificity criteria are presented in the experiments described in this study. In conclusion, we observed that the method used here for submitochondrial localization has a reliability of at least 80–85% and, thus, is superior to any biochemical subfractionation experiment in which additional sources of error attributable to in vitro manipulation of the organelles may grossly blur the analysis.
Submitochondrial location of Tim23 and Cox2
For further validation of the method used in this study, we analyzed the distribution of two proteins, Tim23 and Cox2, which were suspected to be enriched in the IBM or the CM, respectively. Tim23 is a component of the TIM23 translocase that functions in cooperation with the TOM complex. Thus, it should be located preferentially in the IBM. In three independent experiments using an antibody raised against the N terminus of Tim23 on a single preparation of fixed yeast cells, on average, 59.5 ± 3.2% (n 1 = 180; n 2 = 259; n 3 = 294; n total = 733) of gold particles per unit membrane length were found to be in the OM/IBM zone and, correspondingly, 40.5% were found to be in the CM (Fig. 2, A and E; Fig. S1 N, available at http://www.jcb.org/cgi/content/full/jcb.200605138/DC1). Therefore, Tim23 is enriched in the IBM, but is not exclusively located to this compartment under steady-state growth conditions during respiration. In addition, these data show that our approach is highly reproducible. Furthermore, a large proportion of the signal for the N terminus of Tim23 appears to be located at the outer face of the OM (Fig. 2 A; Fig. S1, N and O), supporting the reported two membrane–spanning topology of Tim23 with its N terminus crossing the OM (Donzeau et al., 2000). As a control, we determined the submitochondrial location of a fusion protein in which GFP was fused to the N terminus of Tim23. With this fusion protein we aimed to artificially anchor Tim23 stably and not dynamically to the OM. The fusion protein can fully replace the essential endogenous Tim23 protein, as indicated by its normal growth (unpublished data). First, we determined the submitochondrial location of GFP-Tim23 by biochemical means. Isolated mitochondria under isotonic conditions from the GFP-Tim23–expressing strain were either incubated with proteinase K or not, and then subjected to SDS-PAGE and immunoblotting. The N-terminal GFP part was degraded almost completely by the added protease, similar to the OM protein Tom40, whereas the inner membrane protein Aac2 was not (Fig. 2 B). The remaining fragment representing Tim23 lacking GFP was detected using an antibody raised against the Tim23 in proteinase K treated mitochondria (unpublished data). Thus, the N-terminal GFP moiety was largely located at the outer face of the OM and the inner membrane protein Tim23 is spanning the OM. Upon quantitative immuno-EM using an antibody raised against GFP 87.4% (n = 214) of gold particles per unit membrane length were found in the OM/IBM zone (Fig. 2, C and E). This is highly consistent with the aforementioned biochemical data and shows that GFP-Tim23 is highly enriched in the IBM. It further suggests that, in wild-type, the N terminus of Tim23 is dynamically, rather than stably, spanning the OM.
Figure 2.

Analysis and validation of subcompartmental localization of Tim23 and Cox2. (A) Distribution of Tim23 in wild-type cells under respiratory growth conditions was determined as described in Fig. 1 and Materials and methods. (B) Mitochondria from a GFP-Tim23–expressing strain were or were not treated with proteinase K and analyzed by Western blot analysis with indicated antibodies. (C) Distribution of GFP-Tim23 after immunogold labeling with antibodies raised against GFP. (D) Distribution of Cox2 in wild-type cells under respiratory growth conditions determined as described in Fig. 1. Numbers at x and y axes represent distances in nanometers. (E) Quantification of the distribution of Cox2, Tim23, and GFP-Tim23 in the OM/IBM and the CM zone. Signals were corrected for background and normalized per unit membrane length (see Materials and methods). For Cox2 and Tim23, the experiment was repeated three times using identical fixed cell material. Error bars represent the SD.
Cox2 is a subunit of cytochrome c oxidase (COX, complex IV) and is predicted to be present at the main site of OXPHOS. In three independent experiments using an antibody raised against the C terminus of Cox2 on a single preparation of fixed yeast cells, on average, 59.4 ± 6.8% (n 1 = 220; n 2 = 199; n 3 = 162; n total = 581) of all gold particles per unit membrane length were found to be in the CM zone and 40.6% were found in the OM/IBM zone (Fig. 2, D and E; Fig. S1 B). It appears that Cox2 is enriched in the CM, but it is not exclusively located in this subcompartment. Further, we checked whether the SD increased when yeast cells were grown and processed in separate experiments. In the three experiments performed, we observed 63.4 ± 3.9% (n 1 = 733; n 2 = 226; n 3 = 295; n total = 1,254) of Tim23 in the OM/IBM (Fig. S1 O; Table S1).
In conclusion, the method applied allows the localization of protein complexes in the subdomains of the inner mitochondrial membrane with high precision. Furthermore, Tim23 is preferentially located in the IBM and Cox2 is enriched in the CM, but both proteins do not show an exclusive location.
The CM is the major site of OXPHOS
In a further step, we determined the distribution of seven subunits from a total of four membrane-spanning protein complexes involved in OXPHOS under respiratory growth conditions. All subunits were found to be enriched in CMs (Fig. 3 A). The highest enrichment observed for any protein in this study was that for Core1 (complex III), showing 67.1% (n = 357) of gold particles per unit membrane length in the CM zone (Fig. 3 A and Fig. 1 D). Another subunit of the same complex, Core2, showed a very similar distribution of gold particles in the CM (66.6%; n = 266; Fig. 3 A; Fig. S1 Q). The subunit Cox2 of the cytochrome c oxidase (complex IV), and two subunits (Su e and Su g) of the F1FO-ATP synthase (complex V), the subunit Rip (Fe/S-Rieske protein; complex III), and the ADP/ATP carrier were found enriched in the CM to a similar extent (Fig. 3 A; Fig. S1). Therefore, the CM is the major site of OXPHOS, albeit not the exclusive one. Complex I is not present in Saccharomyces cerevisiae and therefore could not be localized. For the F1FO-ATP synthase, we used two inner membrane subunits (Su e and Su g) that are required for dimerization and oligomerization of monomers of F1FO-ATP synthase and that are present in the dimeric and oligomeric forms of this complex (Arnold et al., 1998). The majority of subunits e and g were found in the CM (55.4%, n = 375 and 60.1%, n = 327, respectively; Fig. 3 A; Fig. S1, vK and L) suggesting that F1FO-ATP synthase, the oligomeric forms in particular, are predominantly located in the CM. Again, two subunits of the same oligomeric complex showed a similar distribution.
Figure 3.

Subcompartmental organization of mitochondrial proteins. Quantification of the distribution of mitochondrial proteins in wild-type cells under respiratory growth conditions was performed as described in Figs. 1 and 2 and Materials and methods. (A) Proteins involved in OXPHOS. (B) Proteins involved in the insertion of nuclear- and mitochondrial-encoded proteins.
Insertion of nuclear- and mitochondrial-encoded proteins into the inner membrane
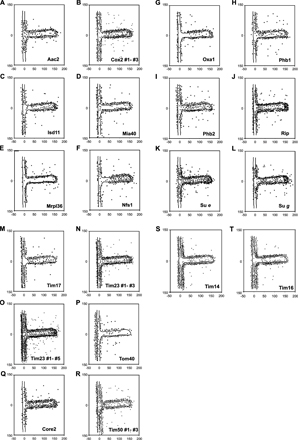
We addressed the question of where nuclear-encoded proteins imported from the cytosol and mitochondrial-encoded proteins from the matrix are inserted into the inner membrane. First, we determined the distribution of proteins involved in mitochondrial biogenesis: Tom20, Tom40, Tim50, Tim14, Tim16, Tim23, Tim17, Mia40, Oxa1, and Mrpl36 (Fig. 3 B; Fig. 1 E; Fig. 2 A; and Fig. S1, D, E, G, M–P, and R–T). The aforementioned enrichment of the subunit Tim23 of the TIM23 complex was corroborated by sublocalization of Tim17, Tim50, Tim14, and Tim16 (Fig. 3 B; Fig. S1, M and R–T). These other essential and tightly associated subunits of the same complex, showed a very similar extent of enrichment in the IBM (59.4%, n = 217) of gold particles per unit membrane length in the OM/IBM zone for Tim17, 59.5% (n = 733) for Tim23, 66.1 ± 6.5% (n 1 = 240; n 2 = 280; n 3 = 333; n total = 853) for Tim50, 66.9% (n = 875) for Tim14, and 64.2% (n = 847) for Tim16. Not surprisingly, both subunits of the TOM complex analyzed, Tom20 and Tom40, are strongly enriched in the OM, reaching 84.9% (n = 213) and 77.2% (n = 179), respectively, of gold particles per unit membrane length in the OM/IBM zone (Fig. 1, E; Fig. 3 B; and Fig. S1 P).
An enrichment in the CM was observed for the mitochondrial large ribosomal subunit protein Mrpl36 and for Oxa1, showing 59.4% (n = 196) and 58.9% (n = 174) of gold particles per unit membrane length in the CM zone, respectively (Fig. 3 B; Fig. S1, E and G). Ribosomes bind to the C terminus of Oxa1 (Jia et al., 2003; Szyrach et al., 2003), explaining their similar distribution. This is also consistent with a much earlier report showing that mitochondrial ribosomes in yeast are present in the matrix, near the CM, and aligned at the IBM (Watson, 1972). Collectively, these results support the view that mitochondrially translated proteins are synthesized preferentially close to the cristae and are also inserted at this location in the inner membrane.
Mia40 is the initial import receptor for small cysteine-containing proteins in the intermembrane space of mitochondria (Chacinska et al., 2004; Mesecke et al., 2005; Terziyska et al., 2005). Mia40 was found to be enriched in the IBM, showing 55.5% (n = 214) of gold particles per unit membrane length in this subcompartment (Fig. 3 B; Fig. S1 D). This is in line with the proposed role of this protein in early trapping of incoming substrate proteins by disulfide bridge formation.
Distribution of components of various pathways in mitochondria
A physiological process that renders mitochondria essential for cell viability is iron–sulfur cluster biogenesis. This process involves several matrix-located proteins (for review see Lill and Kispal, 2000). We determined whether iron–sulfur biogenesis generally occurs near one of the subcompartments of the inner membrane. Two proteins involved in this process were analyzed Isd11 and Nfs1. These two proteins were reported to fractionate with membrane vesicles after sonication of isolated mitochondria at low salt concentrations (Adam et al., 2006; Wiedemann et al., 2006). Immuno-EM demonstrated both proteins to be closely attached to the inner membrane and enriched in the CM (Fig. 4 A; Fig. S1, C and F). For Isd11 57.1% (n = 200) and for Nfs1 66.0% (n = 204) of gold particles per unit membrane length are found in the CM zone. This suggests that at least some steps in iron–sulfur biogenesis occur predominantly at the CM.
Figure 4.

Subcompartmental localization of Mgm1 involved in mitochondrial fusion. Quantification of the distribution of mitochondrial proteins in wild-type cells under respiratory growth conditions was as described in Figs. 1 and 2 and Materials and methods. (A) Proteins involved in iron–sulfur biogenesis, Isd11 and Nfs1. (B) Proteins involved in protein degradation in mitochondria, prohibitin 1 (Phb1) and prohibitin 2 (Phb2). (C) Distribution of the mitochondrial fusion protein Mgm1. Inset, quantification of Mgm1 density in the OM/IBM and the CM zone. Numbers at x and y axes represent distances in nanometers.
We investigated the submitochondrial location of components involved in protein degradation. Prohibitin 1 (Phb1) and 2 (Phb2) are not proteases themselves, but they were reported to be in a supercomplex with the m-AAA protease Yta10/Yta12 (Steglich et al., 1999). We observed for Phb1 58.0% (n = 210) and for Phb2 52.0% (n = 250) of gold particles per unit membrane length in the CM (Fig. 4, B; Fig. S1, H and I). Phb1 appears more enriched in the CM than Phb2, which almost seems evenly distributed within the inner membrane. No immunogold signal was observed for Phb1 in a Δphb1 strain and for Phb2 in a Δphb2 strain showing the high specificity of each antibody. Because both subunits are in the same complex with the m-AAA protease Yta10/Yta12, we propose that protein degradation mediated by this complex occurs both in the CM and the IBM.
Mgm1 is a dynamin-like protein in mitochondria that is required for mitochondrial fusion (Sesaki et al., 2003; Wong et al., 2003). It exists in two isoforms, both of which are required for function (Herlan et al., 2003, 2004), and was found to interact with two OM proteins, Fzo1 and Ugo1, which are both essential for mitochondrial fusion themselves (Sesaki et al., 2003; Wong et al., 2003). Using an antibody recognizing both isoforms, we found Mgm1 to be enriched in the IBM, showing 61.2% (n = 317) of gold particles per unit membrane length (Fig. 4 C). This is in line with the proposed function of Mgm1 in mitochondrial fusion, and furthermore, suggests a role in coordinating the fusion of the outer and the inner membranes.
Dynamics of location of mitochondrial protein complexes
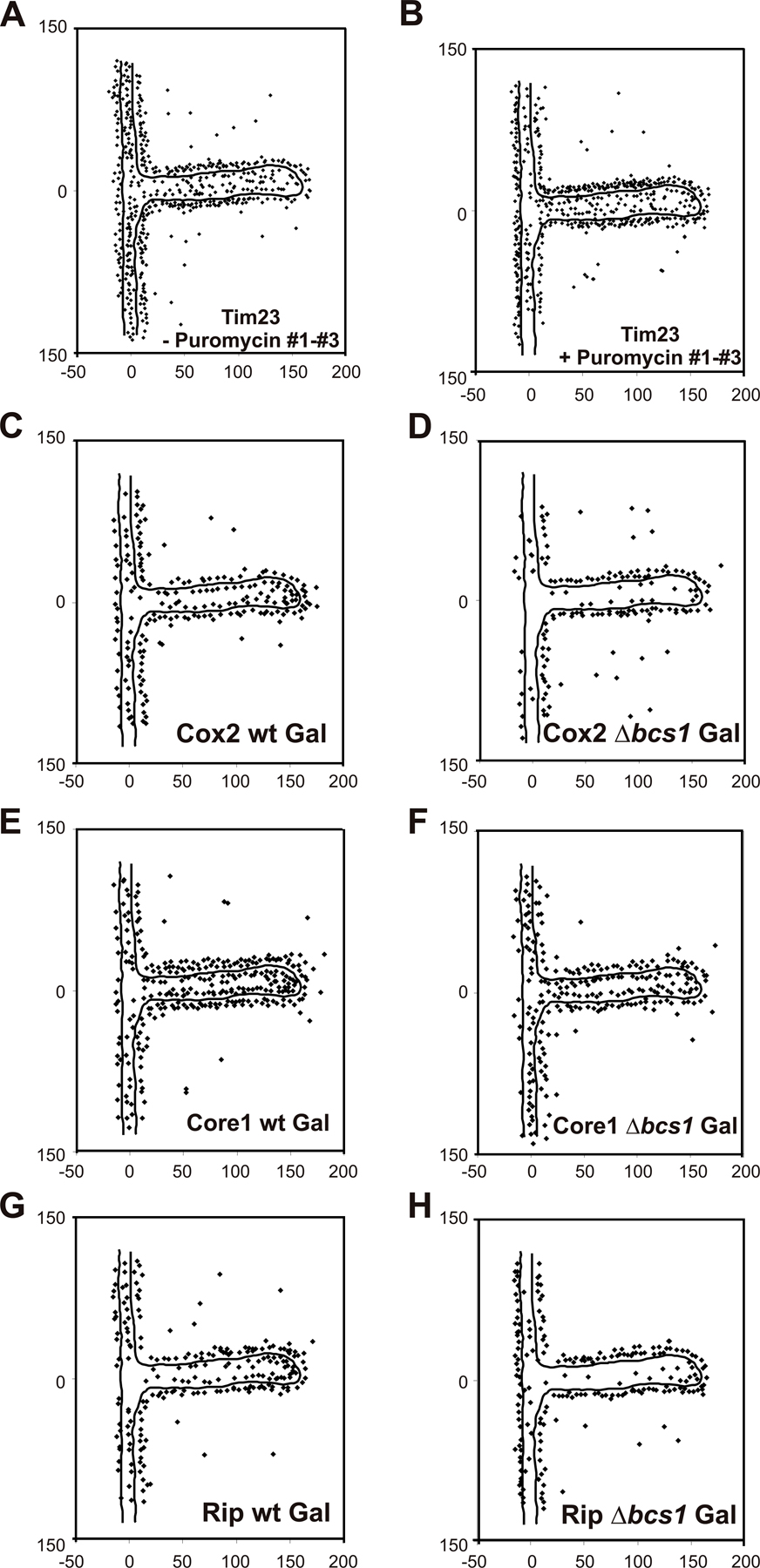
Tim23 in yeast was proposed to span the outer and the inner mitochondrial membranes (Donzeau et al., 2000). This raises the question of whether this topology is static or dynamic. We asked whether Tim23 can redistribute between the two subcompartments depending on the activity of the protein import machinery. We treated a yeast strain overexpressing mtGFP with the translation inhibitor puromycin and followed the distribution of Tim23 by quantitative immuno-EM. When mtGFP was overexpressed, Tim23 was enriched in the OM/IBM zone showing 67.2% ± 3.1 (n 1 = 197; n 2 = 366, n 3 = 378; n total = 941) of gold particles in this zone. This enrichment was slightly higher than under steady-state conditions in wild-type cells, without overexpressing a mitochondrial protein (see above). Upon inhibition of protein translation and, consequently, protein import, Tim23 redistributed from the IBM to the CM (Fig. 5 A; and Fig. S2, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200605138/DC1). After 30 min of puromycin treatment, the density of Tim23 was nearly equal for the IBM (52.4 ± 2.6%; n 1 = 180; n 2 = 248, n 3 = 246; n total = 674) and the CM (47.6 ± 2.6% ; n 1 = 180; n 2 = 248, n 3 = 246; n total = 674). The distribution of an inner membrane protein between IBM and CM is apparently dynamic, and changes with the physiological state of mitochondria. Furthermore, it suggests that the CM can act as a reservoir for proteins that otherwise mainly fulfill their function in close apposition to the OM.
Figure 5.

Dynamic redistribution of mitochondrial proteins involved in protein translocation and synthesis. Cells overexpressing mtGFP were or were not treated with puromycin for 30 min before fixation and immuno-EM. (A) Quantification of Tim23 distribution before or after 30 min of puromycin treatment. The experiment was repeated three times using identical fixed cell material. Error bars represent the SD. (B) Distribution of Mrpl36 before the addition of puromycin. (C) Distribution of Mrpl36 30 min after the addition of puromycin. (D) Location of Mrpl36 in the OM/IBM, CM, or matrix before and 30 min after the addition of puromycin. Numbers at x and y axes represent distances in nanometers.
We asked whether membrane association of mitochondrial ribosomes is also dynamic. In the same experiment, we followed Mrpl36, which is a ribosomal protein of the large subunit of mitochondrial ribosomes. After puromycin treatment, a partial detachment of this protein from the inner mitochondrial membrane was observed, accompanied by an increase in the matrix (Fig. 5, B–D). This is consistent with a recent study showing that upon addition of puromycin only a fraction of Mrpl36 remained associated with the inner membrane (Ott et al., 2006). Our observation suggests that the entire mitochondrial ribosome, the large ribosomal subunit, or Mrpl36 alone partially detaches from the inner membrane upon block of translation and release of nascent chains.
Localization of assembly intermediates of respiratory complex III and IV
Where in the inner membrane do newly inserted subunits become associated to form assembly intermediates and, eventually, complexes and supracomplexes? To address these questions, we analyzed the submitochondrial distribution of partially assembled complexes in a mutant with a deficiency in the formation of complex III. That mutant lacked the factor Bcs1, which is required for a late assembly step of complex III and for formation of the respiratory chain supracomplex III2IV2 (Cruciat et al., 1999, 2000). Mutant strains and wild-type control were grown on fermentable carbon source, as deficiency of fully assembled complex III leads to the inability to grow on nonfermentable carbon sources. Quantitative immuno-EM was performed using antibodies against Rip and Core1 (both complex III), and against Cox2 (complex IV). In wild-type cells, the Core1 and Rip proteins were found strongly enriched in the CM, with only 29.9% (n = 292) and 38.3% (n = 214) of gold particles per unit membrane length in the IBM (Fig. 6; Fig. S2, E and G). In the Δbcs1 deletion mutant, both the Core1 protein, which is present in the partially assembled complex III, and the nonassembled Rip protein were found less enriched in the CM. 40.3% (n = 237) and 47.2% (n = 202) of Core1 and Rip protein, respectively, per unit membrane length were present in the IBM (Fig. 6; Fig. S2, F and H). Apparently, both subunits accumulated more in the IBM when assembly of complex III was impaired. On the other hand, Cox2 was more enriched in the CM in the Δbcs1 deletion strain (65.3%; n = 153), as compared with the wild-type strain (54.7%; n = 205; Fig. 6 and Fig. S2, C and D). In the case of complex III, unassembled or partially assembled subunits accumulate in the IBM, but after full assembly, of complex III they end up mostly in the CM. When the supracomplex of III and IV was not formed, the assembled complex IV accumulated in the CM. This may be explained by the fact that less complex III is present in the CM, and therefore more space is available for complex IV. In summary, we show that assembly intermediates and fully assembled proteins of a respiratory complex distribute differently between the subcompartments. The specific location of single subunits may influence or regulate the assembly of such complexes. Further, these results suggest that assembly of respiratory complexes involves migration in the inner membrane.
Figure 6.

Subcompartmental distribution of assembly intermediates of complex III and IV. Wild-type and Δbcs1 cells were grown on complete media containing 2% galactose, chemically fixed, cryosectioned, and immunogold labeled for Cox2, Core1, and Rip. Quantification of the distribution of mitochondrial proteins was as described in Figs. 1 and 2 and Materials and methods. In the Δbcs1 strain, complex III is only partially assembled, and the supracomplexes III2IV2 are not formed.
Discussion
The inner membrane of mitochondria consists of two morphologically distinct subdomains. One subdomain, the IBM, is tightly apposed to the OM, and the other one forms the CM. These two domains are connected by narrow structures called the cristae junctions. We have addressed the question of whether the morphological differences are reflected in the protein composition of the two domains. For this we have established a procedure for quantitative immuno-EM using yeast cells, an approach that is the least disturbing to the native organization of the membranes. We report on the localization of 20 different proteins in the two subdomains of the inner membrane. We show that the proteins indeed have different characteristic distributions between IBM and CM. For instance, the five subunits of the TIM23 complex investigated were more abundant, relative to unit membrane length, in the IBM than in the CM. This is consistent with the reported two membrane–spanning topology of Tim23 (Donzeau et al., 2000) and earlier reports on the presence of translocation contact sites (Schwaiger et al., 1987; Pon et al., 1989; Rassow et al., 1989; Schulke et al., 1997). Our data further corroborate earlier studies, using both similar and different approaches, such as biochemical fractionation or immunocytochemistry, regarding the preferential location of complexes involved in OXPHOS within the CM (Perotti et al., 1983; Ohlendieck et al., 1986; Gilkerson et al., 2003). The level of respiratory complex III in our study was roughly twofold higher in the CM than in the IBM. This is similar to the reported 2.2–2.6-fold higher density in CM, as compared with IBM of bovine heart mitochondria (Gilkerson et al., 2003).
At least two major possibilities can be envisaged to explain how such diversity is generated and maintained at a molecular level. One is that IBM and CM are two distinct membrane compartments that are separated by the rather narrow cristae junctions, which might serve as strict barriers for lateral diffusion of membrane components, as suggested for ions, metabolites, and small proteins in the intermembrane space (Mannella et al., 1994, 2001; Frey et al., 2002). A second explanation is that there are no such barriers, or only selective barriers or filters that allow more or less free lateral diffusion, and that diversity is generated in a different way. Possible mechanisms might involve differential topological effects, as the IBM is only facing the OM, whereas the CMs are facing each other.
Our results would favor, perhaps surprisingly, the second explanation. First, the distributions observed in our study are, in quantitative terms, not all or none. Rather, all proteins investigated are present in both domains of the inner membrane at different levels. Second, components of the mitochondrial protein import machinery are preferentially found in the IBM and are known to interact with components involved in import in the OM, specifically the TOM complex. Components of the OXPHOS are, not surprisingly, present at higher levels in the CM, which is apparently generated for accommodating these abundant complexes and supracomplexes. Third, our experiments demonstrate a dynamic behavior of certain components in the context of their function. Tim23, which is preferentially present in the IBM under normal conditions, redistributes into the CM when it is no longer engaged in the translocation of polypeptide chains.
What could be the mechanisms that are responsible for establishing an uneven but dynamic distribution between IBM and CM? In cases of proteins localized preferentially in the IBM, contacts of a direct or indirect nature would be a way to generate sequestration of proteins. Indeed, Tim23 in yeast has been shown to interact with the OM with a peripheral segment (Donzeau et al., 2000), thereby generating a bias toward a preferential location in the IBM. As Tim23 interacts tightly with Tim17, the latter component is also maintained in the IBM. By a hierarchy of interactions, whole pathways could be recruited to the IBM. Such interactions have also been established for components of the mitochondrial fusion and fission machinery. For instance, Mgm1 was reported to contact Fzo1 and Ugo1, which are OM proteins involved in the fusion of mitochondria (Sesaki et al., 2003; Wong et al., 2003). Further organizing determinants of the IBM are the morphological contact sites that link it tightly to the OM. These types of contacts are observed in electron micrographs of virtually all mitochondria investigated (van der Klei et al., 1994; Reichert and Neupert, 2002). They appear to play a major role, as they guarantee the presence of permanent linkages and may help to bring together components of both membranes that have to find each other. Unfortunately, the protein components involved have not been identified thus far.
For the cristae, a different principle of stabilizing organization can be proposed. The main components of the CM are the complexes of OXPHOS that associate with each other to form supracomplexes of megadalton molecular mass. A specific role has been assigned in various studies to the F1FO–ATP synthase. This large complex was suggested to form oligomeric supracomplexes that are required for cristae formation (Giraud et al., 2002; Paumard et al., 2002; Bornhovd et al., 2006). In this way, a scaffold would be created to keep not only the F1FO-ATP synthase but also respiratory complexes in the cristae. Other proteins may join this macromolecular network, retaining themselves in the CM. Such an organization could trap supracomplexes within the CM because of their high molecular weight and prevents them from diffusion to the IBM through narrow cristae junctions.
A major conclusion from our study is that both subcompartments are dynamic in the sense that distribution of proteins can change upon changes of the physiological state of the cell. The observed dynamic changes are correlated with the function of the proteins investigated. For instance, Tim23 redistributed from its primary site of action during protein translocation, the IBM, to the CM when protein synthesis and consequently protein import were stopped. In such a situation, the cristae could act as a reservoir for proteins not needed for activity in the IBM. Also the mitochondrial ribosomal protein Mrpl36 moved from its site of action, in this case, from the CM to the matrix, when nascent chains were released from ribosomes.
The observed dynamic nature of the subcompartmentalization has particular relevance in the context of the biogenesis of mitochondria. Newly synthesized proteins are imported into the mitochondrial inner membrane from the cytosol and from the matrix. The complexes involved in OXPHOS, which make up the larger part of the inner membrane proteins, are made from ribosomes present both outside and inside mitochondria. Thus, questions of where they are inserted into the membranes and where they meet to assemble to functional complexes arise. Our data show that the insertion of imported proteins does occur preferentially into the IBM, and that the insertion of mitochondrial translation products occurs mostly into the CM. On the other hand, movement of assembly intermediates between the subcompartments was suggested in our experiments. This again suggests that there are not exclusive sites of complex formation. The lateral movement of assembly intermediates may be facilitated by assembly factors described in mitochondria that shield and protect aggregation-prone surfaces. Furthermore, the preferential accumulation of certain subunits of complex III and IV assembly intermediates correlates well with the location of the components responsible for their import. This suggests that the site of insertion of individual subunits also plays a role, possibly a kinetic one, in the pathway of multisubunit complex formation.
The structure and function of cristae junctions deserves particular attention in further attempts to understand the organization of the mitochondrial inner membrane. They apparently are not absolute barriers, but they could well represent kinetic or diffusion barriers for individual proteins and mark as specific gates. In the future, it is of the utmost importance to learn whether they influence the traffic of proteins between the domains of the inner membrane.
Materials and methods
Strains and growth conditions
S. cerevisiae strains used are wild-type D273-10B; wild-type W303 (Mat α, leu2, trp1, ura3, his3, and ade2) strain; Tim23/Δtim23 (Mat a/α, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, ura3Δ0/ura3Δ0, LYS2/lys2Δ0, MET15/met15Δ0, and TIM23/tim23∷Kan; Open Biosystems); Δbcs1 (W303, Mat α, leu2, trp1, ura3, ade2, and Bcs1∷HIS3; Cruciat et al., 1999); and Oxa1-331 (YPH499, Mat α ade2-1, his3-1,15, leu2-3,112, trp1-1, and ura3-1; Szyrach et al., 2003). Δtim23 + pRS315-GFP-Tim23 (Mat α, his3Δ1 leu2Δ0, ura3Δ0, LYS2, met15Δ0, tim23∷Kan). Culturing of yeast was performed according to standard protocols at 30°C on complete liquid media containing 2% lactate or 2% galactose. Mitochondria were prepared according to Sirrenberg et al. (1996).
Recombinant DNA techniques and plasmid constructions
The GFP-Tim23 construct was obtained by PCR amplification of the complete open reading frame from genomic yeast DNA using the primers Tim23up (5′-AAAGGATCCATGTCGTGGCTTTTTGGAGA-3′) and Tim23down (5′-CCCAAGCTTTCATTTTTCAAGTAGTCTTTTCTTGACAC-3′). The Tim23-promoter region was amplified by PCR using the primers Prom23up (5′-CCTGAGCTCACTGTGACGTCG-3′) and Prom23down (5′-CCCGGTACCGATTGTGTGTGATCTGTTAAAC-3′) on genomic yeast DNA. To amplify the GFP pYX242 mtGFP (Westermann and Neupert, 2000) was used as a template. The primers GFPup (5′-CCCCGGTACCATGAGTAAAGGAGAAGAAC-3′) and GFPdown (5′-CCCCGGATCCTGCTCCTGCTGATCCTCCTTTGTATAGTTCATCCATGC-3′). The resulting PCR-fragments were cloned into pRS315 yeast expression vector in three single steps. The correctness was confirmed by DNA sequencing, and the plasmid was transformed into the heterozygous diploid Tim23/Δtim23 strain. The transformed strain was sporulated, and haploid spores were tested for expression of GFP-Tim23 and used for further analysis.
Quantitative immuno-EM
Mitochondrial membrane proteins were localized by postembedding immunogold labeling of chemically fixed cryosectioned whole yeast cells. Wild-type (D273-10B) cells were grown on complete liquid media containing 2% lactate at 30°C (if not stated differently) and fixed during early log phase in a mixture of freshly prepared 4% formaldehyde and 0.5% glutaraldehyde in 0.1 M sodium citrate buffer adjusted to growth conditions for temperature and pH. Cells were washed with phosphate buffered saline, incubated with 1% sodium metaperiodate for 1 h, immersed in 25% polyvinyl-pyrrolidone (PVP, K15/MW 10000; Fluka) and 1.6 M sucrose (Tokuyasu, 1989) at 30°C for 2–3 h, mounted on specimen holders, frozen in liquid nitrogen, and sectioned at −115°C with an ultracryotome Ultracut S attached with a FCS unit (Leica). Ultrathin, thawed cryosections were prepared with glass/diamond knives and placed on formvar/carbon-coated copper grids (200 mesh, hexagonal). Labeling with IgG primary antibodies and secondary antibody–gold complexes (10 nm; Dianova) was performed as previously described (Kärgel et al., 1996). Finally, the sections were stained and stabilized by a 1:1 mixture of 3% tungstosilicic acid hydrate (Fluka) and 2.5% polyvinyl alcohol (MW 10000; Sigma-Aldrich), and analyzed by standard transmission EM. Specific epitope recognition was ensured by initially optimizing the dilution of each antibody used in such a way that mitochondria were still labeled but giving no or negligible cytosolic background (see Table S2, available at http://www.jcb.org/cgi/content/full/jcb.200605138/DC1, for details). Because labeling occurs only at the surface of the 50–70-nm-thick sections, and because of the low antibody concentration, only a weak signal for endogenously expressed proteins is found. To overcome this disadvantage, we developed a novel tool in which an in silico accumulation of signals onto a single draw-to-scale model representing part of OM, IBM, and CM (Fig. 1 A) was performed. The location of all gold-particles found in mitochondria showing clearly resolvable CMs connected by cristae junctions to the IBM on electron micrographs (22,000× enlargement) were transferred to this model using a microfiche device with 24× enlargement. For each antigen, on average, 250 gold particles were plotted, producing a graphical representation of the average distribution of the protein under investigation. To calibrate the positions of the gold particles, a point of origin and an internal size standard was used. To digitalize the distribution of the gold-particles, Photoshop CS2 (Adobe) and Image J (National Institutes of Health) programs were used. The calibrated x/y-values for each gold particle were used to reconstruct the particle distribution with the program Delta Graph 4.0.1. The result of this procedure is a statistically significant distribution-pattern of the mitochondrial protein investigated. The accuracy of the method is in the range of 10 nm. The quantification and assignment to one of the three predefined zones was done with Excel 2002 (Microsoft). Assignment to the matrix was done for gold particles that are in the background zone, but only those located on the matrix side of the inner membrane. Because mostly integral membrane proteins were analyzed, the background signal for each antibody in the OM/IBM and the CM zone was assumed to be represented by those gold particles found experimentally in the background zone. Therefore, we subtracted the number of gold particles expected to be background of a particular zone from the number of gold particles found in this zone. The relative areas of the OM/IBM, CM, and background zones in our model are 1.23 to 1.0 to 3.48 and were taken into account in this background subtraction. From this the number of gold particles per unit, membrane length of CM or OM/IBM was calculated, taking into account that the length of the CM in these sections was, on average, 1.5-fold longer than that of the IBM under the growth conditions used in these experiments. For galactose-grown cells, the length of the CM was identical to the length of the IBM. Finally, the relative density of gold particles in the CM or the OM/IBM (percentage of gold particles per unit membrane length) can be obtained by dividing the number of gold particles in the CM or the OM/IBM, respectively, by the sum of the densities of gold particles in the CM and the OM/IBM. Therefore, by first subtracting the normalized background determined for each experiment and normalizing for membrane length, we obtain a normalized percentage of gold particles per OM/IBM or CM membrane length. This allows us to compare numerous different proteins that vary markedly in their expression level and/or their reactivity with their corresponding antibody, in respect to a possible enrichment in one or the other subcompartment.
Puromycin treatment of yeast cells
Wild-type cells (w303α) transformed with the mtGFP expression plasmid pVT100U-mtGFP (Westermann and Neupert, 2000) were grown on selective liquid media containing 2% lactate. In the exponential growing phase, 100 μg/ml puromycin was added for various time periods before fixation of the cells.
Online supplemental material
Fig. S1 shows the distribution of gold particles after immuno-EM of mitochondrial proteins. Fig. S2 shows the dynamics of the distribution of mitochondrial proteins. Table S1 shows the quantitative immuno-EM of mitochondrial proteins. Table S2 shows the antibodies and dilutions used.
Supplementary Material
Acknowledgments
We thank Christiane Kotthoff and Margit Vogel for excellent technical assistance, and Drs. Thomas Langer, Kai Hell, Doron Rapaport, Johannes Herrmann, Stefan Paschen, and Gareth Griffiths for helpful discussions and/or antibodies.
This work was supported by the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich SFB 594/project B8, and by the Fonds der Chemischen Industrie.
F. Vogel and C. Bornhövd contributed equally to this paper.
Abbreviations used in this paper: CM, cristae membrane; IBM, inner boundary membrane; mtGFP, matrix targeted GFP; OM, outer membrane; OXPHOS, oxidative phosphorylation.
References
- Adam, A.C., C. Bornhövd, H. Prokisch, W. Neupert, and K. Hell. 2006. The Nfs1 interacting protein Isd11 has an essential role in Fe/S cluster biogenesis in mitochondria. EMBO J. 25:174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardail, D., J.P. Privat, M. Egret-Charlier, C. Levrat, F. Lerme, and P. Louisot. 1990. Mitochondrial contact sites. Lipid composition and dynamics. J. Biol. Chem. 265:18797–18802. [PubMed] [Google Scholar]
- Arnold, I., K. Pfeiffer, W. Neupert, R.A. Stuart, and H. Schagger. 1998. Yeast mitochondrial F1F0-ATP synthase exists as a dimer: identification of three dimer-specific subunits. EMBO J. 17:7170–7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornhovd, C., F. Vogel, W. Neupert, and A.S. Reichert. 2006. Mitochondrial membrane potential is dependent on the oligomeric state of F1FO-ATP synthase supracomplexes. J. Biol. Chem. 281:13990–13998. [DOI] [PubMed] [Google Scholar]
- Chacinska, A., S. Pfannschmidt, N. Wiedemann, V. Kozjak, L.K. Sanjuan Szklarz, A. Schulze-Specking, K.N. Truscott, B. Guiard, C. Meisinger, and N. Pfanner. 2004. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J. 23:3735–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciat, C.M., K. Hell, H. Folsch, W. Neupert, and R.A. Stuart. 1999. Bcs1p, an AAA-family member, is a chaperone for the assembly of the cytochrome bc(1) complex. EMBO J. 18:5226–5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciat, C.M., S. Brunner, F. Baumann, W. Neupert, and R.A. Stuart. 2000. The cytochrome bc1 and cytochrome c oxidase complexes associate to form a single supracomplex in yeast mitochondria. J. Biol. Chem. 275:18093–18098. [DOI] [PubMed] [Google Scholar]
- Daems, W.T., and E. Wisse. 1966. Shape and attachment of the cristae mitochondriales in mouse hepatic cell mitochondria. J. Ultrastruct. Res. 16:123–140. [DOI] [PubMed] [Google Scholar]
- Dolder, M., S. Wendt, and T. Wallimann. 2001. Mitochondrial creatine kinase in contact sites: interaction with porin and adenine nucleotide translocase, role in permeability transition and sensitivity to oxidative damage. Biol. Signals Recept. 10:93–111. [DOI] [PubMed] [Google Scholar]
- Donzeau, M., K. Kaldi, A. Adam, S. Paschen, G. Wanner, B. Guiard, M.F. Bauer, W. Neupert, and M. Brunner. 2000. Tim23 links the inner and outer mitochondrial membranes. Cell. 101:401–412. [DOI] [PubMed] [Google Scholar]
- Egner, A., S. Jakobs, and S.W. Hell. 2002. Fast 100-nm resolution three-dimensional microscope reveals structural plasticity of mitochondria in live yeast. Proc. Natl. Acad. Sci. USA. 99:3370–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey, T.G., and C.A. Mannella. 2000. The internal structure of mitochondria. Trends Biochem. Sci. 25:319–324. [DOI] [PubMed] [Google Scholar]
- Frey, T.G., C.W. Renken, and G.A. Perkins. 2002. Insight into mitochondrial structure and function from electron tomography. Biochim. Biophys. Acta. 1555:196–203. [DOI] [PubMed] [Google Scholar]
- Gilkerson, R.W., J.M. Selker, and R.A. Capaldi. 2003. The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett. 546:355–358. [DOI] [PubMed] [Google Scholar]
- Giraud, M.F., P. Paumard, V. Soubannier, J. Vaillier, G. Arselin, B. Salin, J. Schaeffer, D. Brethes, J.P. di Rago, and J. Velours. 2002. Is there a relationship between the supramolecular organization of the mitochondrial ATP synthase and the formation of cristae? Biochim. Biophys. Acta. 1555:174–180. [DOI] [PubMed] [Google Scholar]
- Herlan, M., C. Bornhövd, K. Hell, W. Neupert, and A.S. Reichert. 2004. Alternative topogenesis of Mgm1 and mitochondrial morphology depend on ATP and a functional import motor. J. Cell Biol. 165:167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlan, M., F. Vogel, C. Bornhövd, W. Neupert, and A.S. Reichert. 2003. Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J. Biol. Chem. 278:27781–27788. [DOI] [PubMed] [Google Scholar]
- Jia, L., M. Dienhart, M. Schramp, M. McCauley, K. Hell, and R.A. Stuart. 2003. Yeast Oxa1 interacts with mitochondrial ribosomes: the importance of the C-terminal region of Oxa1. EMBO J. 22:6438–6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kärgel, E., R. Menzel, H. Honeck, F. Vogel, A. Bohmer, and W.H. Schunck. 1996. Candida maltosa NADPH-cytochrome P450 reductase: cloning of a full-length cDNA, heterologous expression in S. cerevisiae and function of the N-terminal region for membrane anchoring and proliferation of the endoplasmic reticulum. Yeast. 12:333–348. [DOI] [PubMed] [Google Scholar]
- Lill, R., and G. Kispal. 2000. Maturation of cellular Fe-S proteins: an essential function of mitochondria. Trends Biochem. Sci. 25:352–356. [DOI] [PubMed] [Google Scholar]
- Mannella, C.A., M. Marko, P. Penczek, D. Barnard, and J. Frank. 1994. The internal compartmentation of rat-liver mitochondria: tomographic study using the high-voltage transmission electron microscope. Microsc. Res. Tech. 27:278–283. [DOI] [PubMed] [Google Scholar]
- Mannella, C.A., D.R. Pfeiffer, P.C. Bradshaw, I.I. Moraru, B. Slepchenko, L.M. Loew, C.E. Hsieh, K. Buttle, and M. Marko. 2001. Topology of the mitochondrial inner membrane: dynamics and bioenergetic implications. IUBMB Life. 52:93–100. [DOI] [PubMed] [Google Scholar]
- Mesecke, N., N. Terziyska, C. Kozany, F. Baumann, W. Neupert, K. Hell, and J.M. Herrmann. 2005. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 121:1059–1069. [DOI] [PubMed] [Google Scholar]
- Munn, E.A. 1974. The Structure of Mitochondria. Academic Press, London, New York. 465 pp.
- Nicastro, D., A.S. Frangakis, D. Typke, and W. Baumeister. 2000. Cryo-electron tomography of neurospora mitochondria. J. Struct. Biol. 129:48–56. [DOI] [PubMed] [Google Scholar]
- Ohlendieck, K., I. Riesinger, V. Adams, J. Krause, and D. Brdiczka. 1986. Enrichment and biochemical characterization of boundary membrane contact sites from rat-liver mitochondria. Biochim. Biophys. Acta. 860:672–689. [DOI] [PubMed] [Google Scholar]
- Okamoto, K., and J.M. Shaw. 2005. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu. Rev. Genet. 39:503–536. [DOI] [PubMed] [Google Scholar]
- Ott, M., M. Prestele, H. Bauerschmitt, S. Funes, N. Bonnefoy, and J.M. Herrmann. 2006. Mba1, a membrane-associated ribosome receptor in mitochondria. EMBO J. 25:1603–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumard, P., J. Vaillier, B. Coulary, J. Schaeffer, V. Soubannier, D.M. Mueller, D. Brethes, J.P. di Rago, and J. Velours. 2002. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 21:221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins, G., C. Renken, M.E. Martone, S.J. Young, M. Ellisman, and T. Frey. 1997. Electron tomography of neuronal mitochondria: three- dimensional structure and organization of cristae and membrane contacts. J. Struct. Biol. 119:260–272. [DOI] [PubMed] [Google Scholar]
- Perkins, G.A., J.Y. Song, L. Tarsa, T.J. Deerinck, M.H. Ellisman, and T.G. Frey. 1998. Electron tomography of mitochondria from brown adipocytes reveals crista junctions. J. Bioenerg. Biomembr. 30:431–442. [DOI] [PubMed] [Google Scholar]
- Perotti, M.E., W.A. Anderson, and H. Swift. 1983. Quantitative cytochemistry of the diaminobenzidine cytochrome oxidase reaction product in mitochondria of cardiac muscle and pancreas. J. Histochem. Cytochem. 31:351–365. [DOI] [PubMed] [Google Scholar]
- Pon, L., T. Moll, D. Vestweber, B. Marshallsay, and G. Schatz. 1989. Protein import into mitochondria: ATP-dependent protein translocation activity in a submitochondrial fraction enriched in membrane contact sites and specific proteins. J. Cell Biol. 109:2603–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassow, J., B. Guiard, U. Wienhues, V. Herzog, F.U. Hartl, and W. Neupert. 1989. Translocation arrest by reversible folding of a precursor protein imported into mitochondria. A means to quantitate translocation contact sites. J. Cell Biol. 109:1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert, A.S., and W. Neupert. 2002. Contact sites between the outer and inner membrane of mitochondria-role in protein transport. Biochim. Biophys. Acta. 1592:41–49. [DOI] [PubMed] [Google Scholar]
- Renken, C., G. Siragusa, G. Perkins, L. Washington, J. Nulton, P. Salamon, and T.G. Frey. 2002. A thermodynamic model describing the nature of the crista junction: a structural motif in the mitochondrion. J. Struct. Biol. 138:137–144. [DOI] [PubMed] [Google Scholar]
- Scheffler, I.E. 1999. Mitochondria. Wiley-Liss, Canada. 384 pp.
- Schulke, N., N.B. Sepuri, and D. Pain. 1997. In vivo zippering of inner and outer mitochondrial membranes by a stable translocation intermediate. Proc. Natl. Acad. Sci. USA. 94:7314–7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaiger, M., V. Herzog, and W. Neupert. 1987. Characterization of translocation contact sites involved in the import of mitochondrial proteins. J. Cell Biol. 105:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano, L., M. Ashiya, K. Buttle, S. Weiler, S.A. Oakes, C.A. Mannella, and S.J. Korsmeyer. 2002. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell. 2:55–67. [DOI] [PubMed] [Google Scholar]
- Sesaki, H., S.M. Southard, M.P. Yaffe, and R.E. Jensen. 2003. Mgm1p, a dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol. Biol. Cell. 14:2342–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simbeni, R., L. Pon, E. Zinser, F. Paltauf, and G. Daum. 1991. Mitochondrial membrane contact sites of yeast. Characterization of lipid components and possible involvement in intramitochondrial translocation of phospholipids. J. Biol. Chem. 266:10047–10049. [PubMed] [Google Scholar]
- Sirrenberg, C., M.F. Bauer, B. Guiard, W. Neupert, and M. Brunner. 1996. Import of carrier proteins into the mitochondrial inner membrane mediated by Tim22. Nature. 384:582–585. [DOI] [PubMed] [Google Scholar]
- Steglich, G., W. Neupert, and T. Langer. 1999. Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Mol. Cell. Biol. 19:3435–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyrach, G., M. Ott, N. Bonnefoy, W. Neupert, and J.M. Herrmann. 2003. Ribosome binding to the Oxa1 complex facilitates co-translational protein insertion in mitochondria. EMBO J. 22:6448–6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terziyska, N., T. Lutz, C. Kozany, D. Mokranjac, N. Mesecke, W. Neupert, J.M. Herrmann, and K. Hell. 2005. Mia40, a novel factor for protein import into the intermembrane space of mitochondria is able to bind metal ions. FEBS Lett. 579:179–184. [DOI] [PubMed] [Google Scholar]
- Tokuyasu, K.T. 1989. Use of poly(vinylpyrrolidone) and poly(vinyl alcohol) for cryoultramicrotomy. Histochem. J. 21:163–171. [DOI] [PubMed] [Google Scholar]
- van der Klei, I.J., M. Veenhuis, and W. Neupert. 1994. A morphological view on mitochondrial protein targeting. Microsc. Res. Tech. 27:284–293. [DOI] [PubMed] [Google Scholar]
- Watson, K. 1972. The organization of ribosomal granules within mitochondrial structures of aerobic and anaerobic cells of Saccharomyces cerevisiae. J. Cell Biol. 55:721–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann, B., and W. Neupert. 2000. Mitochondria-targeted green fluorescent proteins: convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast. 16:1421–1427. [DOI] [PubMed] [Google Scholar]
- Wiedemann, N., E. Urzica, B. Guiard, H. Muller, C. Lohaus, H.E. Meyer, M.T. Ryan, C. Meisinger, U. Muhlenhoff, R. Lill, and N. Pfanner. 2006. Essential role of Isd11 in mitochondrial iron-sulfur cluster synthesis on Isu scaffold proteins. EMBO J. 25:184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, E.D., J.A. Wagner, S.V. Scott, V. Okreglak, T.J. Holewinske, A. Cassidy-Stone, and J. Nunnari. 2003. The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J. Cell Biol. 160:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}