

Abstract
The yeast Saccharomyces cerevisiae uses two class V myosins to transport cellular material into the bud: Myo2p moves secretory vesicles and organelles, whereas Myo4p transports mRNA. To understand how Myo2p and Myo4p are adapted to transport physically distinct cargos, we characterize Myo2p and Myo4p in yeast extracts, purify active Myo2p and Myo4p from yeast lysates, and analyze their motility. We find several striking differences between Myo2p and Myo4p. First, Myo2p forms a dimer, whereas Myo4p is a monomer. Second, Myo4p generates higher actin filament velocity at lower motor density. Third, single molecules of Myo2p are weakly processive, whereas individual Myo4p motors are nonprocessive. Finally, Myo4p self-assembles into multi-motor complexes capable of processive motility. We show that the unique motility of Myo4p is not due to its motor domain and that the motor domain of Myo2p can transport ASH1 mRNA in vivo. Our results suggest that the oligomeric state of Myo4p is important for its motility and ability to transport mRNA.
Introduction
Cell polarity creates unique functional domains within cells, exemplified by axons and dendrites of neurons and the apical and basolateral surfaces of epithelial cells (Drubin and Nelson, 1996; Caplan, 1997; Horton and Ehlers, 2003). Cells generate polarity, in part, through directed transport of secretory vesicles and mRNAs by cytoskeleton-based motor proteins (St Johnston, 1995; Caplan, 1997; Pruyne and Bretscher, 2000; Lopez de Heredia and Jansen, 2004; Czaplinski and Singer, 2006). Motor proteins involved in the transport of vesicles and mRNAs have been identified, and much is known about how these motor proteins move along actin filaments and microtubules in vitro (Vale and Milligan, 2000). Less clear is how in vitro motility translates into long distance transport of cargo in vivo. In particular, the obvious physical differences between cargo, such as membrane-bound vesicles and RNAs, would seem to necessitate different mechanisms to generate sustained transport in vivo.
To address this question, we analyzed two class V myosins in the budding yeast Saccharomyces cerevisiae: Myo2p and Myo4p. Together, the two myosins transport most of the cellular material into the bud (Bretscher, 2003). Myo2p is required for cell viability and is essential for the transport of secretory vesicles into the bud to facilitate growth (Johnston et al., 1991). Myo2p also transports other membrane-bound cargo, including vacuoles, late Golgi, peroxisomes, and mitochondria (Beach et al., 2000; Catlett et al., 2000; Hoepfner et al., 2001; Rossanese et al., 2001; Itoh et al., 2002). In contrast, Myo4p is nonessential, but its primary biological function is to transport ASH1 mRNA to the bud tip, generating cell fate differences between mother and daughter cells (Haarer et al., 1994; Long et al., 1997; Takizawa et al., 1997). Myo4p has also been implicated in the movement of cortical ER into the bud (Estrada et al., 2003). Myo2p and Myo4p are closely related and are descended from a common ancestral gene, but they cannot functionally substitute for each other, indicating that they have evolved to transport different types of cargo.
To move cargo over appreciable distances, motors must bind tightly to membranes and nucleic acids. Myo2p binds membrane-bound vesicles and organelles through separate domains in its globular C-tail. For example, amino acids 1389–1491 facilitate association with secretory vesicles, whereas amino acids 1131–1345 mediate association with vacuoles (Catlett et al., 2000; Pashkova et al., 2005). These sequences do not appear to bind directly to membranes; rather, protein intermediaries transiently link Myo2p to different membrane-bound cargo. For example, Vac8p and Vac17p mediate the binding of Myo2p to vacuoles, whereas Sec4p likely facilitates the binding of Myo2p to vesicles (Wagner et al., 2002; Tang et al., 2003). The interaction between Myo2p and vacuoles is dissolved by the proteolysis of Vac17p (Tang et al., 2003). In contrast, Myo4p binds ASH1 mRNA through She3p (Takizawa and Vale, 2000). Myo4p directly binds the N terminus of She3p, and the C terminus of She3p associates with ASH1 mRNA (Bohl et al., 2000). Another RNA-binding protein, She2p, facilitates a specific interaction between She3p and sequences in ASH1 mRNA called zipcodes that are sufficient to localize an RNA (Bohl et al., 2000; Long et al., 2000).
In S. cerevisiae, vesicles and mRNA move rapidly and continuously into the bud. Myo2p moves secretory vesicles at rates around 3 μm/s, and Myo4p transports RNA at 0.25–0.4 μm/s (Bertrand et al., 1998; Schott et al., 2002). How Myo2p and Myo4p facilitate long distance transport in vivo is unclear, as an initial analysis of Myo2p and Myo4p motility indicated that both are nonprocessive (Reck-Peterson et al., 2001). Processive motors have a high probability of remaining associated with a filament through many enzymatic cycles, allowing them to move considerable distances before falling off a filament. In contrast, nonprocessive motors have a low probability of maintaining an association with a filament during their enzymatic cycles, preventing individual nonprocessive motors from moving along a filament. Groups of nonprocessive motors bound to the same cargo might generate sustained movement by providing multiple attachments to a filament, thereby increasing the probability that the cargo remains associated with a filament. Vesicles and organelles contain ample surface area to bind multiple motors, and recent work has shown that vesicles and some organelles use multiple motors to generate bidirectional transport, as well as increase the distance of sustained movement (Miller and Sheetz, 2000; Levi et al., 2006; Gross et al., 2007). Whether a similar mechanism works for mRNA transport is unclear. ASH1 mRNA contains four zipcodes, but a single zipcode is sufficient to generate continuous transport of an RNA to the bud tip (Chartrand et al., 1999). Moreover, active zipcodes can be as small as 25 nucleotides, clearly limiting the number of Myo4p motors that could bind (Jambhekar et al., 2005; Olivier et al., 2005). The spatial restriction of zipcode suggests that Myo4p likely utilizes a different mechanism than Myo2p to generate continuous movement of RNA.
Here, we have compared the structure and motility of Myo2p and Myo4p and discovered two substantial differences. First, Myo4p is a monomeric motor, whereas Myo2p, like all other class V myosins, is a dimer. Second, the in vitro motility of Myo2p and Myo4p were very different. In an actin filament gliding assay, as Myo2p density decreased, filament velocity decreased, indicative of a nonprocessive motor. In contrast, as Myo4p density decreased, filament velocity increased, suggesting that Myo4p is more efficient in smaller groups. Single- molecule analysis demonstrated that Myo2p motors are weakly processive, and although individual Myo4p motors are nonprocessive, ensembles of Myo4p can move processively. To determine whether the motor domain of Myo4p is responsible for the unique motility of Myo4p, we constructed chimeras of Myo2p and Myo4p. We found that a Myo2p-motor/Myo4p-tail chimera (Myo2/4p) is a monomer with motility similar to Myo4p, whereas a Myo4p-motor/Myo2p-tail chimera (Myo4/2p) is a dimer with motility similar to Myo2p. Moreover, Myo2/4p restored localization of ASH1 mRNA to the bud tip in myo4Δ cells, and Myo4/2p restored vesicle transport in a myo2 mutant. Our results suggest that the monomeric structure of Myo4p is important for regulating its motility and transporting mRNAs in vivo.
Results
Myo4p is a monomeric class V myosin
Class V myosin heavy chains are predicted to dimerize through coiled-coil interactions in their proximal tail domain (Espreafico et al., 1992). For vertebrate myosin Va, dimerization allows the two motor domains to coordinate their enzymatic cycles to produce fast and processive movement along actin filaments (Purcell et al., 2002, 2005; Baker et al., 2004; Veigel et al., 2005). The pair-coil algorithm predicts a robust region of coiled-coil in the myosin Va C-tail, and a similar stretch of coiled-coil is found in the C-tail of Myo2p (Fig. 1 A). In contrast, Myo4p contains a limited stretch of amino acids (aa 990–1027) predicted to form a coiled-coil, and such a short coiled-coil domain may be insufficient to facilitate dimerization of Myo4p heavy chains (Fig. 1 A). Because of the importance of dimerization in motor function, we investigated whether Myo4p exists as a monomer or dimer.
Figure 1.

Myo4p is a monomeric, class V myosin in cell extracts. (A) Paircoil predictions for myosin Va, Myo2p, and Myo4p. Myo4p has a predicted coiled-coil domain of only 30 amino acids, considerably shorter than predicted coiled-coil domains in myosin Va or Myo2p. (B) Myo4p-HA does not coprecipitate with Myo4p-GFP. MYO4-HA was expressed on a CEN/ARS plasmid in YPT87. Myo4p-GFP was immunoprecipitated from yeast lysates, and supernatants and pellets were subsequently analyzed by Western blot. Myo4p-HA was not detected in the Myo4p-GFP immunoprecipitate, but She3p-HA was clearly present. S3 is a high-speed supernatant and represents the starting material for the immunoprecipitations. (C) Myo2p-HA coprecipitates with Myo2p-1/2TAP. MYO2-HA and MYO4-HA were expressed on CEN/ARS plasmids in YPT117 and YPT118, respectively. Myo2p-1/2TAP and Myo4p-1/2TAP were immunoprecipitated from cell lysates with IgG beads, and supernatants and pellets were subsequently analyzed by Western blot. Myo2p-HA was evident in immunoprecipitates of Myo2p-Tap1/2, but Myo4p-HA was not detected in Myo4p-Tap1/2 immunoprecipitates. (D) Chemical cross-linking indicates that Myo4p is a monomer. Cell lysates from YPT68 and YPT70 were treated with 50 mM EDC for 1 h at RT and then analyzed by Western blot. EDC-treated Myo4p-GFP migrated as a larger complex than untreated Myo4p-GFP, but smaller than EDC-treated Myo2p. Note that a single Myo4p-GFP polypeptide is ∼15 kD larger than a single Myo2p polypeptide, so a cross-linked Myo4p-GFP dimer would be expected to be larger than a cross-linked Myo2p dimer. She3p-HA cross-links with Myo4p-GFP. EDC-treated Myo4p-GFP in she3Δ extracts showed a minimal shift in size compared with untreated Myo4p-GFP.
We first tested for Myo4p dimers using a coimmunoprecipitation assay. Two versions of Myo4p with different C-terminal tags were expressed in the same cell. MYO4-GFP was integrated and under the control of its native promoter, whereas MYO4-HA was expressed from a low copy CEN/ARS plasmid. Anti-GFP antibodies were used to immunoprecipitate Myo4p-GFP, and the amount of Myo4p-HA in the immunoprecipitates was determined by Western blot. We did not detect Myo4p-HA in immunoprecipitates of Myo4p-GFP, suggesting that the two heavy chains do not interact. (Fig. 1 B). As a control, we probed the Myo4p-GFP immunoprecipitates for She3p-HA, which binds directly to Myo4p. She3p-HA was readily detected in Myo4p-GFP immunoprecipitates (Fig. 1 B), indicating that most Myo4p heavy chains associate with She3p, but not with another Myo4p heavy chain.
To demonstrate that the coimmunoprecipitation assay can detect myosin dimers, we analyzed Myo2p. MYO2 or MYO4 were tagged at their C termini with a 1/2TAP module (TEV cleavage site and IgG-binding domain) and coexpressed with MYO2-HA or MYO4-HA, respectively. Myo2p-1/2TAP or MYO4p-1/2TAP were adsorbed from cell extracts onto IgG resin, and the amount of Myo2p-HA or Myo4p-HA in the immunoprecipitates was determined by Western blot. Myo2p-HA was clearly present in the immunoprecipitates of Myo2p-1/2TAP, but we failed to detect Myo4p-HA in the immunoprecipitates of Myo4p-1/2TAP (Fig. 1 C). We achieved similar results using other combinations of tagged Myo2p and Myo4p (unpublished data). The results demonstrated that the coimmunoprecipitation assay can detect myosin dimers, and therefore, our inability to coprecipitate two Myo4p heavy chains suggests that Myo4p does not form dimers in cell extracts.
Myo4p is a monomer by EDC cross-linking
One limitation of the coimmunoprecipitation assay is that weak or transient Myo4p dimers may dissociate during the assay. To capture these potential dimers, we added the zero-length chemical cross-linker 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) to yeast lysates, and then determined by Western blot whether EDC increased the molecular weight of Myo4p to a size consistent with a dimer. We performed the cross-linking in cell lysates from MYO4-GFP cells because GFP increases the molecular weight of the Myo4p heavy chain to ∼15 kD more than Myo2p. Therefore, a cross-linked Myo4p dimer should be larger and migrate more slowly than a cross-linked Myo2p dimer. MYO4-GFP cell lysates were incubated with or without 50 mM EDC for 1 h at room temperature. Subsequent Western blot analysis revealed that EDC-treated Myo4p-GFP migrated as a larger complex compared with untreated Myo4p-GFP (Fig. 1 D). Reprobing the same blot for Myo2p revealed that EDC-treated Myo2p showed a dramatic increase in molecular weight compared with untreated Myo2p (Fig. 1 D). Importantly, EDC-treated Myo4p-GFP was smaller than EDC-treated Myo2p, indicating that the cross-linked Myo4p-GFP complex was not Myo4-GFP dimers. Instead, the EDC-treated Myo4- GFP complex probably reflects Myo4p-GFP cross-linked to She3p. In support of this conclusion, She3p was detected at the same location as EDC-treated Myo4p on Western blots (Fig. 1 D). Second, in she3Δ cells, EDC-treated Myo4p-GFP did not exhibit a considerable size shift, whereas in the same extracts EDC-treated Myo2p still migrated as a large complex (Fig. 1 D and unpublished data). Thus, cross-linking failed to capture Myo4p dimers and indicated that the bulk of Myo4p in cell extracts exists as a monomer coupled to She3p.
Hydrodynamic analysis of Myo2p and Myo4p
To test further the oligomeric state of Myo4p, we partially purified native Myo2p and Myo4p from cell extracts and performed velocity sedimentation analysis. Myo2p-1/2TAP and Myo4p-1/2TAP were adsorbed onto IgG resin from cell lysates and then eluted with TEV protease. Both eluates were enriched for either Myo2p or Myo4p and showed robust activity in an actin filament gliding assay (Fig. 3 A; and Videos 1 and 2, available at http://www.jcb.org/cgi/content/full/jcb.200707080/DC1). The TEV eluates were analyzed by velocity sedimentation on 5–20% sucrose gradients. Silver stain and Western blot analysis of the sucrose gradient fractions revealed that Myo2p sedimented as a larger complex than Myo4p (Fig. 2 A). Based on the sedimentation of four protein standards, we estimate sedimentation values of 9.1 ± 0.6S for Myo2p and 7.6 ± 0.3S for Myo4p (Fig. 2 A). Interestingly, the sedimentation value of Myo2p is similar to myosin V (10S), which forms a dimer, and the sedimentation value of Myo4p is similar to myosin IXb (7.3S), a monomeric myosin (Post et al., 2002; Wang et al., 2004).
Figure 3.

Profile and oligomeric state of purified myosins. (A) Silver stains of TEV-eluted fractions and ATP released actin-affinity purified (AA pure) fractions of Myo2p, Myo4p, Myo2/4p, and Myo4/2p. Note that She3p only copurifies with myosins containing the C-tail of Myo4p. (B) Purified Myo2p is a dimer, whereas purified Myo4p is a monomer. Actin affinity purified myosins were cross-linked with 40 mM EDC and NHS for 1 h at RT. Cross-linked proteins were run on the same gel and analyzed by Western blot. Cross-linked Phosphorylase b was used as a high molecular weight marker. The bulk of cross-linked Myo4p migrated between 270 and 300 kD, while Myo2p migrated between 400 and 450 kD. When overexposed (right panel), a second, much larger Myo4p band that is likely cross-linked Myo4p dimers is detected. (C) Purified Myo4/2p is a dimer, whereas purified Myo2/4p is a monomer. Actin affinity purified myosins were cross-linked and analyzed by Western blot as in B. The Myo4/2p chimera migrates at the same location as Myo2p, while the Myo2/4p chimera migrates at the same size as Myo4p. She3p cross-links with Myo4p and the Myo2/4p chimera.
Figure 2.

Hydrodynamic and EM analysis of partially purified myosins. (A) TEV-eluted Myo4p, Myo2p, Myo2/4p, and Myo4/2p were analyzed by velocity sedimentation in 5–20% sucrose gradients. Fractions were collected from the bottom of the gradient and analyzed by silver stain and Western blot. Parallel gradients containing the protein standards catalase, aldolase, albumin, and ovalbumin were used to generate a sedimentation coefficient curve and obtain sedimentation coefficient values of 9.1 ± 0.6S for Myo2p (n = 5), 7.6 ± 0.3S for Myo4p/She3p (n = 8), 7.4 ± 0.2S for Myo4p she3Δ, (n = 4), 9.1 ± 0.4S Myo4/2p (n = 2), and 7.3S for Myo2/4p. Note that She2p disassociates from Myo4p and She3p in the sucrose gradient. (B) EM images indicate that Myo2p is a dimer and Myo4p is a monomer. Partially purified (TEV eluted) Myo2p-GFP and Myo4p-GFP were analyzed by rotary shadowing electron microscopy. Representative images of Myo2p-GFP and Myo4p-GFP are shown. Myo2p-GFP images contain many examples of three globular domains linked into a Y-shaped structure. For Myo4p-GFP, the predominant image is of a single dot, likely the motor domain. In a few examples (left-most images), both the head and neck domains are visible. In very rare instances (bottom right), what might be a Myo4p-GFP dimer is visible.
EM analysis of Myo2p and Myo4p
To confirm visually the oligomeric state of Myo2p and Myo4p, we imaged both motors by rotary metal shadowing. Images of Myo2p contained many examples of a Y-shaped molecule with globular domains at each end (Fig. 2 B). Previously published images of myosin V dimers revealed a similar structure and suggest that two of the globular domains in our images correspond to the motor domains of Myo2p and the third to the C tail (Krementsov et al., 2004). The images thus confirm that Myo2p is a dimer. In contrast, most images of Myo4p revealed a single globular domain, occasionally with an extended tail (Fig. 2 B). Rarely, we observed two globular domains in proximity, but the spacing of the heads was noticeably different than Myo2p. The density of images of double-headed motor in several fields of Myo2p and Myo4p was 3.8 /μm2 and 0.4/μm2, respectively, and the density of images of single globular domains of Myo2p and Myo4p was 12/μm2 and 10.8/μm2, respectively (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200707080/DC1). The number of single globular domains may be inflated, as dust and debris often appear similar to a single globular domain. Thus, evidence from coimmunoprecipitation, cross-linking, hydrodynamic analysis, and electron microscopy indicated that Myo4p is a monomeric myosin, sharply distinguishing it from other class V myosins.
Purification of active Myo4p and Myo2p
Like other class V myosins, Myo4p moves cargo, in this case mRNA, rapidly and continuously within cells. To gain a better understanding of how monomeric Myo4p transports ASH1 mRNA in vivo, we purified native Myo4p and analyzed its motility in vitro. We also purified Myo2p to compare the motility of Myo4p to a known dimer. TEV-eluted Myo2p and Myo4p fractions were highly enriched for each myosin, but contained additional proteins (Fig. 3 A). Most of these were not associated with the myosins, as they did not cofractionate in the sucrose gradients (Fig. 2). TEV-eluted Myo4p contained both She3p and She2p, but only She3p cofractionated with Myo4p on sucrose gradients, suggesting that She2p had dissociated from Myo4p/She3p. The weak interaction between She2p and Myo4p/She3p indicated that only a fraction of purified Myo4p/She3p was associated with She2p. To avoid analyzing two different species of Myo4p, we purified Myo4p from she2Δ cells.
To further purify Myo4p and Myo2p, we used an actin bind and release step. A similar method using microtubules was recently used to purify dynein from S. cerevisiae (Reck-Peterson et al., 2006). TEV-eluted myosins were incubated with phalloidin-stabilized F-actin in the absence of ATP to promote binding to actin filaments. Actin filaments and attached myosins were sedimented by centrifugation and then incubated with buffer containing ATP to release myosins from actin filaments. The actin filaments were again sedimented by centrifugation, and the supernatant (ATP-release) was saved. Silver stain analysis of the ATP-releases revealed that Myo4p and Myo2p were highly enriched and pure (Fig. 3 A). Both fractions contained myosin light chains (∼15 kD), and the Myo4p fraction contained She3p. In addition, a small amount of actin was present representing short filaments that had not completely sedimented. Myo2p appeared to be clipped during purification, as the protein bands between 75 and 150 kD that were apparent by silver stain were detected with anti-Myo2p-tail antibody by Western blot (unpublished data). Thus, the two-step purification yielded very pure preparations of native Myo4p and Myo2p.
Purified Myo4p is a monomer
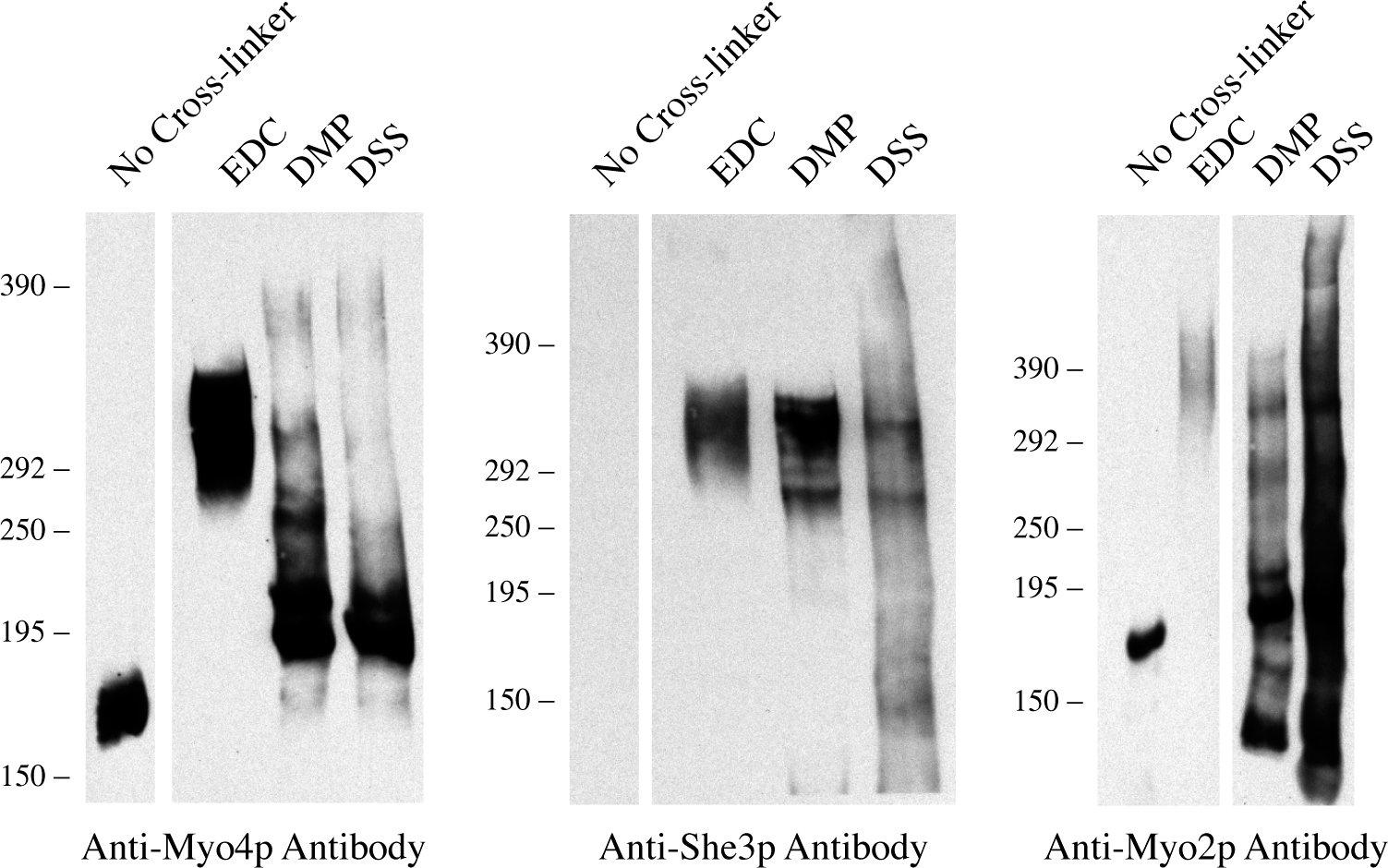
To demonstrate that purified Myo4p and Myo2p maintain their oligomeric status of monomer and dimer, respectively, we incubated both purified myosins with EDC. EDC-treated and untreated samples were run on the same gel and detected by Western blot with antibodies against the C-tails of Myo4p and Myo2p (Fig. 3 B). EDC-treated Myo2p migrated as a large band between 400 and 450 kD, which fits well with the theoretical molecular weight of two cross-linked Myo2p heavy chains and some of the associated myosin light chains. EDC-treated Myo4p migrated between 270 and 325 kD, which approximates a single Myo4p heavy chain cross-linked to She3p and the light chains. Interestingly, when the film was overexposed, a second higher molecular weight Myo4p band was detected. This band migrated slower than cross-linked Myo2p, suggesting that a small amount of Myo4p had formed into dimers. Reprobing the blot with anti-She3p antibodies revealed that She3p co-migrated with EDC-treated Myo4p (Fig. 3 C). We also tested cross-linkers with different reaction groups and longer spacers than EDC, but they cross-linked Myo2p less efficiently than EDC and none generated Myo4p dimers (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200707080/DC1). We conclude that the majority of purified Myo4p exists in a monomeric state bound to She3p, but a small fraction of Myo4p forms dimers when concentrated.
Myo4p and Myo2p exhibit different motility properties
We analyzed the motility and processivity of purified Myo2p and Myo4p in an actin filament gliding assay. Nonprocessive motors exhibit decreasing filament velocity as motor density decreases, whereas processive motors show little change in velocity over several orders of motor density (Rock et al., 2000). In addition, processive motors generate filament movement at densities of 1–2 motors/μm2. Purified Myo2p behaved as a nonprocessive motor (Fig. 4 A). At high motor densities, Myo2p moved actin filaments at velocities of 2–2.5 μm/s, which decreased as motor density decreased. Below ∼50 motors/μm2, actin filaments did not land on coverslips. The maximum velocity of purified Myo2p is consistent with the velocity measured for secretory vesicles in vivo. Our analysis of the motility of purified Myo2p confirms previous work on Myo2p motility in cell lysates (Reck-Peterson et al., 2001).
Figure 4.

Motility analysis of purified myosins. Actin gliding assays were performed by directly adsorbing purified myosins onto the surface of a glass coverslip in a motility chamber. Fluorescently labeled actin filaments were added and movement was recorded within the first 5 min of adding actin filaments to the motility chambers, except at low motor densities, when filament landing was slower. Motor density was determined by quantitative Western blots of the amount of motor input and the amount that passed through the chamber without binding. Myosins were diluted before addition to the motility chambers. (A) The velocities of Myo2p and Myo4p respond differently to changes in motor density. The velocity of actin filaments decreases as Myo2p density decreases, indicative of a nonprocessive motor. In contrast, the velocity of actin filaments increases as Myo4p density decreases. (B) The decreased filament velocity at higher Myo4p density is not due to She3p. The velocity vs. motor density profiles of Myo4p with She3p (top panel) and without She3p (bottom panel) are similar. (C) The velocity versus motor density profile of Myo2/4p resembles Myo4p, whereas the profile of Myo4/2p is similar to Myo2p. Note that the maximum velocity of Myo4/2p is comparable to Myo4p and the maximum velocity of Myo2/4p is equivalent to Myo2p. Shown are representative curves for each myosin, each point is the average filament velocity (μm/s ± SD) for all filaments analyzed in a chamber.
In contrast, Myo4p produced an actin velocity curve that was the inverse of Myo2p (Fig. 4 A). At the highest Myo4p densities, actin filaments moved at 0.45–0.65 μm/s, but as Myo4p density decreased, filament velocity increased to a maximum of 1.5–1.75 μm/s. This unique velocity curve is unlikely to result from inactive or damaged Myo4p, as only active myosin should be purified with the actin affinity step. Further, we detected very few nonmotile actin filaments on the coverslips, which indicates healthy active motors (Videos 1–4). In contrast to our results, an earlier study of Myo4p showed that Myo4p generated a velocity curve similar to Myo2p in actin gliding assays (Reck-Peterson et al., 2001). Two differences in methodology may account for this discrepancy. First, Reck-Peterson et al. (2001) studied Myo4p-GFP in cell lysates, whereas we used purified Myo4p. Second, in the actin gliding assay, Reck-Peterson et al. tethered Myo4-GFP to coverslips with anti-GFP antibodies, whereas we directly adsorbed Myo4p to coverslips. Bivalent antibodies have the potential to bind two Myo4p-GFP motors and perhaps stabilize Myo4p dimers. Although the velocity curve of Myo4p is different from the nonprocessive curve of Myo2p, we suspect that Myo4p is also nonprocessive, as below Myo4p densities of ∼100 motors/μm2, filaments did not land on the coverslips.
Because Myo4p co-purifies with She3p, we tested whether She3p affected the ability of Myo4p to move actin filaments. We purified Myo4p-1/2TAP from she3Δ cells and analyzed Myo4p motility. We observed the same inverse relation between filament velocity and Myo4p density, indicating that She3p was not affecting Myo4p activity (Fig. 4 B). The slight difference in the lowest density of Myo4p that supports filament gliding between figures is likely due to variability in the amount of nonfunctional motor in different preparations of Myo4p and errors in calculating motor density in the flow cells.
Single-molecule analysis of Myo2p-GFP and Myo4p-GFP motility
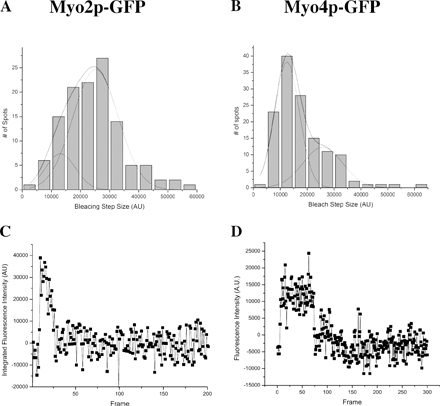
To test more definitively whether Myo2p and Myo4p move processively, we analyzed the motility of purified Myo2p-GFP and Myo4p-GFP on actin bound to the coverslip surface by total internal reflection fluorescence (TIRF) microscopy. First, we measured the total fluorescence intensity and the photobleaching of individual spots to determine how many GFP molecules exist in each. The initial intensity of a spot minus the background after photobleaching was measured and plotted as a histogram (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200707080/DC1). The data for Myo2p-GFP and Myo4p-GFP were each plotted as separate histograms and each were fitted with a two-peak Gaussian function corresponding to a population with a large intensity (25500 a.u.) and a smaller intensity (13,000 a.u.), probably representing spots with two and one GFP molecules, respectively. This result revealed that most Myo2p-GFP spots contained two GFPs, whereas the majority of Myo4p-GFP spots contained a single GFP, indicating that Myo2p-GFP is a dimer and Myo4p-GFP is a monomer (Fig. S3). In the presence of ATP, most Myo2p-GFP spots failed to move along actin filaments, and instead either released from the filaments or bleached. At low (300 nM) ATP, a small fraction of Myo2p-GFP (∼2%) moved processively for distances less than 0.5 μm, and fluorescence imaging with one-nanometer accuracy (FIONA) analysis revealed a trend toward step of 36 and 72 nm (Fig. S4). One interpretation of the results is that Myo2p-GFP is an intermediate duty ratio motor with a low probability of remaining associated with a filament over a few enzymatic cycles, and thus, in a population of Myo2p motors a small fraction will move processively for short distances. This conclusion is supported by previous published work, and we suggest that Myo2p is a weakly processive motor (Reck-Peterson et al., 2001). In contrast, none of the Myo4p-GFP spots with fluorescent intensity equivalent to one or two GFPs moved processively at any ATP concentration. Interestingly, we observed several Myo4p-GFP spots with fluorescent intensity equivalent to at least 3 GFPs, and of these, ∼20% moved processively (Fig. 5 B), taking a range of step sizes (Fig. S4). The variability in step size in this case may be related to the presence of multiple, simultaneously interacting myosin molecules with actin, which might dampen the measured step size (Leduc et al., 2007). Back steps of Myo4p-GFP were occasionally observed at 100 nM ATP, and back-and-forth movement of Myo4p-GFP was also observed at high (1 mM) ATP. The results suggest that Myo4p is nonprocessive as a monomer, but at a low frequency assembles into ensembles capable of processive movement.
Figure 5.

Myo2p is weakly processive and Myo4p ensembles move processively. Stepping traces and photobleaching of Myo2p-GFP and Myo4p-GFP as measure by FIONA. (A) Myo2p-GFP. (a–c) Stepping traces of representative Myo2p-GFP spots measure by FIONA and (d – f) respective photobleaching of the spots. Only 2% of spots show processive movement illustrated in (f). (B) Myo4p-GFP. (a–c) Stepping traces of representative Myo4p-GFP spots measure by FIONA and (d–f) respective photobleaching of the spots. Monomers (a, d) and potential dimers (b and e) of Myo4p-GFP do not move processively, but ensembles (c and f) of at least three Myo4p-GFPs move processively. Note that Myo2p-GFP and Myo4p-GFP were imaged with different cameras resulting in different spot intensities.
Myo2p-Myo4p chimeras
The strikingly different velocity curve of Myo4p as compared with other myosins could be due to unique properties of the Myo4p motor domain or in its oligomeric state (i.e., monomer versus dimer). To distinguish between these possibilities, we constructed chimeras consisting of the motor domain from one myosin fused to the IQ and C-tail domains of the other myosin. Myo2/4p (Myo2 motor − Myo4 IQ + C-tail) was expressed with a C-terminal 1/2TAP tag in myo4Δ cells and purified as described for Myo4p-1/2TAP. The Myo4/2p chimera (Myo4 motor − Myo2 IQ + C-tail) was expressed with a C-terminal 1/2TAP tag in MYO2-GFP cells and purified as described for Myo2p-1/2TAP with one additional step. Because Myo4/2p contains the coiled-coil domain of Myo2p, a small amount of Myo4/2p-1/2TAP may dimerize with Myo2p-GFP. To eliminate these mixed dimers from purified Myo4/2p, we incubated the TEV eluate with beads coated with anti-GFP antibodies. The unbound fraction was then subjected to the actin affinity step to further purify Myo4/2p.
The structure and protein composition of the purified chimeras resembled the myosin from which they obtained their C-tail. The protein profile of purified Myo2/4p contained She3p and myosin light chains (Fig. 3 A). EDC cross-linking of purified Myo2/4p increased its molecular weight similarly to purified Myo4p, and She3p was evident in the cross-linked complex (Fig. 3 C). Myo2/4p had a velocity sedimentation coefficient of 7.3S, similar to Myo4p (Fig. 2). These results indicated that the Myo2/4p chimera, like Myo4p, is a monomer that binds directly to She3p, confirming previous results that She3p interacts with the C-tail of Myo4p (Bohl et al., 2000). In contrast, the Myo4/2p chimera did not co-purify She3p, and EDC-treated Myo4/2p migrated at a similar molecular weight as EDC-treated Myo2p (Fig. 3, A and C). In addition, velocity sedimentation analysis of purified Myo4/2p revealed a sedimentation coefficient of 9.1S, identical to Myo2p (Fig. 2). Thus, the Myo4/2p chimera, like Myo2p, is a dimer.
To determine whether the motor domain or oligomeric state of Myo4p limits its ability to move actin filaments at high density, we analyzed Myo4/2p and Myo2/4p in the actin filament gliding assay. Myo4/2p behaved similarly to Myo2p: as the density of Myo4/2p decreased, the velocity of actin filaments decreased (Fig. 4 C). This profile suggests that Myo4/2p, like Myo4p, is a nonprocessive motor. Interestingly, Krementsova et al. (2006) engineered a myosin chimera that contained the Myo4p motor and myosin V C-tail. In contrast to Myo4/2p, their myosin chimera was processive. In addition, they found that their chimera exhibited longer run lengths in lower ionic strength buffers. To determine whether Myo4/2p was processive at lower salt concentrations, we tested the motility of Myo4/2p in the actin filament gliding assay in buffer containing 25 mM KCl. There was no difference in the velocity curve at 25 mM KCl compared with higher salt buffers, indicating that the difference between the chimeras is not due to buffer conditions (unpublished data). Instead, the results suggest that subtle differences in the coiled-coil domains of Myo2p and myosin V may be crucial for generating processivity. Similar to Myo4p, Myo2/4p showed an inverse relationship between motor density and velocity: as Myo2/4p density decreased, actin filament velocity increased (Fig. 4 C). Importantly, the maximum velocity of Myo2/4p was similar to Myo2p, and Myo4/2p velocity was similar to Myo4p, suggesting that the basic enzymatic properties of the motors had not substantially changed in the chimeras. Thus, the results indicate that the slowing of actin filaments at higher Myo4p density is not due to the properties of the Myo4p motor domain. Rather, they suggest that the monomeric status of Myo4p allows it to move filaments more efficiently than Myo2p at low motor densities.
The motor domains of Myo2p and Myo4p are functionally interchangeable
We next examined whether the myosin chimeras could functionally replace the wild-type myosins. To determine whether Myo2/4p localizes Ash1p to the daughter cell nucleus, we used a growth assay that is sensitive to the localization of Ash1p. pHO-ADE2 cells grow in the absence of adenine, because Ash1p localizes exclusively to the daughter cell, and its absence from the mother cell allows for expression of ADE2 and synthesis of adenine. pHO-ADE2, myo4Δ cells fail to grow without adenine because Ash1p accumulates in the nuclei of both mother and daughter cells where it represses expression of ADE2. We expressed from low copy plasmids either MYO4 or MYO2/4 in pHO-ADE2, myo4Δ cells. Compared with cells containing only an empty vector, both MYO4 and MYO2/4 restored growth in the absence of adenine, suggesting that Myo2/4p was sufficient to localize Ash1p to the daughter cell (Fig. 6 A). To confirm that Myo2/4p localizes ASH1 mRNA to the bud tip, we examined the same cells by FISH with probes against ASH1 mRNA. The results showed that ASH1 mRNA localized to the bud tip in 66% of cells expressing MYO4 and 62% of cells expressing MYO2/4 (Fig. 6 B). In contrast, none of the cells containing only an empty vector showed localized ASH1 mRNA. Thus, Myo2/4p can functionally substitute for Myo4p to transport ASH1 mRNA to the bud tip.
Figure 6.

The motor domains of Myo2p and Myo4p are interchangeable. (A) Myo2/4p localizes Ash1p to the daughter cell. Four tenfold serial dilutions of YPT148, YPT149, and YPT150 at OD600 ∼0.3 were made and spotted onto either SC −leu + adenine or SC −leu −adenine. Plates were incubated for 2 d at 30°C. (B) Myo2/4p localizes ASH1 mRNA to the bud tip. FISH analysis of YPT148, YPT149, YPT150 with probes against ASH1 mRNA shows that both Myo4p and Myo2/4p localized ASH1 mRNA to the bud tip. Bar = 5 μm. (C) Myo4/2p partially rescues growth of myo2-66 cells. YPT168, YPT171, and YPT172 were incubated on Sc −leu at 25, 30, or 37°C. Myo4/2p fully restored growth at 30°C but not at 37°C. (D) Myo4/2p localizes GFP-Sec4p at the restrictive temperature. YPT173, YPT174, and YPT175 were incubated in SC −leu for 5 h at 25, 30, or 37°C. Cells were spotted onto glass slides and visualized by fluorescence microscopy. Bar = 5 μm.
To determine whether Myo4/2p could substitute for Myo2p, we expressed either MYO2 or MYO4/2 on low copy plasmids in myo2-66 cells, which contain a temperature-sensitive mutation in the MYO2 motor domain, rendering the motor inactive at elevated temperature (Johnston et al., 1991). Because MYO2 is essential, myo2-66 cells do not grow at elevated temperatures. All strains, myo2-66, myo2-66 + MYO2, and myo2-66 + MYO4/2 grew well at 25°C, but only MYO2 and MYO4/2 grew robustly at 30°C (Fig. 6 C). Although MYO4/2 rescued growth at 30°C, it grew poorly at 37°C compared with MYO2. To determine whether Myo4/2p mediated vesicle transport to the bud at restrictive temperatures, we examined the distribution of Sec4p, which accumulates on secretory vesicles (Goud et al., 1988). All strains, myo2-66, myo2-66 + MYO2, and myo2-66 + MYO4/2, localized GFP-Sec4p to buds at 25°C (unpublished data). But only Myo2p and Myo4/2p restored localization of GPF-Sec4p to buds of myo2-66 cells at 30°C (Fig. 6 D). At 37°C, GFP-Sec4p was detected in buds of cells expressing Myo4/2p, but GFP-Sec4p localization was stronger in cells expressing Myo2p. The velocity of Myo4/2p was about half of Myo2p (Fig. 4, A and C), which may explain the weaker localization of GFP-Sec4p in MYO4/2 cells. The results demonstrate that Myo4/2p mediates vesicle transport, though less efficiently than Myo2p.
Discussion
Although a large body of information exists on the mechanism of molecular motor movement, limited work has been done to address how the activity of a motor translates to successful cargo transport in vivo (Forkey et al., 2003). In particular, the physical and biochemical differences between membrane-bound vesicles and mRNAs would seem to require different properties in a motor protein. We have analyzed two closely related class V myosins, Myo2p and Myo4p, which transport secretory vesicles and mRNA, respectively, in S. cerevisiae. We find two important differences between Myo2p and Myo4p. First, unlike Myo2p and other known class V myosins, Myo4p is a monomer (Reck-Peterson et al., 2000). Second, Myo2p and Myo4p differ in their movement along actin filaments. Myo2p behaves as a nonprocessive motor in actin gliding assays and as a weakly processive motor in single-molecule assays. In this regard, it is worth noting that myosin V from Drosophila melanogaster was also shown to be a low duty ratio motor that probably does not move processively (Toth et al., 2005). The motility of Myo4p is unique. In an actin gliding assay, the velocity of Myo4p increases as motor density decreases. Further, single-molecule analysis suggests that although individual Myo4p motors are nonprocessive, ensembles of Myo4p move processively. Finally, the motor domain of Myo2p can substitute for Myo4p motor to localize ASH1 mRNA, indicating that the unique motility of Myo4p is not due to differences in its motor domain.
How do the properties of Myo2p and Myo4p allow them to transport vesicles and mRNA, respectively, in vivo? Myo2p is at best a weakly processive motor, and the run lengths of the few processive Myo2p motors were at most ∼0.5 μm, far shorter than the length of a yeast cell. Thus, a single Myo2p is unlikely to maintain transport of cargo to the bud tip. The key, therefore, to generating sustained movement of cargo is likely the presence of multiple Myo2p motors on a vesicle or organelle, increasing the probability that the vesicle or organelle remains associated with a filament during transport to the bud. That Myo2p is weakly processive suggests that as few as two or three motors may be sufficient to sustain transport to the bud, and even the smallest vesicle should provide ample surface area to bind multiple motors. Although the number of molecules of Myo2p on a vesicle or vacuole has not been quantified, the expression levels of Myo2p (∼4,000/cell) and Vac8p (∼5,000/cell), which links Myo2p to vacuoles, offer sufficient amounts of both proteins to bind multiple Myo2p motors to every vacuole (Ghaemmaghami et al., 2003). An important question is whether motors bound to the same cargo and filament coordinate their enzymatic cycles to ensure sustained and rapid transport in vivo. We found that at high densities of Myo2p, when multiple motors are attached to a filament, filament velocity is comparable to the rate of vesicle transport in vivo, suggesting that Myo2p motors can coordinate their activities in vitro to generate rapid transport.
How Myo4p generates sustained movement of RNA in vivo is likely more complicated. Myo4p is clearly nonprocessive as a single-headed molecule, implying that multiple motors are needed to sustain transport of RNA in vivo. The small size of an active zipcode, however, seems to preclude the attachment of multiple motors. Our results suggest a potential solution. Ensembles of three or more Myo4p motors move processively, so instead of Myo4p motors binding to multiple sites along an mRNA, like Myo2p on a vesicle, Myo4p may assemble into a multimotor complex that can be linked to a single zipcode within an mRNA. How Myo4p assembles into ensembles is unclear, as there is no evidence that other class V myosins form multimotor complexes. The primary structural difference between Myo4p and other class V myosins is that Myo4p is a monomer, suggesting the possibility that as a monomer Myo4p is capable of assembling into larger order complexes. She3p may be an important component, as it binds tightly and directly to Myo4p. She3p binds the C-tail of Myo4p, including the coiled-coil region (Bohl et al., 2000). She3p also contains an extensive stretch of sequence with a strong probability of forming a coiled-coil. Intriguingly, the multicoil program predicts six heptad repeats to form a three-stranded coiled-coil. One explanation, then, why Myo4p is a monomer is that the C-tail evolved to form association with She3p rather than with itself. Finally, She2p, which links Myo4p/She3p to ASH1 mRNA, is a dimer and could potentially bind two copies of Myo4/She3p to facilitate assembly of Myo4p ensembles.
Another unusual aspect of Myo4p motility is that the velocity of actin filaments was threefold higher at low density of motors compared with high density. Interestingly, the in vitro velocity of Myo4p at high density is similar to the in vivo velocity of RNA particles (Bertrand et al., 1998). What generates this unique motility of Myo4p is unclear. A chimera of the Myo2p motor and C-tail of Myo4p exhibited a similar motility to Myo4p, suggesting that either the C-tail of Myo4p regulates the activity of the motor or that the monomeric state of the motor protein affects its activity. Recent work on myosin Va has shown that the C-tail folds over to bind the motor domain and repress its activity, but our results question whether such a mechanism explains the behavior of Myo4p (Krementsov et al., 2004; Wang et al., 2004). First, velocity sedimentation analysis of myosin Va reveals a 14S version, representing the C-tail bound to the motor, and an 11S species, which is myosin Va extended. Our analysis of Myo4p and Myo2p did not reveal more compact versions of either myosin, suggesting that both are fully extended. Second, the slowing of Myo4p velocity is concentration dependent, whereas the velocity of myosin Va is not affected by concentration but instead regulated by Ca2+ (Krementsov et al., 2004). Although our results do not rule out a role for the C-tail in regulating the activity of the Myo4p motor, we suggest that the monomeric state of Myo4p is the likely reason for its unusual motility.
To determine the molecular mechanisms of active transport complexes, future work will need to be directed at reconstituting and analyzing active myosin–cargo complexes. Toward this aim, we have developed methods to purify native and active Myo2p and Myo4p. RNA transport is especially ripe for reconstitution, as the components that link Myo4p to RNA have been identified, and the cargo can be readily synthesized and modified in vitro. Thus, we can address questions about the minimal number of Myo4p motors needed to generate sustained transport and whether multiple zipcodes increase the robustness of RNA transport. Similarly, it is now possible to attempt to reconstitute vesicle or vacuole transport in vitro. In addition to the number of Myo2p motors needed for cargo movement, an important question is whether an even distribution or clustering of Myo2p on the surface of a vesicle facilitates sustained transport. Finally, the use of myosin chimeras will enable us to dissect further those features of Myo2p and Myo4p that are unique for vesicle and RNA transport, respectively, and will lead to a broader understanding of how motor proteins have evolved to transport different types of cargo.
Materials and methods
Strains and plasmids
All strains used in this study are listed in Table I. MYO4-GFP, MYO4-TAP, MYO4-1/2TAP, MYO2-GFP, MYO2-TAP, and MYO2-1/2TAP were made by PCR-mediated gene modification. MYO4-HA and SHE3-HA were made by gene replacement of endogenous MYO4 and SHE3, respectively. The Myo4-HA plasmid was made by inserting a single HA sequence into an engineered BamHI site just upstream of the stop codon. The She3-HA plasmid was made by inserting a single HA sequence in an engineered NheI site just upstream of the stop codon. Both Myo4-HA and She3-HA were subcloned into CEN ARS vectors to allow for expression from a low copy plasmid. pHO-ADE-HO 3′ UTR has been described elsewhere (Irie et al., 2002), and pHO-ADE-HO 3′ UTR myo4Δ was made by PCR-mediated gene disruption. To make the Myo2/4 plasmid, PCR products of MYO2 5′ UTR and promoter (-579 to +3), MYO4 motor domain (+4 to +2334), MYO2 IQ + C-tail (+2347 to +4722), and MYO2 3′ UTR (stop codon + 370 nucleotides of downstream sequence) were ligated using unique restriction sites and inserted into pRS315. To make the Myo4/2 plasmid, PCR products of MYO4 5′ UTR and promoter (−687 to +3), MYO2 motor domain (+4 to +2346), MYO4 IQ + C-tail (+2335 to +4413), and MYO4 3′ UTR (stop codon + 193 nucleotides of downstream sequence) were ligated using unique restriction sites and inserted into pRS315. 1/2 TAP versions of both proteins were made by inserting a PCR product containing the sequence encoding a TEV protease site and IgG-binding domain into unique restriction sites just upstream of the stop codon in MYO2/4 and MYO4/2.
Table I.
Strains used in this study
| Strain | Genotype |
|---|---|
| W303 | MATa ura3 trp1 leu2 his3 ade2 GAL |
| YPT3 | myo4Δ |
| YPT68 | MYO4-GFP |
| YPT70 | MYO4-GFP she3Δ |
| YPT87 | MYO4-GFP SHE3-HA |
| YPT117 | MYO2-1/2TAP |
| YPT118 | MYO4-1/2TAP |
| YPT119 | MYO4-1/2TAP she3Δ |
| YPT120 | MYO4-1/2TAP she2Δ |
| YPT138 | myo4Δ pHO-ADE2-HO 3′ UTR |
| YPT140 | MATa ura3 leu2 his6 ade1 myo2-66 |
| YPT148 | myo4Δ pHO-ADE2-HO 3′ UTR + pRS315 |
| YPT149 | myo4Δ pHO-ADE2-HO 3′ UTR + MYO4 (LEU2) |
| YPT150 | myo4Δ pHO-ADE2-HO 3′ UTR + MYO2/4 (LEU2) |
| YPT168 | MATa ura3 leu2 his6 ade1 myo2-66 + pRS315 |
| YPT171 | MATa ura3 leu2 his6 ade1 myo2-66 + MYO4/2-HA (LEU2) |
| YPT172 | MATa ura3 leu2 his6 ade1 myo2-66 + MYO2-HA (LEU2) |
| YPT173 | MATa ura3 leu2 his6 ade1 myo2-66 GFP-SEC4 + pRS315 |
| YPT174 | MATa
ura3 leu2 his6 ade1 myo2-66 GFP-SEC4 + MYO4/2-HA (LEU2) |
| YPT175 | MATa
ura3 leu2 his6 ade1 myo2-66 GFP-SEC4 + MYO2-HA (LEU2) |
Reagents and antibodies
9E10 (anti-myc) and HA.11 (anti-HA) were purchased from Covance Research Products. JL8 (anti-GFP) was purchased from CLONTECH Laboratories, Inc. Antisera against She2p, She3p, Myo4p, GFP, and HA were prepared by Cocalico Biologicals. Antibodies were affinity purified using either purified 6× His She2p, 6× His She3p 197–425, or GST-Myo4p C-tail (1075 to end). GFP antibodies were affinity purified on a membrane containing GFP. HA antibodies were affinity purified on a column containing HA peptide. ECL reagents, protein molecular weight standards, IgG Sepharose 6 Fast Flow, and protein G Sepharose 4 Fast Flow were purchased from GE Healthcare. 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were purchased from Pierce Chemical Co. Cross-linked Phosphorylase b, glucose oxidase, catalase, phalloidin, and TRITC-phalloidin were purchased from Sigma- Aldrich. AcTEV protease was purchased from Invitrogen. Actin was purchased from Cytoskeleton, Inc. Anti-Myo2p tail antibody was a gift from Mark Mooseker (Yale University, New Haven, CT).
Coimmunoprecipitation
Yeast cultures were grown in YPD or selective media to mid-log phase. Cells were harvested by centrifugation, pellets were washed twice with cool H2O, and resuspended in 3× cell pellet volume of lysis buffer A (20 mM Imidazol, pH 7.2, 150 mM KCl, 2.5 mM MgCl2, 1 mM EGTA, 1 mM EDTA, 2 μg/ml of aprotinin, leupeptin, and pepstatin, 400 μM AEBSF, 5 mM ATP, and 2 mM DTT). Lysates were made by bead-beating 4× 45 s, with 60 s in ice water between cycles. The lysate was centrifuged for 5 min at 2,000 rcf, and supernatant was collected and centrifuged for 20 min at 85,000 rpm in a TLA120.2 rotor. The high speed supernatant (S3) was used for coimmunoprecipitation experiments. S3 was incubated at 4°C for 1 h with either free antibody or antibody coupled to beads. Free antibody was subsequently linked to protein G beads for 1 h at 4°C. Beads were isolated by centrifugation, washed 4× with 50× bead volume of wash buffer (150 mM KOAc, 25 mM Hepes-KOH, pH 7.5, 2 mM MgOAc, and 0.1% IGEPAL), resuspended in 1× SDS-PAGE sample buffer, and boiled. Supernatants were mixed with 5× SDS-PAGE sample buffer and boiled.
EDC/NHS cross-linking
EDC and NHS were dissolved in H2O just before use at a concentration of 1 M. For crude S3 lysates, only EDC was added to a final concentration of 50 mM. For TEV and ATP-released actin affinity-purified myosin, both EDC and NHS were added to a final concentration of 40 mM. Samples were incubated for 1 h at room temperature and reactions were quenched by addition of 5× sample buffer. The samples were boiled and run on either 3.5 or 5% denaturing gels and then analyzed by Western blot. Bio-Rad all-blue precision plus and Sigma-Aldrich cross-linked Phosporylase b were used as size standards. We detected Phosphorylase b by staining Western blot membranes with ponceau S before blocking.
Myosin purification
A modified TAP tag (1/2TAP) lacking the calmodulin binding domain was used for myosin purification. The tag was added to the C terminus of all myosins. 1/2TAP-tagged Myo2p and Myo4p were integrated, whereas 1/2TAP-tagged MYO2/4 and MYO4/2 were expressed from low copy plasmids. Cultures were grown to mid-log phase in YPD and harvested by centrifugation. Cell pellets were washed twice with cool H2O, resuspended in 1/5 pellet vol lysis buffer A, frozen in liquid N2, and stored at −80°C. Frozen cells were lysed by blending in a Waring blender with liquid nitrogen. Lysed cells were thawed in 1.5× cell pellet vol of ice-cold lysis buffer A containing 0.3 mg/ml RNase A on a hot plate set to low with constant stirring. Thawed lysates were centrifuged for 10 min at 20 g, supernatants were collected, and centrifuged for 1 h at 50,000 rpm in a Ti70 rotor. Cleared lysate was passed over IgG sepharose columns at 4°C. Columns were washed with 250× column vol of lysis buffer without protease inhibitors or ATP. Washed beads were resuspended in 1× column vol lysis buffer without protease inhibitors or ATP containing 50–100 U TEV protease and incubated overnight at 4°C. Columns were eluted and washed with lysis buffer A without protease inhibitors or ATP. Elution fractions were used in the assays described or further purified. TEV eluted myosin was further purified by incubating with phalloidin-stabilized actin filaments (final actin concentration 0.5 μM) for 10–15 min at 4°C. The myosin bound to actin was added onto two-step sucrose gradients (100 μl 60% and 300 μl 10% sucrose) and centrifuged for 45 min at 45,000 rpm in a SW 50.1 rotor. The top of the gradient was discarded, leaving the bottom 250 μl containing the myosin bound to actin. ATP was added to the myosin and actin (final ATP concentration 10 mM) and then immediately centrifuged for 25 min at 85,000 rpm in a TLA 120.2 rotor. The supernatant containing myosin released from actin was collected and used for motility assays and EDC cross-linking.
Sucrose gradients
TEV-eluted myosin was added to the top of 5–20% 5-ml sucrose gradients. Sucrose was dissolved in buffer A. Gradients were spun for 16–20 h at 40,000 rpm in a SW50.1 rotor. Fractions were collected from the bottom of the gradients with a peristaltic pump. Parallel gradients containing ovalbumin, albumin, aldolase, and catalase protein standards were used to calculate S values for each myosin.
Rotary shadowing EM
Myo2p-GFP and Myo4p-GFP were diluted to 0.1 mg/ml in 0.5 M ammonium acetate and mixed thoroughly with glycerol. The samples were sprayed onto a mica chip and loaded into the vacuum chamber of RFD-9010 CR Freeze Etching Equipment (RMC Products). Platinum was deposited by rotary shadowing at an angle of 9° with 3 KV and 70 mA. Carbon was deposited to reinforce the replica at an angle of 90° with 2.5 KV and 90 mA. The replicas were prepared by floating on clean water and lifted onto formvar-carbon coated cupper grids and observed in an electron microscope (JEM-1200EX II; JEOL).
Actin gliding assays
Actin gliding assays were preformed as described in Rock et al. (2000) with the following changes. Grease-coated coverslips were used as spacers to make the motility chambers. Buffer A without protease inhibitors or ATP was used for all washes. Coverslips were not coated with nitrocellulose. Chambers were pre-blocked with 0.01 mg/ml BSA in buffer A, washed, and purified myosin was added. The myosin was incubated in the chamber for 30+ min on wet ice. Motility buffer (10 mM imidazole, pH 7.2, 75 mM KCl, 1 mM EGTA, 2.5 mM MgCl2, 10 mM Mg-ATP, 8 mM DTT, 0.2 mg/ml glucose oxidase, and 0.1 mg/ml catalase) was mixed with TRITC-phalloidin–stabilized actin filaments (8 nM final) and dextrose (0.25% final). Unlabeled actin was not used to block dead myosin. Actin filaments were observed on a fluorescence microscope (TE2000; Nikon) equipped with a 100×/NA 1.4 lens (Nikon), and images were acquired with an ORCA-ER CCD camera (Hamamatsu) using IPLab software (Scanalytics, Inc.). Videos were taken within the first 5 min of adding actin in motility buffer, except at low motor densities when actin landing was slower. Filaments were tracked manually from frame to frame to compute velocity.
Single-molecule motility assay
Single-molecule motility assay and FIONA analysis using TIRF microscopy was performed and quantified as described previously (Yildiz et al., 2003; Snyder et al., 2004; Sakamoto et al., 2005). Fluorescence intensities of individual spots were measured as follows. Myosin solutions without ATP in single-motility buffer were added into a flow-chamber that had actin filaments bound to the surface, and were incubated for 2 min at room temperature. The actin filaments were a copolymer with 10% biotin-labeled actin + 90% unlabeled actin (Sakamoto et al., 2000). They were bound to the coverslip surface via a biotin-avidin sandwich. The free myosin was washed out with 600 μl single-molecule buffer (SMB: 20 mM MOPS, pH 7.2, 5 mM MgCl2, 0.1 M EGTA, and 50 mM KCl) containing 1 mg/ml bovine serum albumin, 50 mM DTT, and an oxygen scavenging system (125 mg/ml glucose, 1,665 units glucose oxidase (G-7076; Sigma-Aldrich), and 26,000 units Catalase (106810; Roche). The flow-chamber was then placed on a microscope (IX70; Olympus) and observed through a 60×/NA 1.45 lens (Olympus) in TIRF mode at 25°C. Two relay lenses of 1.5 and 2.5× were also used to achieve a total magnification of 225. Images were captured with either a Micromax CCD (Roper Scientific) or an Andor EMCCD DV 897 using Andor iQ software. The surface of the flow chambers was first checked to determine whether myosin molecules were bound to actin filaments. A manual shutter to block the light was closed and the stage was moved to a different field. To prevent drifting of stage, the shutter was closed 2–4 min. Image acquisition started when the shutter was reopened.
Data analysis of intensity and photobleaching of individual spots
To analyze spot intensity and photobleaching, an 8 × 8 pixel region of interest (ROI) was applied to each spot and the integrated intensities for all frames were determined with MetaMorph (Molecular Devices). Background signal intensities were subtracted from the measured intensities following photobleaching of each 8 × 8 pixel ROI.
FISH
YPT148, YPT149, and YPT150 were grown overnight in minimal media lacking leucine. The overnight cultures were diluted into fresh minimal media lacking leucine and incubated at 30°C until achieving OD600 ∼0.5. Cells were fixed in 4% formaldehyde for 45 min at room temperature, washed, and then spheroplasted. Spheroplasts were adsorbed onto poly-l-lysine coated glass coverslips and incubated in hybridization mix (50% formamide, 5× SSC 1 mg/ml, yeast tRNA,100 μg/ml Heparin, 1× Denhardt's Solution, 0.1% Tween 20, 0.1% Triton X-100, and 5 mM EDTA, pH 8.0) for 1 h at room temperature. Samples were incubated in hybridization mix containing an anti-sense, Dig-labeled RNA probe against ASH1 mRNA overnight at 37°C. Samples were washed in 0.2× SSC and then incubated with mouse anti-Dig in 0.05 M Tris-Cl, pH 7.5, 0.15 M NaCl, and 5% fetal bovine serum for 30 min at 37°C. Samples were washed in 0.05 M Tris-Cl, pH 7.5, 0.15 M NaCl, and 0.05% Tween 20 and then incubated with goat anti–mouse Alexa488 in 0.05 M Tris-Cl, pH 7.5, 0.15 M NaCl, and 5% fetal bovine serum for 1 h at room temperature. Samples were then washed in 0.05 M Tris-Cl, pH 7.5, 0.15 M NaCl, and 0.05% Tween 20 and then mounted on glass slides. Samples were observed at room temperature on a fluorescence microscope (TE2000; Nikon) equipped with a 100×/NA1.4 lens (Nikon), and images were acquired with an ORCA-ER CCD camera (Hamamatsu) using IPLab software (Scanalytics, Inc.). Cropping and brightness/contrast adjustments were made in Adobe Photoshop.
Growth assays
For Ash1p localization, YPT148, YPT149, and YPT150 were grown overnight in synthetic media lacking leucine (SC −leu) The cultures were adjusted to OD600 ∼0.3 and then four tenfold serial dilutions were made from the culture. 5 μl of the starting culture and each dilution were spotted onto SC −leu plates with or without 25 mg/l adenine. The plates were incubated at 30°C for 3 d. For rescue of myo2-66 growth, YPT168, YPT171, and YPT172 were grown overnight in SC −leu. The cultures were diluted to OD600 ∼0.3 and four tenfold serial dilutions were made from the culture. 5 μl of the starting culture and each dilution were spotted onto SC −leu plates and incubated for 2 d at either 25, 30, or 37°C.
GFP-Sec4p localization
YPT173, YPT174, and YPT175 were grown overnight in SC −leu. Cultures were diluted into fresh SC −leu and incubated for 6 h at 25, 30, or 37°C. Cells were fixed in 4% formaldehyde, washed, and mounted on glass slides. Samples were observed at room temperature on a fluorescence microscope (Nikon TE2000) equipped with a 63×/NA1.4 lens (Nikon), and images were acquired with an ORCA-ER CCD camera (Hamamatsu) using IPLab software (Scanalytics, Inc.). Cropping and brightness/contrast adjustments were made in Adobe Photoshop.
Online supplemental material
Videos 1–4 are representative actin gliding assay movies. Videos 1 and 3 are TEV-eluted and ATP-released actin affinity-purified Myo2p, respectively. Videos 2 and 4 are TEV-eluted and ATP-released actin affinity-purified Myo4p, respectively. Collection and display rates are both 2.5 frames/s. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200707080/DC1.
Supplementary Material
Acknowledgments
We would like to thank Mark Mooseker, Rania Zaarour, and Chris O'Connell for generous gifts of reagents and advice. We would like to thank Paul Forscher and Dave Van Goor for initial help with TIRF microscopy.
This work was supported by grants from the Searle Scholars Foundation (to P.A. Takizawa), National Institutes of Health (GM073734, to P.A Takizawa), and NSF Graduate Research Fellowship (to B.D. Dunn).
Abbreviations used in this paper: EDC, 1-Ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride; FIONA, fluorescence imaging with one-nanometer accuracy; Myo2/4p, Myo2p-motor/Myo4p-tail chimera; Myo4/2p, Myo4p-motor/Myo2p-tail chimera; NHS, N-hydroxysuccinimide; TIRF, total internal reflection fluorescence.
References
- Baker, J.E., E.B. Krementsova, G.G. Kennedy, A. Armstrong, K.M. Trybus, and D.M. Warshaw. 2004. Myosin V processivity: multiple kinetic pathways for head-to-head coordination. Proc. Natl. Acad. Sci. USA. 101:5542–5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach, D.L., J. Thibodeaux, P. Maddox, E. Yeh, and K. Bloom. 2000. The role of the proteins Kar9 and Myo2 in orienting the mitotic spindle of budding yeast. Curr. Biol. 10:1497–1506. [DOI] [PubMed] [Google Scholar]
- Bertrand, E., P. Chartrand, M. Schaefer, S.M. Shenoy, R.H. Singer, and R.M. Long. 1998. Localization of ASH1 mRNA particles in living yeast. Mol. Cell. 2:437–445. [DOI] [PubMed] [Google Scholar]
- Bohl, F., C. Kruse, A. Frank, D. Ferring, and R.P. Jansen. 2000. She2p, a novel RNA-binding protein tethers ASH1 mRNA to the Myo4p myosin motor via She3p. EMBO J. 19:5514–5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher, A. 2003. Polarized growth and organelle segregation in yeast: the tracks, motors, and receptors. J. Cell Biol. 160:811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan, M.J. 1997. Membrane polarity in epithelial cells: protein sorting and establishment of polarized domains. Am. J. Physiol. 272:F425–F429. [DOI] [PubMed] [Google Scholar]
- Catlett, N.L., J.E. Duex, F. Tang, and L.S. Weisman. 2000. Two distinct regions in a yeast myosin-V tail domain are required for the movement of different cargoes. J. Cell Biol. 150:513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartrand, P., X.H. Meng, R.H. Singer, and R.M. Long. 1999. Structural elements required for the localization of ASH1 mRNA and of a green fluorescent protein reporter particle in vivo. Curr. Biol. 9:333–336. [DOI] [PubMed] [Google Scholar]
- Czaplinski, K., and R.H. Singer. 2006. Pathways for mRNA localization in the cytoplasm. Trends Biochem. Sci. 31:687–693. [DOI] [PubMed] [Google Scholar]
- Drubin, D.G., and W.J. Nelson. 1996. Origins of cell polarity. Cell. 84:335–344. [DOI] [PubMed] [Google Scholar]
- Espreafico, E.M., R.E. Cheney, M. Matteoli, A.A. Nascimento, P.V. De Camilli, R.E. Larson, and M.S. Mooseker. 1992. Primary structure and cellular localization of chicken brain myosin-V (p190), an unconventional myosin with calmodulin light chains. J. Cell Biol. 119:1541–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada, P., J. Kim, J. Coleman, L. Walker, B. Dunn, P. Takizawa, P. Novick, and S. Ferro-Novick. 2003. Myo4p and She3p are required for cortical ER inheritance in Saccharomyces cerevisiae. J. Cell Biol. 163:1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forkey, J.N., M.E. Quinlan, M.A. Shaw, J.E. Corrie, and Y.E. Goldman. 2003. Three-dimensional structural dynamics of myosin V by single-molecule fluorescence polarization. Nature. 422:399–404. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami, S., W.K. Huh, K. Bower, R.W. Howson, A. Belle, N. Dephoure, E.K. O'Shea, and J.S. Weissman. 2003. Global analysis of protein expression in yeast. Nature. 425:737–741. [DOI] [PubMed] [Google Scholar]
- Goud, B., A. Salminen, N.C. Walworth, and P.J. Novick. 1988. A GTP-binding protein required for secretion rapidly associates with secretory vesicles and the plasma membrane in yeast. Cell. 53:753–768. [DOI] [PubMed] [Google Scholar]
- Gross, S.P., M. Vershinin, and G.T. Shubeita. 2007. Cargo transport: two motors are sometimes better than one. Curr. Biol. 17:R478–R486. [DOI] [PubMed] [Google Scholar]
- Haarer, B.K., A. Petzold, S.H. Lillie, and S.S. Brown. 1994. Identification of MYO4, a second class V myosin gene in yeast. J. Cell Sci. 107(Pt 4):1055–1064. [DOI] [PubMed]
- Hoepfner, D., M. van den Berg, P. Philippsen, H.F. Tabak, and E.H. Hettema. 2001. A role for Vps1p, actin, and the Myo2p motor in peroxisome abundance and inheritance in Saccharomyces cerevisiae. J. Cell Biol. 155:979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton, A.C., and M.D. Ehlers. 2003. Neuronal polarity and trafficking. Neuron. 40:277–295. [DOI] [PubMed] [Google Scholar]
- Itoh, T., A. Watabe, E.A. Toh, and Y. Matsui. 2002. Complex formation with Ypt11p, a rab-type small GTPase, is essential to facilitate the function of Myo2p, a class V myosin, in mitochondrial distribution in Saccharomyces cerevisiae. Mol. Cell. Biol. 22:7744–7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie, K., T. Taduchi, P.A. Takizawa, R.D. Vale, K. Matsumoto, and I. Herskowitz. 2002. The Khd1 protein, which has three KH RNA-binding motifs, is required for proper localization of ASH1 mRNA in yeast. EMBO J. 21:1158–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jambhekar, A., K. McDermott, K. Sorber, K.A. Shepard, R.D. Vale, P.A. Takizawa, and J.L. DeRisi. 2005. Unbiased selection of localization elements reveals cis-acting determinants of mRNA bud localization in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 102:18005–18010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, G.C., J.A. Prendergast, and R.A. Singer. 1991. The Saccharomyces cerevisiae MYO2 gene encodes an essential myosin for vectorial transport of vesicles. J. Cell Biol. 113:539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krementsov, D.N., E.B. Krementsova, and K.M. Trybus. 2004. Myosin V: regulation by calcium, calmodulin, and the tail domain. J. Cell Biol. 164:877–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krementsova, E.B., A.R. Hodges, H. Lu, and K.M. Trybus. 2006. Processivity of chimeric class V myosins. J. Biol. Chem. 281:6079–6086. [DOI] [PubMed] [Google Scholar]
- Leduc, C., F. Ruhnow, J. Howard, and S. Diez. 2007. From the cover: detection of fractional steps in cargo movement by the collective operation of kinesin-1 motors. Proc. Natl. Acad. Sci. USA. 104:10847–10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi, V., A.S. Serpinskaya, E. Gratton, and V. Gelfand. 2006. Organelle transport along microtubules in Xenopus melanophores: evidence for cooperation between multiple motors. Biophys. J. 90:318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, R.M., R.H. Singer, X. Meng, I. Gonzalez, K. Nasmyth, and R.P. Jansen. 1997. Mating type switching in yeast controlled by asymmetric localization of ASH1 mRNA. Science. 277:383–387. [DOI] [PubMed] [Google Scholar]
- Long, R.M., W. Gu, E. Lorimer, R.H. Singer, and P. Chartrand. 2000. She2p is a novel RNA-binding protein that recruits the Myo4p-She3p complex to ASH1 mRNA. EMBO J. 19:6592–6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Heredia, M., and R.P. Jansen. 2004. mRNA localization and the cytoskeleton. Curr. Opin. Cell Biol. 16:80–85. [DOI] [PubMed] [Google Scholar]
- Miller, K.E., and M.P. Sheetz. 2000. Characterization of myosin V binding to brain vesicles. J. Biol. Chem. 275:2598–2606. [DOI] [PubMed] [Google Scholar]
- Olivier, C., G. Poirier, P. Gendron, A. Boisgontier, F. Major, and P. Chartrand. 2005. Identification of a conserved RNA motif essential for She2p recognition and mRNA localization to the yeast bud. Mol. Cell. Biol. 25:4752–4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashkova, N., N.L. Catlett, J.L. Novak, G. Wu, R. Lu, R.E. Cohen, and L.S. Weisman. 2005. Myosin V attachment to cargo requires the tight association of two functional subdomains. J. Cell Biol. 168:359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post, P.L., M.J. Tyska, C.B. O'Connell, K. Johung, A. Hayward, and M.S. Mooseker. 2002. Myosin-IXb is a single-headed and processive motor. J. Biol. Chem. 277:11679–11683. [DOI] [PubMed] [Google Scholar]
- Pruyne, D., and A. Bretscher. 2000. Polarization of cell growth in yeast. I. Establishment and maintenance of polarity states. J. Cell Sci. 113:365–375. [DOI] [PubMed] [Google Scholar]
- Purcell, T.J., C. Morris, J.A. Spudich, and H.L. Sweeney. 2002. Role of the lever arm in the processive stepping of myosin V. Proc. Natl. Acad. Sci. USA. 99:14159–14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell, T.J., H.L. Sweeney, and J.A. Spudich. 2005. A force-dependent state controls the coordination of processive myosin V. Proc. Natl. Acad. Sci. USA. 102:13873–13878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reck-Peterson, S.L., D.W. Provance Jr., M.S. Mooseker, and J.A. Mercer. 2000. Class V myosins. Biochim. Biophys. Acta. 1496:36–51. [DOI] [PubMed] [Google Scholar]
- Reck-Peterson, S.L., M.J. Tyska, P.J. Novick, and M.S. Mooseker. 2001. The yeast class V myosins, Myo2p and Myo4p, are nonprocessive actin-based motors. J. Cell Biol. 153:1121–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reck-Peterson, S.L., A. Yildiz, A.P. Carter, A. Gennerich, N. Zhang, and R.D. Vale. 2006. Single-molecule analysis of dynein processivity and stepping behavior. Cell. 126:335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock, R.S., M. Rief, A.D. Mehta, and J.A. Spudich. 2000. In vitro assays of processive myosin motors. Methods. 22:373–381. [DOI] [PubMed] [Google Scholar]
- Rossanese, O.W., C.A. Reinke, B.J. Bevis, A.T. Hammond, I.B. Sears, J. O'Connor, and B.S. Glick. 2001. A role for actin, Cdc1p, and Myo2p in the inheritance of late Golgi elements in Saccharomyces cerevisiae. J. Cell Biol. 153:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto, T., I. Amitani, E. Yokota, and T. Ando. 2000. Direct observation of processive movement by individual myosin V molecules. Biochem. Biophys. Res. Commun. 272:586–590. [DOI] [PubMed] [Google Scholar]
- Sakamoto, T., A. Yildez, P.R. Selvin, and J.R. Sellers. 2005. Step-size is determined by neck length in myosin V. Biochemistry. 44:16203–16210. [DOI] [PubMed] [Google Scholar]
- Schott, D.H., R.N. Collins, and A. Bretscher. 2002. Secretory vesicle transport velocity in living cells depends on the myosin-V lever arm length. J. Cell Biol. 156:35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder, G.E., T. Sakamoto, J.A. Hammer III, J.R. Sellers, and P.R. Selvin. 2004. Nanometer localization of single green fluorescent proteins: evidence that myosin V walks hand-over-hand via telemark configuration. Biophys. J. 87:1776–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Johnston, D. 1995. The intracellular localization of messenger RNAs. Cell. 81:161–170. [DOI] [PubMed] [Google Scholar]
- Takizawa, P.A., and R.D. Vale. 2000. The myosin motor, Myo4p, binds Ash1 mRNA via the adapter protein, She3p. Proc. Natl. Acad. Sci. USA. 97:5273–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa, P.A., A. Sil, J.R. Swedlow, I. Herskowitz, and R.D. Vale. 1997. Actin-dependent localization of an RNA encoding a cell-fate determinant in yeast. Nature. 389:90–93. [DOI] [PubMed] [Google Scholar]
- Tang, F., E.J. Kauffman, J.L. Novak, J.J. Nau, N.L. Catlett, and L.S. Weisman. 2003. Regulated degradation of a class V myosin receptor directs movement of the yeast vacuole. Nature. 422:87–92. [DOI] [PubMed] [Google Scholar]
- Toth, J., M. Kovacs, F. Wang, L. Nyitray, and J.R. Sellers. 2005. Myosin V from Drosophila reveals diversity of motor mechanisms within the myosin V family. J. Biol. Chem. 280:30594–30603. [DOI] [PubMed] [Google Scholar]
- Vale, R.D., and R.A. Milligan. 2000. The way things move: looking under the hood of molecular motor proteins. Science. 288:88–95. [DOI] [PubMed] [Google Scholar]
- Veigel, C., S. Schmitz, F. Wang, and J.R. Sellers. 2005. Load-dependent kinetics of myosin-V can explain its high processivity. Nat. Cell Biol. 7:861–869. [DOI] [PubMed] [Google Scholar]
- Wagner, W., P. Bielli, S. Wacha, and A. Ragnini-Wilson. 2002. Mlc1p promotes septum closure during cytokinesis via the IQ motifs of the vesicle motor Myo2p. EMBO J. 21:6397–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, F., K. Thirumurugan, W.F. Stafford, J.A. Hammer III, P.J. Knight, and J.R. Sellers. 2004. Regulated conformation of myosin V. J. Biol. Chem. 279:2333–2336. [DOI] [PubMed] [Google Scholar]
- Yildiz, A., J.N. Forkey, S.A. McKinney, T. Ha, Y.E. Goldman, and P.R. Selvin. 2003. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 300:2061–2065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}