

Abstract
Endothelial cell apoptosis plays a critical role in the disruption of blood vessels mediated by natural inhibitors of angiogenesis and by anti-vascular drugs. However, the proportion of endothelial cells required to mediate a significant decrease in microvessel density is unknown. A system based on an inducible caspase (iCaspase-9) offers a unique opportunity to address this question. The dimerizer drug AP20187 induces apoptosis of human dermal microvascular endothelial cells stably transduced with iCaspase-9 (HDMEC-iCaspase-9), but not control cells (HDMEC-LXSN). Here, we generated blood vessels containing several HDMEC-iCaspase-9:HDMEC-LXSN ratios, and developed a mathematical modeling involving a system of differential equations to evaluate experimentally inaccessible ratios. A significant decrease in capillary sprouts was observed when at least 17% of the endothelial cells underwent apoptosis in vitro. Exposure to vascular endothelial growth factor (VEGF165) did not prevent apoptosis of HDMEC-iCaspase-9, but increased the apoptotic requirement for sprout disruption. In vivo experiments showed the requirement of at least 22% apoptotic endothelial cells for a significant decrease in microvascular density. The combined use of biological experimentation with mathematical modeling allowed us to conclude that apoptosis of a relatively small proportion of endothelial cells is sufficient to mediate a significant decrease in microvessel density.
Introduction
The fundamental observation that cancer is an angiogenesis-dependent disease [1-3] has provided the conceptual framework for the development of anti-angiogenic and anti-vascular agents that aim the blockade of pro-survival signals and/or the induction of endothelial cell apoptosis as a means to disrupt microvessels and treat cancer [4-9]. Several studies have demonstrated that VEGF is a potent inducer of survival of endothelial cells [10-15]. Blockade of pro-survival signals such as VEGF was shown to be sufficient to induce apoptosis of angiogenic endothelial cells [16,17]. This knowledge resulted in the development of Avastin (Bevacizumab, an humanized monoclonal anti-VEGF antibody) and Macugen (Pegaptanib, an aptamer inhibitor of VEGF), two anti-angiogenic drugs approved by FDA for treatment of cancer and age-related macular degeneration respectively [18,19]. Alternatively, anti-angiogenic strategies based on direct inhibitors of angiogenesis have also been extensively investigated [20,21]. Molecules such as thrombospondin-1 (TSP1), Endostatin, and Angiostatin disrupt angiogenic vessels through a mechanism that involves the induction of endothelial cell apoptosis, leading to consequent decreases in tissue vascularization [22-27].
One of the lingering questions in regards to the evaluation of natural inhibitors of angiogenesis and anti-vascular agents is what the minimum requirement of vascular endothelial cell apoptosis required for microvascular regression. Notably, correlations between levels of endothelial cell apoptosis induced by anti-vascular strategies in vitro, and microvascular regression in vivo, are difficult to predict. Artificial death switches such as inducible Caspases might be useful for the study of the process of microvascular regression. Inducible caspases were developed and characterized in the nineties as a means to induce selective and controlled apoptosis of cells upon exposure to dimerizer drugs [28]. They are constituted by the catalytic domain of the Caspase fused to an FKBP-based domain that allows for dimerization upon exposure to specific dimerizer drugs. These dimerizer drugs (e.g. AP20187) are not toxic to untransduced cells, while being effective in inducing apoptosis of cells expressing the fusion protein [29,30]. One of the features of inducible Caspases is that they do not seem to present the bystander effects that have been reported for other suicide genes such as the herpes simplex virus thymidine kinase gene [31].
We have generated and characterized human dermal microvascular endothelial cells (HDMEC) stably expressing iCaspase-9 (HDMEC-iCaspase-9) [32-34]. These cells undergo apoptosis in vitro, and blood vessels lined with these cells are disrupted in vivo, when iCaspase-9-expressing cells are exposed to the dimerizer drug AP20187 [32]. Notably, the dimerizer drug does not induce apoptosis of empty vector control or parental endothelial cells [32,33]. We reasoned that the ability to induce the apoptosis of a pre-determined cohort of endothelial cells while sparing the remaining endothelial cells lining the walls of blood vessels could be an useful tool to understand how much endothelial cell apoptosis blood vessels withstand before a significant drop in microvessel density is observed. Here, we engineered capillary sprouts in vitro, and human blood vessels in immunodeficient mice, with varying proportions of iCaspase-9 expressing endothelial cells and empty vector controls. Mathematical modeling using a system of differential equations was developed to evaluate experimentally inaccessible ratios. This experimental strategy based on the combination of angiogenesis assays with mathematical modeling allowed for the determination of the minimal proportion of endothelial cells that need to undergo apoptosis to cause a significant decrease in microvessel density.
Materials and Methods
Primary endothelial cells stably expressing iCaspase-9
The HDMEC-iCaspase-9 cells were generated as described previously [32]. Briefly, recombinant retroviruses were generated in PA317 cells (gift from D. Miller), and used to infect HDMEC. Viral supernatants were collected and diluted at 1/5 in the endothelial cell growth medium (EGM2-MV, Cambrex, Walkersville, MD), and HDMEC (Cambrex) were transduced by three consecutive 4 h followed by a 12 h incubation in the viral dilution supplemented with 4 μg/ml polybrene (Sigma, Saint Louis, MO) at 37°C. Starting 2 days after retroviral infection, transduced HDMEC were selected with 250 μg/ml G418 (Mediatech, Herndon, VA) for a minimum of 2 weeks.
Capillary tube assay
Varying ratios of HDMEC-iCaspase-9 and HDMEC-LXSN were seeded in 6-well plates containing 1.5 ml layer of gelled type I collagen (Cohesion, Palo Alto, CA). The total number of cells (1.5 × 105 cells) was maintained constant in all experimental conditions. Cells were allowed to attach to the collagen for 24 h, then cultured in EGM2-MV containing 50 ng/ml VEGF165 (R & D Systems, Minneapolis, MN) for 4 days, followed by additional 7 days with EGM2-MV containing 0 or 100 nM AP20187 (Ariad) alone, or together with 50 ng/ml VEGF165. Cells exposed to VEGF165 in absence of AP20187 throughout the experiment were used as positive controls for sprouting. The numbers of capillary tubes in 6 random microscope fields per well (100x) were counted daily in triplicate wells per condition. The images were taken with a Leica DM IRB microscope, and recorded in TIFF by an attached camera (Leica DC500) using Leica FireCam software (version 1.2.0).
Spheroid-based endothelial sprouting assay
Spheroid-based endothelial sprouting assay [35,36] was performed with three ratios of HDMEC-LXSN:HDMEC-iCaspase-9. Briefly, endothelial cell spheroids were generated by seeding 750 endothelial cells in EGM2-MV (Cambrex) containing 0.25% (w/v) methylcellulose (Sigma) in Greiner non-adherent round bottom 96-well plates (VWR, Suwanee, GA). After 18 h incubation at 37°C (5%) CO2, endothelial cell spheroids were centrifuged at 400 g, re-suspended in type I collagen (Cohesion), and immediately transferred into pre-warmed plates. After gel polymerization, spheroids were cultured for 12 h in EGM2-MV (Cambrex). Cell culture medium was removed, and spheroids were cultured in EGM2-MV containing 100 nM of AP20187 (Ariad) or vehicle for 4 additional days. Images were taken with a Leica DM IRB microscope, and recorded in TIFF by an attached camera (Leica DC500) using Leica FireCam software (version 1.2.0).
Flow cytometry
HDMEC-iCaspase-9 and/or HDMEC-LXSN cells were cultured with EGM2-MV supplemented with 0-100 nM AP20187 for 96 h, in presence or absence of 50 ng/ml VEGF165. For quantification of apoptosis, cells were harvested, washed and incubated in 50 μg/ml propidium iodide for 30 min. Apoptotic cells were quantified by flow cytometry, as described [32]. To determine the percentage of cells expressing iCaspase-9, cells were fixed with 2% paraformaldehyde in PBS, washed with 0.03% saponin in PBS, then incubated in monoclonal anti-HA antibody (Santa Cruz, Santa Cruz, CA) solution for 30 min at room temperature, followed by 30 min incubation in FITC conjugated anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA) solution. After washing in 0.03% saponin twice, cells were suspended in 500 μl PBS and sorted by flow cytometry. Normal mouse IgG (Santa Cruz) was used as negative control.
Trypan blue exclusion assay
To evaluate the effect of VEGF on the viability endothelial cells treated with AP20187, the Trypan blue exclusion assay was used. Briefly, varying ratios of HDMEC-iCasp-9 and HDMEC-LXSN cells (2.5 × 105 cells/well) were seeded in 24-well plates and cultured in EGM2-MV (Cambrex) supplemented with 0-100 ng/ml VEGF165 (R & D Systems) and 100 nM AP20187 (Ariad), or vehicle control. After 96 h, cells were trypsinised and stained with 0.4% (w/v) Trypan blue (ICN Pharmaceuticals, Inc. USA). The number of cells excluding Trypan blue was counted under light microscopy in a blinded fashion. Triplicate wells per condition were evaluated and the data is representative of three independent experiments.
Western blots
After a 96-hour exposure to 0-100 nM AP20187, HDMEC-iCaspase-9 or HDMEC-LXSN were harvested, whole-cell lysates were prepared as described [32,33], resolved by 12% SDS-PAGE, and transferred to a nitrocellulose membrane. The membranes were probed with 1 μg/ml monoclonal anti-Caspase-9 antibody (Oncogene, La Jolla, CA) overnight at 4°C and incubated with HRP conjugated anti-mouse IgG (Jackson ImmunoResearch) followed by exposure to an enhanced chemiluminescent substrate for detection of HRP (Pierce, Rockford, IL). Western blots with monoclonal anti-GAPDH antibody (Chemicon, Temecula, CA) were used for loading controls.
In vivo model of human angiogenesis
The poly-L-lactic acid (PLLA, Boehringer Ingelheim Pharma GmbH & Co. KG, Ingelheim, Germany) biodegradable scaffolds were prepared as previously described [37,38]. Briefly, 1 × 106 HDMEC-iCaspase-9 and/or HDMEC-LXSN were resuspended in 36 μl of a mixture (1:1) of Growth Factor-Reduced Matrigel (BD Bioscience, Bedford, MA) and EGM2-MV (Cambrex), seeded on a scaffold, and incubated for 30 min at 37°C. Male 6 to 7 week old SCID mice (Charles River Laboratory, Wilmington, MA) were anesthetized with ketamine/xylazine, and 2 scaffolds were implanted subcutaneously in the dorsum of each mouse. Starting 11 days after implantation, mice received one daily intraperitoneal injection of 2 mg/kg AP20187 in a solution of 10% PEG 400 and 1.7% Tween 20 for 3 consecutive days. The implants were retrieved and fixed in 10% neutral buffered formalin overnight at 4°C, and processed for histology as described previously [38]. The care and treatment of mice were in accordance with University of Michigan’s institutional guidelines.
Immunohistochemistry
Sections were deparaffinized in xylene, rehydrated, and washed in TBS-T, pH 8.0, then incubated in antigen retrieval solution (Dakocytomation, Dako, Carpinteria, CA) for 20 min at 90-95°C. A polyclonal rabbit anti-human Factor VIII antibody (Lab Vision, Fremont, CA) was used to localize the microvascular networks formed by the implanted human endothelial cells. Color development was performed with Dako EnVision + system kit (AEC, Dakocytomation) according to the manufacturer’s instructions. The number of microvessels in 10 random fields per implant was counted under a light microscope (200x) in 6 implants from independent mice per condition. The images were taken with a Nikon E800 microscope, recorded in TIFF by an attached camera (SPOT RT Slider; SPOT DIAGNOSTIC) using SPOT RT 3.0 Software.
Statistical analysis
One-way ANOVA followed by the Student-Newman-Keuls post-hoc tests were used for all the biological assays described here (SigmaStat 2.0 software, SPSS, Chicago, IL). Statistical significance was determined at p≤0.05.
Results
Characterization of endothelial cells stably transduced with iCaspase-9
We used recombinant retroviruses to generate new pools of primary human dermal microvascular endothelial cells (HDMEC) stably expressing iCaspase-9 (HDMEC-iCaspase-9) and empty vector controls (HDMEC-LXSN), as described [32]. We observed that the recombinant iCaspase-9 protein was detected in all HDMEC-iCaspase-9 cell pools tested here, and that the levels of endogenous Caspase-9 were not affected by the transduction of iCaspase-9 (Fig. 1A). Flow cytometry demonstrated levels of dimerizer drug (AP20187)-induced apoptosis that seem to correlate with the iCaspase-9 expression levels (Figs. 1A and B). Previous experiments have revealed that the optimal concentration of AP20187 for induction of HDMEC-iCaspase-9 apoptosis is 100 nM, and therefore this concentration was kept constant in the current study [32]. ZD5 was the pool selected for this study, since it presented the highest drug-induced apoptosis levels (about 90%) while maintaining baseline apoptosis similar to controls. Flow cytometry studies confirmed that nearly all cells in pool ZD5 were iCaspase-9 positive (Figs. 1C and D). Western blots demonstrated the appearance of a cleaved (processed) iCaspase-9 band 12 hours after exposure to AP20187, and complete processing of iCaspase-9 after 48 hours (Fig. 1E).
Fig. 1. Characterization of iCaspase-9 transduced primary endothelial cells.

(A) Expression of iCaspase-9 and endogenous caspase-9 in several pools of human dermal microvascular endothelial cell stably transduced with iCaspase-9 (HDMEC-iCasp-9) or empty vector (HDMEC-LXSN). Positive controls were 293T cells transfected with iCaspase-9. (B) Percentage of apoptotic cells analyzed by flow cytometry after propidium iodide staining. Cells were exposed to 100 nM AP20187, or vehicle, for 4 days. (C and D) Percentage of cells expressing iCaspase-9 (ZD5 pool) was evaluated by flow cytometry for HA. (E) AP20187-induced processing of iCaspase-9 in HDMEC-iCaspase-9 (ZD5) cells. Western blotting with caspase-9 antibody of cells exposed to 100 nM AP20187 for 0-48 h.
To establish the baseline information for microvessel disruption in vitro, HDMEC-iCaspase-9 or HDMEC-LXSN cells were seeded on type I collagen in 6-well plates, and cultured in endothelial cell growth medium (EGM2-MV) supplemented with 50 ng/ml VEGF165 for 4 days to induce sprouting. Thereafter, cells were exposed for 7 days to one of the following conditions: A) 50 ng/ml VEGF165; B) 50 ng/ml VEGF165 + 100 nM AP20187; or C) 100 nM AP20187. We observed that exposure to VEGF165 alone enhanced the number of capillary-like sprouts on collagen matrices, and that both HDMEC-iCaspase-9 and HDMEC-LXSN presented similar numbers of sprouts throughout the duration of the experiment (Figs. 2A and B). Treatment with AP20187 decreased the number of sprouts in collagen matrices populated with HDMEC-iCaspase-9 cells (Figs. 2E, F, H and I), but not when control HDMEC-LXSN cells were tested (Figs. 2D, F, G, and I). VEGF165 did not prevent the disruption of capillary sprouts when iCaspase-9 was activated in endothelial cells (Figs. 2E and F). Indeed, 2 days after initiation of AP20187 treatment, the number of sprouts was already lower in the HDMEC-iCaspase-9 group, as compared to controls (Fig. 2F). To confirm the results obtained with the capillary sprouting assays, we performed the spheroid-based endothelial sprouting assay (Fig. 3), as described [35,36]. We observed the differentiation of sprouts starting 12 hours after plating the endothelial cell spheroids in collagen (Fig. 3). At that time, AP20187 (or vehicle) was added and the spheroids were observed for additional 72 hours. We observed that AP20187 treatment eliminated sprouts when the spheroids were prepared with HDMEC-iCaspase-9 cells, and reduced the number of sprouts in spheroids prepared with a 3:3 ratio of HDMEC-iCaspase-9:HDMEC-LXSN. In contrast, sprouts were maintained when spheroids prepared control HDMEC-LXSN cells were exposed to AP20187, or when spheroids prepared with HDMEC-iCaspase-9 were treated with vehicle (Fig. 3). Taken together, these results demonstrate that the iCaspase-9/AP20187 is a tightly regulated model system to study the effect of endothelial cell apoptosis on microvascular density.
Fig. 2. Time course for microvessel disruption induced by endothelial cell apoptosis.

Photomicrographs (200x) of representative fields of capillary sprouts developed by HDMEC-LXSN (A, D and G) or HDMEC-iCaspase-9 cells (B, E and H) in type I collagen matrices. Cells were cultured for 4 days in endothelial growth medium supplemented with 50 ng/ml VEGF, followed by 7 days culture in medium supplemented with 50 ng/ml VEGF165 alone (A-C), 50 ng/ml VEGF165 + 100 nM AP20187 (D-F), or 100 nM AP20187 alone (G-I). Arrowheads point to capillary sprouts. (C, F and I) Graphs depict the number of sprouts per microscopic field (100x). Horizontal bars depict the duration of treatment with 50 ng/ml VEGF165. (F and I) Black arrows indicate the beginning of treatment with AP20187. At daily intervals, the number of sprouts was counted in 6 random fields from triplicate wells per condition, from three independent experiments. * P<0.05.
Fig. 3. Time course for microvessel disruption in the spheroid-based endothelial sprouting assay.

Photomicrographs (100x) of representative spheroids prepared with varying ratios of HDMEC-iCaspase-9:HDMEC-LXSN, i.e. 0:6, 3:3, or 6:0. All endothelial cell spheroids were initially cultured in collagen type I for 12 hours prior to treatment with AP20187 (or vehicle control). The number of hours depicted in the figure represents the length of time that the spheroids were treated with 100 nM AP20187, or vehicle control.
Requirement of endothelial cell apoptosis for capillary tube disruption in vitro
To study the proportion of endothelial cells that need to undergo apoptosis to disrupt capillary tubes in vitro, HDMEC-iCaspase-9 and HDMEC-LXSN were mixed in varying ratios (i.e. 6:0, 5:1, 4:2, 3:3, 2:4, 1:5, 0:6) prior to plating on collagen matrices. After 4 days in angiogenic culture conditions that allowed for the induction of capillary sprouts (i.e. EGM2-MV supplemented with 50 ng/ml VEGF165), cells were exposed to 50 ng/ml VEGF165, 100 nM AP20187, or to 50 ng/ml VEGF165 + 100 nM AP20187 for additional 7 days. In presence of VEGF165, exposure to AP20187 resulted in a significant decrease (*, p≤0.05) in number of capillary tubes when cells were mixed at a minimum ratio of 2:4 HDMEC-iCaspase-9:HDMEC-LXSN, or higher proportions of HDMEC-iCaspase-9 cells, as compared to endothelial cells exposed to VEGF165 only (Figs. 4A-E). In contrast, with all HDMEC-iCaspase-9:HDMEC-LXSN ratios (except for 6:0) we observed more sprouts (+, p≤0.05) in the VEGF165 + AP20187 group than in the AP20187 alone group (Figs. 4B-G). These data suggested that VEGF165 has a protective effect and somehow inhibited the ability of endothelial cell apoptosis to disrupt sprouts in vitro. In attempt to investigate a potential mechanism for the protective effect observed with VEGF165, we stained cells with propidium iodide and performed flow cytometry studies. We observed that VEGF165 did not affect the ability of AP20187 to mediate endothelial cell death at each one of the HDMEC-iCaspase-9:HDMEC-LXSN ratios tested here (Fig. 5A). However, cell cycle analysis demonstrated an increased proportion of viable cells in “S” phase in VEGF165-treated cultures (indicating an increase in cell proliferation), as compared to VEGF165-negative controls (Fig. 5B). To further evaluate the protective function of VEGF against AP20187-induced death of HDMEC-iCaspase-9, we performed a dose-dependency assay (Figs. 5C and D). We observed that concentrations of 50 ng/ml VEGF165 or higher were correlated with increased numbers of non-apoptotic endothelial cells, as compared to 1 ng/ml VEGF or untreated controls (Fig. 5D).
Fig. 4. Endothelial cell apoptosis and consequent capillary tube disruption in presence of VEGF.

Cells were cultured for 4 days in endothelial growth medium supplemented with 50 ng/ml VEGF165, followed by 7 days culture in medium supplemented with 50 ng/ml VEGF165 alone (squares), 50 ng/ml VEGF165 + 100 nM AP20187 (triangles), or 100 nM AP20187 alone (circles). HDMEC-iCaspase-9 and control HDMEC-LXSN were seeded at 7 different ratios: (A) 6:0, (B) 5:1, (C) 4:2, (D) 3:3, (E) 2:4, (F) 1:5 and (G), 0:6 HDMEC-iCaspase-9:HDMEC-LXSN. Arrows indicate the end of the initial period of 4 days in which all cultures were treated with VEGF to induce sprouts. At daily intervals, the number of sprouts was counted in 6 random fields from triplicate wells per condition, from three independent experiments. Vehicle control for AP20187 was EtOH and for VEGF165 was H2O. *P<0.05, reflects the comparison between the group that received 50 ng/ml VEGF165 + vehicle (squares), and the cells that were treated with 50 ng/ml VEGF165 + 100 nM AP20187 (triangles). +P<0.05, reflects the comparison between the group that received 100 nM AP20187 + vehicle (open circles), and the cells that were treated with 50 ng/ml VEGF165 + 100 nM AP20187 (triangles).
Fig. 5. Effect of VEGF on iCaspase-9 induced endothelial cell apoptosis and DNA synthesis.

(A) Percentage of apoptotic cells analyzed by flow cytometry after propidium iodide staining. Several ratios of HDMEC-iCaspase-9:HDMEC-LXSN were cultured for 96 h in EGM2-MV containing 100 nM AP20187 alone or together with 50 ng/ml VEGF165. Vehicle control for AP20187 was EtOH and for VEGF165 was H2O. (B) Percentage of cells in the “S” phase of cell cycle analyzed by flow cytometry after propidium iodide staining. Cells were treated as above. The statistical analyses were performed using the “vehicle + AP20187” group (white bar) as reference for each individual HDMEC-iCaspase-9:HDMEC-LXSN ratio (*P<0.05). (C and D) Dose dependency assay for the effect of VEGF165 on endothelial cell proliferation. (C) Photomicrographs (200x) of representative fields of with varying ratios of HDMEC-iCaspase-9:HDMEC-LXSN, i.e. 0:6, 3:3, or 6:0. Cells were seeded in 24-well plates and cultured in EGM2-MV (Cambrex) supplemented with 0-100 ng/ml VEGF165 and 100 nM AP20187. (D) Graph depicts the number of endothelial cells after 96 hours, according to HDMEC-iCaspase-9:HDMEC-LXSN ratio and concentration of VEGF165 in the culture medium, from triplicate wells per condition.
Requirement of endothelial cell apoptosis for a decrease in microvessel density in vivo
To investigate the proportion of apoptotic endothelial cells required to mediate a decrease in microvascular density, we engineered human microvessels in severe combined immunodeficient (SCID) mice, as described [38]. The same ratios of HDMEC-iCaspase-9:HDMEC-LXSN used for the in vitro studies described above were also used here. It is known that scaffolds seeded only with HDMEC-iCaspase-9 or with HDMEC-LXSN presents similar microvascular density before administration of the dimerizer drug AP20187 [32]. Starting eleven days after implantation (time required to form functional human blood vessels in this model), mice received a daily intraperitoneal injection of either vehicle or 2 mg/kg AP20187, since this concentration was determined previously to be effective in inducing HDMEC-iCaspase-9 apoptosis in vivo [32,33]. Immunostaining with anti-Factor VIII antibody was used to identify the microvessels in paraffin embedded sections, and demonstrated a direct relationship between increasing proportions of HDMEC-iCaspase-9 cells with decreasing numbers of blood vessels at the end of the experiment (Figs. 6A-G). A significant decrease in microvessel density was observed when the scaffolds were seeded with a 2:4 HDMEC-iCaspase-9:HDMEC-LXSN ratio, or higher (Fig. 6H).
Fig. 6. Activation of iCaspase-9 induced capillary tube disruption in vivo.
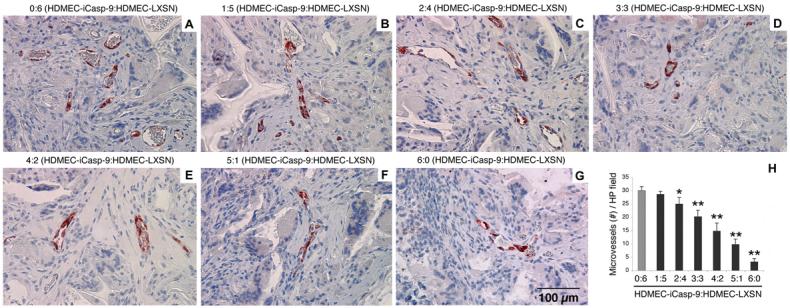
(A-G) Representative microscopic fields (400x) depicting microvessels within the biodegradable scaffolds that were implanted subcutaneously in SCID mice. Prior to seeding on the scaffolds, we mixed the HDMEC-iCaspase-9 and HDMEC-LXSN in several ratios: (A) 0:6, (B) 1:5, (C) 2:4, (D) 3:3, (E) 4:2, (F) 5:1 and (G) 6:0, respectively. Starting 11 days after implantation, and continuing 3 consecutive days thereafter, mice daily received one daily intraperitoneal injection of 2 mg/kg AP20187. Microvessels were identified with immunostaining with Factor VIII antibody (positive cells are labeled in red). (H) Graph depicting the number of microvessels in implants populated with the HDMEC-iCaspase-9:HDMEC-LXSN ratios described above. Microvessels were counted in 10 random microscopic fields (200x) in each of 5 implants per condition from independent mice. The statistical analyses were performed using the microvessel density for implants containing only control HDMEC-LXSN (gray bar) as reference (* P<0.05; ** P<0.001).
Mathematical modeling for determination of exact proportion of apoptotic endothelial cells required for a significant decrease in microvessel density
The in vitro results suggest that an initial ratio of 2:4 HDMEC-iCaspase-9:HDMEC-LXSN, i.e. killing approximately 30% of the endothelial cells by day 8, leads to significant (p=0.002) reduction in vessel density. However, the next experimental ratio tested here (i.e. 1:5 HDMEC-iCaspase-9:HDMEC-LXSN) results in a p-value of 0.495, which does not reflect a significant reduction in vessel density. In order to more rigorously investigate the proportion of endothelial cells that must be killed to obtain a significant reduction in blood vessel density, a mathematical model of the experimental procedure described above was formulated. The mathematical model consists of a system of five differential equations describing the temporal changes in the two populations of endothelial cells - HDMEC-LSXN and HDMEC-iCaspase-9 - the angiogenic factors VEGF and CXCL8, and the capillary tubes. Briefly, prior to treatment, the endothelial cells are assumed to follow the following statement of balance:
Rate of change of endothelial cells = VEGF & CXCL8-dependent proliferation - VEGF-dependentdeath - VEGF& CXCL8-dependent vascular inclusion
The proliferation, death, and vascular inclusion rates depend upon the amount of VEGF and/or CXLC8that is bound to cell-surface receptors. CXCL8 expression is included in the equation because we have demonstrated that VEGF induces CXCL8 expression in endothelial cells in a process mediated by Bcl-2 and NF-kB [39]. The equations for VEGF and CXCL8 follow the approach of [40] and start from biochemically motivated reaction diagrams that describe the molecular events associated with VEGFR2 dimerization and the monomeric binding of CXCL8 to CXCR1 and/or CXCR2 on endothelial cells. These diagrams are translated into differential equations using the Law of Mass Action and simplified (when appropriate) using the traditional quasi-steady state assumptions. Finally, the capillaries are formed from endothelial cells at a rate estimated by fitting the model to the experimental data in Fig. 4. This type of mathematical formulation can be used to predict the evolution of these species with time under circumstances that are difficult to test experimentally. The full model equations are in supplemental information along with the details of parameter estimation.
The mathematical model is used to focus on the experimentally inaccessible ratios between 2:4 and 1:5 HDMEC-iCaspase-9:HDMEC-LXSN in order to precisely determine the percentage of cells that must be killed so that a significant reduction in vessel density is obtained. The model predicts that starting with 19.39% HDMEC-iCaspase-9 and 80.61% HDMEC-LXSN cells leads to a significant reduction in vessel density (p=0.0498) upon AP20187 treatment (Fig. 7A). When corrected for the fact that 90% of the HDMEC-iCaspase-9 cells are dead 4 days after exposure to AP20187 in vitro (Fig. 1B), the critical percentage of apoptotic endothelial cells required to mediate a significant decrease in capillary sprouting is 17.45%. In addition, we used the mathematical model to investigate the efficacy of AP20187 treatment under varying VEGF concentrations in vitro. The percentage of apoptotic cells is fixed at 17.45%, i.e. at a level at which a significant reduction in vessel density is predicted in the presence of 50 ng/ml VEGF. After 4 days of treatment with AP20187, the model predicts an increase of 2.6% in the vessel density if the cells are cultured in presence of 100 ng/ml VEGF (as opposed to 50 ng/ml VEGF). In contrast, a decrease of 91% is predicted in vessel density on the fourth day of treatment, if the cells are cultured in the presence of 1 ng/ml VEGF. If no VEGF is added to the system, the decrease in vessel density is 99.9%. Taken together, these data demonstrate that the protective effect of VEGF on in vitro sprouting of endothelial cells is dose-dependent.
Fig. 7. Mathematical modeling of the effect of endothelial cell apoptosis on angiogenesis.

A system of differential equations describing the two populations of endothelial cells (HDMEC-iCaspase-9 and HDMEC-LSXN), angiogenic factors, and capillary tubes was derived for evaluation of inaccessible HDMEC-iCaspase-9:HDMEC-LXSN ratios in vitro and in vivo. (A) Data from the in vitro control experiments in which cells were supplemented with 50 ng/ml of VEGF alone (squares) are plotted together the best fit of the mathematical model (dashed line) to this data as well as the mathematical prediction of sprout number when a culture containing 19.39% HDMEC-iCaspase-9 and 80.61% HDMEC-LXSN are exposed to 50 ng/ml VEGF165 + 100 nM AP20187 (solid line). The model predicts that at this critical percentage (corrected to 17.45% to account for 90% effectiveness of drug) of apoptotic endothelial cells, there is a significant decrease (p = 0.0498) in capillary sprouting in vitro. (B) The single data point from the in vivo control experiments in which no HDMEC-iCaspase-9 cells are present (square) is plotted together with the best fit of the mathematical model (dashed line) as well as the mathematical prediction of sprout number when 24.92% of the cells are HDMEC-iCaspase-9 (solid line). The model predicts that at this critical percentage (corrected to 22.43% to account for 90% effectiveness of drug) of apoptotic endothelial cells, there is a significant decrease (p = 0.0498) in capillary sprouting in vivo. (C) Apoptotic requirement of endothelial cells is plotted versus increasing efficacy of a hypothetical anti-VEGF treatment that is applied continuously from day 11 through the end of the experiment along with 100 nM AP20187. The model predicts that for a level of anti-VEGF therapy at least 15% efficacious, a significant reduction in vessel density is obtained even in absence of HDMEC-iCaspase-9 cells (i.e. cells that will undergo apoptosis upon AP20187 treatment). As the efficacy of anti-VEGF treatment is reduced further, the critical percentage of AP20187-induced apoptotic cells required for a significant decrease in microvessel density increases to a maximum of 22.43% (if no anti-VEGF therapy is administered).
This mathematical model was modified to simulate the in vivo data, by adding background VEGF production by the host. The detailed information and equations have been added as supplemental information. The in vivo experiments indicated that 15-30% of cells needs to be killed for a significant reduction in blood vessel density (Fig. 6). Our mathematical model predicts that starting with 74.08% HDMEC-LXSN and 24.92% HDMEC-iCaspase-9 will cause a significant reduction in vessel density (p=0.0498) (Fig. 7B). When adjusted for the percentage of HDMEC-iCaspase-9 cells that undergo apoptosis when treated with AP20187, the critical percentage for achieving a significant decrease in microvessel density in vivo comes to 22.43%. We can also use the mathematical model to investigate the effect of an anti-VEGF therapy on the apoptotic requirement of endothelial cells (Fig. 7C). In modeling terms, we defined the effect of anti-VEGF therapy to be a reduction in the bioavailability of VEGF to the endothelial cells. This therapy is applied at the same time as AP20187, i.e. starting on day 11, and is continued for three additional days. The model predicts that there is a significant reduction in microvessel density for therapy with an efficacy level greater than 15%, even when we do not plate HDMEC-iCaspase-9 cells, or do not treat them with AP20187. However, for an anti-VEGF therapy that is 10% efficacious, the critical percentage of AP20187-induced endothelial cell apoptosis for achieving a significant reduction in capillary sprouting is predicted as 16.92%. This requirement increases to 19.73% for an anti-VEGF therapy that is only 5% efficacious (Fig. 7C).
Discussion
Improved knowledge of the process of microvascular regression is important for the understanding of mechanisms underlining the maintenance of vascular homeostasis in health, and its loss in disease. The validation of Folkman’s original hypothesis that a tumor is an angiogenesis-dependent disease [1] has led to the development of a number of anti-angiogenic and anti-vascular therapies for cancer treatment over the last years. Despite being quite different in nature, and target molecule, most agents being investigated today have as mechanism of action the induction of endothelial cell apoptosis leading to the disruption of the tumor’s microvascular networks. Here, we used iCaspase-9 to specifically induce apoptosis of a defined proportion of endothelial cells and examine how much endothelial cell death is required to mediate a significant decrease in microvessel density.
A collagen-based capillary sprouting assay was selected for in vitro testing because it allows for a clear distinction between a negative control condition (i.e. HDMEC cultured in EGM2-MV present little or no sprouting), and positive controls (i.e. HDMEC cultured in EGM2-MV supplemented with 50 ng/ml VEGF165 sprout vigorously) [41]. As a consequence, the effect of endothelial cell apoptosis on the number of sprouts could be readily evaluated. Exposure of the endothelial cells to VEGF165 induced a significant number of sprouts in the collagen matrices within 4 days. Both, endothelial cells expressing iCaspase-9 and control cells presented similar sprouting in response to VEGF165 stimulation. We observed that sprouts were disrupted upon activation of iCaspase-9 with the dimerizer drug AP20187, and that the degree of sprout disruption was directly dependent on the proportion of the cells expressing iCaspase-9 lining the sprouts. These results were confirmed with a second experimental model, the spheroid-based endothelial sprouting assay [35,36]. These experiments confirmed that we could induce the disruption of capillary sprouts in vitro in a controlled manner by taking advantage of the caspase-based artificial death switch iCaspase-9 [29].
It is well established that tumor cells secrete a panel of angiogenic factors that enable tumors to acquire and sustain their own vascular network [42,43]. VEGF and bFGF are among the predominant pro-angiogenic factors in tumors, and have been shown to function as strong inducers of endothelial cell survival [44,45]. VEGF promotes endothelial cell survival through upregulation of Bcl-2 expression in a process mediated by VEGFR2 and the PI3K/Akt pathways [10,39,41]. Here, we observed that VEGF165 did not prevent AP20187-induced apoptosis of HDMEC-iCaspase-9 cells. We hypothesize that the lack of VEGF’s protection against apoptosis observed when iCaspase-9 was activated reflects the fact that Caspase-9 functions downstream from Bcl-2 in the activation of the apoptotic pathway [46]. And direct activation of Caspase-9 bypasses the protective effect that is mediated by VEGF and Bcl-2 in endothelial cells. However, VEGF165 did increase the threshold of apoptotic requirement for a significant decrease in microvascular density. We observed a general trend for higher number of sprouts when cells were exposed to VEGF165 and dimerizer drug, as compared to dimerizer drug only. A similar trend was observed when cells are exposed to bFGF (data not shown). These findings were somewhat unexpected since we had observed that VEGF165 does not protect against endothelial cell death induced by activation of iCaspase-9. We hypothesize that the protective effect of VEGF165 is mediated by its ability to enhance proliferation of the cells adjacent to the apoptotic cell and “close the gap” before the vessel is disrupted. This hypothesis was supported by the observation that VEGF165 enhances proliferation of the endothelial cells that were not apoptotic. Since the majority of tumors secrete high levels of VEGF, bFGF, among other angiogenic factors, one concludes that intrinsic difficulties of treating cancer with agents that aim the induction of endothelial cell apoptosis could be exacerbated by the ability of tumor-derived angiogenic factors to enhance the blood vessel’s capability of self-repair and regeneration.
The in vivo investigation was performed with the SCID mouse model of human angiogenesis [36]. In this model, functional human microvessels are generated in biodegradable scaffolds implanted subcutaneously in the dorsal region of SCID mice. Here, varying ratios of HDMEC-iCaspase-9 and HDMEC-LXSN were mixed and seeded in the scaffolds to generate blood vessels lined with different proportions of cells that would undergo apoptosis upon administration of the dimerizer drug. The in vivo results corroborated our in vitro data, and demonstrated that approximately 22% of the neovascular endothelial cells need to undergo apoptosis to mediate a significant decrease in the number of blood vessels. The combination of mathematical modeling with the biological assays made this analysis possible, since it would not have been feasible to test every proportion of HDMEC-iCaspase-9:HDMEC-LXSN in vivo.
Since the financial and the human cost of clinical trials are significant, decisions related to whether or not a new anti-angiogenic therapeutic strategy should be tested in patients are of paramount importance. The results of the present study might be useful as a parameter for decisions in regards to the advancement of anti-vascular agents towards clinical testing. In addition, these studies further our understanding about the intricate biology of vascular regression, and demonstrate that the death of a relatively small proportion of endothelial cells is sufficient for a significant decrease in the microvessel density of a tissue.
Supplementary Material
Acknowledgements
We thank ARIAD Pharmaceuticals (www.ariad.com/regulationkits) for the dimerizer agent AP20187; the Biological Resources Branch of the NIH/NCI for the rhVEGF; and Dr. H. Weber (Spherogenex, Freiburg, Germany) for help with the spheroid-based endothelial sprouting assay. This work was supported by grants R01-DE14601, R01-DE15948, R01-DE16586 from the NIH/NIDCR (JEN), grant from the American Dental Association Health Foundation (JEN), and grants R01-CA70057 (GN) and R01-CA77266 (DMS) from the NIH/NCI.
Grant numbers and sources of support: Grants R01-DE14601, R01-DE15948, R01-DE16586 from the NIH/NIDCR (JEN), grant from the American Dental Association Health Foundation (JEN); and grants R01-CA70057 (GN) and R01-CA77266 (DMS) from the NIH/NCI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Folkman J. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- [2].Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- [3].Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- [4].Zogakis TG, Libutti SK. General aspects of anti-angiogenesis and cancer therapy. Expert. Opin. Biol. Ther. 2001;1:253–275. doi: 10.1517/14712598.1.2.253. [DOI] [PubMed] [Google Scholar]
- [5].Bisacchi D, Benelli R, Vanzetto C, Ferrari N, Tosetti F, Albini A. Anti-angiogenesis and angioprevention: mechanisms, problems and perspectives. Cancer Detect. Prev. 2003;27:229–238. doi: 10.1016/s0361-090x(03)00030-8. [DOI] [PubMed] [Google Scholar]
- [6].Mahendra G, Kumar S, Isayeva T, Mahasreshti PG, Curiel DT, Stockardt CR, Grizzle WE, Alapati V, Singh R, Siegal GP, Meleth S, Ponnazhagan S. Anti-angiogenic cancer gene therapy by adeno-associated virus 2-mediated stable expression of the soluble FMS-like tyrosine kinase-1 receptor. Cancer Gene Ther. 2005;2:26–34. doi: 10.1038/sj.cgt.7700754. [DOI] [PubMed] [Google Scholar]
- [7].Sengupta S, Eavarone D, Capila I, Zhao G, Watson N, Kiziltepe T, Sasisekharan R. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature. 2005;436:568–572. doi: 10.1038/nature03794. [DOI] [PubMed] [Google Scholar]
- [8].Folkman J. Angiogenesis and apoptosis. Semin. Cancer Biol. 2003;13:159–167. doi: 10.1016/s1044-579x(02)00133-5. [DOI] [PubMed] [Google Scholar]
- [9].Chavakis E, Dimmeler S. Regulation of endothelial cell survival and apoptosis during angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2002;22:887–893. doi: 10.1161/01.atv.0000017728.55907.a9. [DOI] [PubMed] [Google Scholar]
- [10].Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- [11].Tran J, Rak J, Sheehan C, Saibil SD, LaCasse E, Korneluk RG, Kerbel RS. Marked induction of the IAP family antiapoptotic proteins survivin and XIAP by VEGF in vascular endothelial cells. Biochem. Biophys. Res. Commun. 1999;264:781–788. doi: 10.1006/bbrc.1999.1589. [DOI] [PubMed] [Google Scholar]
- [12].Nör JE, Christensen J, Mooney DJ, Polverini PJ. Vascular endothelial growth factor (VEGF)-mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl-2 expression. Am. J. Pathol. 1999;154:375–384. doi: 10.1016/S0002-9440(10)65284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kerbel RS, Yu J, Tran J, Man S, Viloria-Petit A, Klement G, Coomber BL, Rak J. Possible mechanisms of acquired resistance to anti-angiogenic drugs: implications for the use of combination therapy approaches. Cancer Metastasis Rev. 2001;20:79–86. doi: 10.1023/a:1013172910858. [DOI] [PubMed] [Google Scholar]
- [14].Glade-Bender J, Cooney EM, Kandel JJ, Yamashiro DJ. Vascular remodeling and clinical resistance to antiangiogenic cancer therapy. Drug Resist. Updat. 2004;7:289–300. doi: 10.1016/j.drup.2004.09.001. [DOI] [PubMed] [Google Scholar]
- [15].Rosen LS. VEGF-targeted therapy: therapeutic potential and recent advances. Oncologist. 2005;10:382–389. doi: 10.1634/theoncologist.10-6-382. [DOI] [PubMed] [Google Scholar]
- [16].Klement G, Huang P, Mayer B, Green SK, Man S, Bohlen P, Hicklin D, Kerbel RS. Differences in therapeutic indexes of combination metronomic chemotherapy and an anti-VEGFR-2 antibody in multidrug-resistant human breast cancer xenografts. Clin. Cancer Res. 2002;8:221–232. [PubMed] [Google Scholar]
- [17].Stupack DG, Cheresh DA. Apoptotic cues from the extracellular matrix: regulators of angiogenesis. Oncogene. 2003;22:9022–9029. doi: 10.1038/sj.onc.1207110. [DOI] [PubMed] [Google Scholar]
- [18].Ng EW, Adamis AP. Targeting angiogenesis, the underlying disorder in neovascular age-related macular degeneration. Can. J. Ophthalmol. 2005;40:352–368. doi: 10.1016/S0008-4182(05)80078-X. [DOI] [PubMed] [Google Scholar]
- [19].Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem. Biophys. Res. Commun. 2005;333:328–335. doi: 10.1016/j.bbrc.2005.05.132. [DOI] [PubMed] [Google Scholar]
- [20].Folkman J, Hahnfeldt P, Hlatky L. The logic of anti-angiogenic gene therapy. In: Friedmann T, editor. The Development of Human Gene Therapy. Cold Spring Harbor Laboratory Press; New York: 1999. pp. 527–543. [Google Scholar]
- [21].Abdollahi A, Lipson KE, Sckell A, Zieher H, Klenke F, Poerschke D, Roth A, Han X, Krix M, Bischof M, Hahnfeldt P, Grone HJ, Debus J, Hlatky L, Huber PE. Combined therapy with direct and indirect angiogenesis inhibition results in enhanced anti-angiogenic and anti-tumor effects. Cancer Res. 2003;63:8890–8898. [PubMed] [Google Scholar]
- [22].Abdollahi A, Hlatky L, Huber PE. Endostatin: the logic of anti-angiogenic therapy. Drug Resist. Updat. 2005;8:59–74. doi: 10.1016/j.drup.2005.03.001. [DOI] [PubMed] [Google Scholar]
- [23].Dhanabal M, Ramchandran R, Waterman MJ, Lu H, Knebelmann B, Segal M, Sukhatme VP. Endostatin induces endothelial cell apoptosis. J. Biol. Chem. 1999;274:11721–11726. doi: 10.1074/jbc.274.17.11721. [DOI] [PubMed] [Google Scholar]
- [24].Rastinejad F, Polverini PJ, Bouck NP. Regulation of the activity of a new inhibitor of angiogenesis by a cancer suppressor gene. Cell. 1989;56:345–355. doi: 10.1016/0092-8674(89)90238-9. [DOI] [PubMed] [Google Scholar]
- [25].O’Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79:315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- [26].O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- [27].Nör JE, Mitra RS, Sutorik MM, Mooney DJ, Castle VP, Polverini PJ. Thrombospondin-1 induces endothelial cell apoptosis and inhibits angiogenesis by activating the caspase death pathway. J. Vasc. Res. 2000;37:209–218. doi: 10.1159/000025733. [DOI] [PubMed] [Google Scholar]
- [28].MacCorkle RA, Freeman KW, Spencer DM. Synthetic activation of caspases: artificial death switches. Proc. Natl. Acad. Sci. U. S. A. 1998;95:3655–3660. doi: 10.1073/pnas.95.7.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fan L, Freeman KW, Khan T, Pham E, Spencer DM. Improved artificial death switches based on caspases and FADD. Hum. Gene Ther. 1999;10:2273–2285. doi: 10.1089/10430349950016924. [DOI] [PubMed] [Google Scholar]
- [30].Xie X, Zhao X, Liu Y, Zhang J, Matusik RJ, Slawin KM, Spencer DM. Adenovirus-mediated tissue-targeted expression of a caspase-9-based artificial death switch for the treatment of prostate cancer. Cancer Res. 2001;61:6795–6804. [PubMed] [Google Scholar]
- [31].Chen CY, Chang YN, Ryan P, Linscott M, McGarrity GJ, Chiang YL. Effect of herpes simplex virus thymidine kinase expression levels on ganciclovir-mediated cytotoxicity and the “bystander effect”. Hum. Gene Ther. 1995;6:1467–1476. doi: 10.1089/hum.1995.6.11-1467. [DOI] [PubMed] [Google Scholar]
- [32].Nör JE, Hu Y, Song W, Spencer DM, Núñez G. Ablation of microvessels in vivo upon dimerization of iCaspase-9. Gene Ther. 2002;9:444–451. doi: 10.1038/sj.gt.3301671. [DOI] [PubMed] [Google Scholar]
- [33].Song W, Sun Q, Dong Z, Spencer DM, Núñez G, Nör JE. Anti-angiogenic gene therapy: disruption of neovascular networks mediated by inducible caspase-9 delivered with a transcriptionally targeted adenoviral vector. Gene Ther. 2005;12:320–329. doi: 10.1038/sj.gt.3302306. [DOI] [PubMed] [Google Scholar]
- [34].Pinsky MS, Song W, Dong Z, Warner K, Zeitlin B, Karl E, Hall DE, Nör JE. Activation of iCaspase-9 in neovessels inhibits oral tumor progression. J. Dent. Res. 2006;85:436–441. doi: 10.1177/154405910608500508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Korff T, Augustin HG. Integration of endothelial cells in multicellular spheroids prevents apoptosis and induces differentiation. J. Cell Biol. 1998;143:1341–1352. doi: 10.1083/jcb.143.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Korff T, Krauss T, Augustin HG. Three-dimensional spheroidal culture of cytotrophoblast cells mimics the phenotype and differentiation of cytotrophoblasts from normal and preeclamptic pregnancies. Exp. Cell Res. 2004;297:15–23. doi: 10.1016/j.yexcr.2004.03.043. [DOI] [PubMed] [Google Scholar]
- [37].Mooney DJ, Sano K, Kaufmann PM, Majahod K, Schloo B, Vacanti JP, Langer R. Long-term engraftment of hepatocytes transplanted on biodegradable polymer sponges. J. Biomed. Mater. Res. 1997;37:413–420. doi: 10.1002/(sici)1097-4636(19971205)37:3<413::aid-jbm12>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- [38].Nör JE, Peters MC, Christensen JB, Sutorik MM, Linn S, Khan MK, Addison CL, Mooney DJ, Polverini PJ. Engineering and characterization of functional human microvessels in immunodeficient mice. Lab. Invest. 2001;81:453–463. doi: 10.1038/labinvest.3780253. [DOI] [PubMed] [Google Scholar]
- [39].Karl E, Warner K, Zeitlin B, Kaneko T, Wurtzel L, Jin T, Chang J, Wang S, Wang CY, Strieter RM, Núñez G, Polverini PJ, Nör JE. Bcl-2 acts in a proangiogenic signaling pathway through nuclear factor-kappaB and CXC chemokines. Cancer Res. 2005;65:5063–5069. doi: 10.1158/0008-5472.CAN-05-0140. [DOI] [PubMed] [Google Scholar]
- [40].Levine HA, Sleeman BD, Nilson-Hamilton M. A mathematical model for the role of pericytes and macrophages in the initiation of angiogenesis. I. The role of protease inhibitors in preventing angiogenesis. Math. Biosci. 2000;168:77–115. doi: 10.1016/s0025-5564(00)00034-1. [DOI] [PubMed] [Google Scholar]
- [41].Nör JE, Polverini PJ. Role of endothelial cell survival and death signals in angiogenesis. Angiogenesis. 1999;3:101–116. doi: 10.1023/A:1009053411094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mukhopadhyay D, Datta K. Multiple regulatory pathways of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in tumors. Semin. Cancer Biol. 2004;14:123–130. doi: 10.1016/j.semcancer.2003.09.019. [DOI] [PubMed] [Google Scholar]
- [43].Presta M, Dell’Era, S. Mitola P, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- [44].Lutsenko SV, Kiselev SM, Severin SE. Molecular mechanisms of tumor angiogenesis. Biochemistry (Mosc) 2003;68:286–300. doi: 10.1023/a:1023002216413. [DOI] [PubMed] [Google Scholar]
- [45].Wary KK. Molecular targets for anti-angiogenic therapy. Curr. Opin. Mol. Ther. 2004;6:54–70. [PubMed] [Google Scholar]
- [46].Nuñez G, Benedict MA, Hu Y, Inohara N. Caspases: the proteases of the apoptotic pathway. Oncogene. 1998;17:3237–3245. doi: 10.1038/sj.onc.1202581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.