

Abstract
Selenoprotein biosynthesis is mediated by tRNASec, which inserts selenocysteine at UGA codons in a complex, context-specific manner. This opal suppressor serves in the conversion of serine to selenocysteine as well. The mouse tRNASec gene (Trsp) maps to a proximal segment of chromosome 7. We constructed mice carrying a targeted deletion of the Trsp gene. The heterozygous mutants were viable, fertile, and appeared normal. Although the level of tRNASec was reduced to about 50%–80% of the wild type in most organs, one of the selenoproteins, glutathione peroxidase, remained unaffected in the levels of its mRNA, protein, and enzyme activity, indicating that the haploid amount of tRNASec is not limiting in its biosynthesis. In contrast, the homozygous mutants died shortly after implantation, and the embryos were resorbed before 6.5 days post coitum. When the preimplantation embryos were placed in culture, however, the trophoectoderm cells showed outgrowths and the inner cell mass cells of the homozygous embryos were able to proliferate. These results indicate that Trsp expression is essential for early development of the embryo, and its lack causes peri-implantation lethality. However, the lethality does not appear to be due to a cell-autonomous function of tRNASec.
Keywords: selenium, selenoprotein, gene targeting, knockout, implantation
Selenocysteine is incorporated into protein as directed by in-frame UGA opal codons present in particular sequence contexts of mRNA encoding a limited number of proteins (for reviews see refs. 1–6). Such selenoproteins include prokaryotic formate dehydrogenases, mammalian glutathione peroxidases (GPx), selenoprotein P, and iodothyronine deiodinase types I and III, selenoprotein W, and human T-cell thioredoxin reductase (7). Selenoprotein synthesis is mediated by a specific tRNA designated as tRNASec (or tRNA[Ser]Sec), which serves as both the site of selenocysteine biosynthesis from serine and as the translational “adaptor” molecule. The details of the process are well characterized in Escherichia coli (5). They are postulated to be similar in eukaryotes (8), but with some distinct differences (6, 9). Best characterized components of selenoprotein synthesis are the tRNASec molecule itself, and the selenium donor, selenophosphate. tRNASec has been isolated and characterized from various organisms (10–13). The human gene maps to chromosome 19 (14, 15), whereas we and others mapped the mouse gene precisely to a proximal region of chromosome 7 (16, 17). The tRNA has characteristic structural features (18–20). The U34 wobble nucleoside of the UGA anticodon is modified to mcm5U, and further to mcm5Um by ribose 2′-O-methylation (21). In contrast to conventional tRNA genes, the promoter region of the tRNASec gene has no active box A element, but its transcription is controlled by an internal box B element and 5′ regulatory elements (17, 22). Decoding of UGA by tRNASec is a rather inefficient process (23), and the tRNASec level was suggested to be one of the limiting factors (24). In some patients of chronic active hepatitis, autoantibodies were detected against a tRNASec protein complex or the tRNA molecule itself (25), suggesting that lack of selenoproteins can be caused not only by dietary selenium deficiency but also by reactive mechanisms. To understand the biological role of selenoproteins, we have knocked out the mouse gene encoding tRNASec (Trsp) by homologous recombination in embryonic stem (ES) cells, constructed germ-line mutant mice, and analyzed the heterozygous and homozygous phenotypes.
MATERIALS AND METHODS
Construction of a Replacement Vector.
A targeting vector (Fig. 1A) was constructed from 129/Sv genomic Trsp fragments (17). The SwaI–XcaI (1.1 kb) and HpaI–PvuII (5.7 kb) fragments were placed upstream and downstream of the phosphoglycerate kinase I promoter/neo (PGKneo) cassette (26), respectively. Finally, the PGK-HSV-tk-bpA cassette was inserted at the upstream end for the negative selection. This cassette was constructed by replacing the neo gene in PGKneo (26) with the HSV-tk gene (27).
Figure 1.
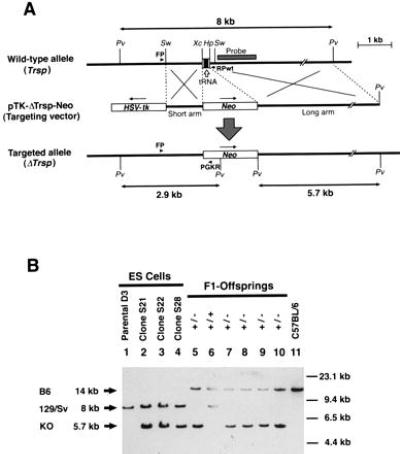
Generation of the Trsp gene knockout mice. (A) Targeting strategy by homologous recombination in ES cells showing the structures of the wild-type Trsp gene (Top), the targeting vector pTK-ΔTrsp-Neo (Middle), and the targeted allele (ΔTrsp) (Bottom). Open arrow points to the genomic segment encoding the tRNASec. Open boxes show the HSV-tk and neo gene cassettes placed in the opposite transcriptional orientations (arrows), with each cassette driven by the PGK promoter and followed by a polyadenylylation signal. The short and long arms indicate the regions of homology between the wild-type allele and the targeting vector, to cause a double crossing-over. The shaded box indicated as “Probe” shows the genomic fragment used for the Southern blot hybridization shown in B. Arrowheads denoted FP, RPwt, and PGKR indicate the PCR primers used for screening the homologous recombinant ES cells. Only relevant restriction sites are shown: Pv, PvuII; Sw, SwaI; Xc, XcaI; and Hp, HpaI. (B) Confirmation of homologous recombination in the ES cell clones, and genotype analysis of the germ-line transmitted knockout mutation in F1 offspring by Southern blot hybridization. Arrows on left indicate the hybridized PvuII fragments derived from the wild-type C57BL/6J allele (B6, 14 kb, Top) and 129/Sv allele (129/Sv, 8 kb, Middle), and the knockout allele (KO, 5.7 kb, Bottom). Molecular size markers are indicated on the right. Lanes 1–4, DNA extracted from ES cell clones were loaded (lane 1, parental ES cell line D3; lanes 2–4, homologous recombinant clones S21, S22, and S28, respectively). Lanes 5–10, tail DNA samples from F1 offspring were loaded. Note that all mice had the wild-type C57BL/6 allele and either the wild-type 129/Sv allele (lane 6) or the knockout allele (lanes 5 and 7–10). Lane 11, DNA of wild-type C57BL/6.
Gene Targeting in ES Cells.
Linearized vector DNA was introduced by electroporation into an ES cell line (D3a2). Homologous recombinant candidates were screened by PCR using primers mTRSP1 (5′-GGT AGG GCT GTT TAT GGA GCG A-3′) and PGKRa (5′-CTA AAG CGC ATG CTC CAG ACT GCC-3′), followed by Southern blot hybridization (Fig. 1B). Chimeras were generated by injecting the ES cell clones into C57BL/6 blastocysts, and transferred to pseudopregnant MCH females (CLEA Japan, Tokyo) as described (28).
Genotyping.
After proteinase K digestion of the tail and embryo [12.5 and 8.5 days post coitum (dpc)] samples, DNA was extracted by phenol and chloroform. DNA of inner cell mass (ICM), and that of 6.5 dpc embryos scraped from frozen sections were prepared by proteinase K digestion only. PCR amplifications were performed using three primers in single reactions: primer mTRSP30 [5′-CCC CTA GGG AGT TGG GCT ACA A-3′, base numbers 1519–1540 by Bösl et al. (17)]; primer PGKRa (see above); and primer mTRSP4 [5′-TTG GTC GGA AAT TCC TGG GAG G-3′, base numbers 2359–2380 by Bösl et al. (17)], for 30 cycles (60 s at 94°C, 90 s at 57°C, 120 s at 72°C).
Histological Analyses.
Decidual swellings dissected at 6.5 dpc were fixed overnight in 4% formaldehyde in phosphate-buffered saline and embedded in the OCT compound (Miles). Frozen sections were prepared at 10-μm thickness and stained with hematoxylin and eosin as described (29).
Culture of Preimplantation Embryos.
Embryos were flushed from uteri at 3.5 dpc. They were cultured on a mouse embryonic fibroblast feeder layer in Hepes-buffered Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, and incubated at 37°C in 5% CO2/95% air, as described (30).
Determination of tRNASec Levels.
This method has been described previously (17).
GPx Assay.
Formation of glutathione disulfide produced upon regeneration of the -SeH group of the enzyme was measured as described (31). Namely, subsequent conversion of glutathione disulfide to glutathione in the presence of glutathione reductase was determined by reading the decrease in A340 of NADPH in a coupled reaction: NADPH + glutathione disulfide → 2glutathione + NADP+.
Western Blot Analysis.
Essentially the same procedures were employed as described previously (28). Briefly, cells were homogenized in the solubilizing buffer by sonication. After boiling for 5 min in the sample buffer containing 100 μM 2-mercaptoethanol and 2% sodium dodecyl sulfate, 100 μg of protein per lane was separated in 10–20% gradient polyacrylamide gels and transferred to Hybond–enhanced chemiluminescence (Amersham) by electroelution. After blocking in 5% skim milk, the membrane was incubated with sheep anti-bovine GPx antibody (Biogenesis, Bournemouth, U.K.), or rabbit anti-bovine GPx antibody [a gift from R. Mizutani (Nagoya City University, Japan)]. Horseradish peroxidase-conjugated anti-sheep IgG (Cappel) or anti-rabbit IgG (Amersham) was used as a secondary antibody. The bands reactive with the antibodies were visualized by the horseradish peroxidase–enhanced chemiluminescence method (Amersham). The purified human GPx was purchased from Sigma.
RESULTS
Generation of Selenocysteine tRNA Gene Knockout Mice.
The selenocysteine tRNASec is encoded by a single gene (Trsp) on mouse chromosome 7 (17). As shown in Fig. 1A, a targeting vector (pTK-ΔTrsp-Neo) was constructed, in which the 0.25 kb XcaI–HpaI fragment of the mouse Trsp gene was deleted, including the entire tRNA coding region, and replaced with a PGKneo cassette (26). The targeting vector was introduced into ES cells by electroporation, and eight independent-candidate ES cell clones were identified by PCR out of 150 G418-resistant clones. Three of four such candidates were verified to be homologous recombinants by Southern blot analysis (Fig. 1B), and injected into C57BL/6 blastocysts. Two ES cell clones resulted in good chimeras with one transmitting the mutation to the germ line (Fig. 1B). Two mutant mouse lines were established by backcrossing the chimera with C57BL/6 (currently N9) and MCH (discontinued after N4).
Embryonic Lethality of Homozygous Knockout Mice.
Genotyping of the offspring derived from intercrosses of the Trsp (+/−) mice revealed that 72 of 183 pups were wild type, whereas the remaining 111 were all heterozygotes (Fig. 1B and Table 1). The absence of homozygous mutants, with heterozygous and wild-type offspring obtained at a ratio of approximately 2:1, suggested that homozygosity for the Trsp null mutation resulted in embryonic lethality. To determine the time of embryonic death, embryos from heterozygous crosses were genotyped at 12.5, 8.5, and 6.5 dpc. For embryos of 6.5 dpc, decidual swellings were sectioned, and the embryonic tissues were scraped off the specimens. Then, the DNA was extracted and genotyped by PCR. As shown in Table 1, all embryos that showed normal growth were either wild type or heterozygotes. At these stages, approximately one-quarter of decidual swellings were empty, with a trace of residual pyknotic cells (data not shown). The majority of them were likely to be derived from implantations of homozygous blastocysts because few empty decidua were found in Trsp (+/+) × (+/−) backcrosses (3 of 30). These results suggest that the Trsp (−/−) embryos induced the decidual reaction upon implantation, but the embryos proper died as early as at the egg cylinder stage. To investigate this phenomenon further, 59 preimplantation embryos (morulae and blastocysts) obtained at 3.5 dpc by Trsp (+/−) intercrosses were analyzed in culture. Six did not hatch out of zona pellucida or their trophoblasts could not attach to the feeder layer as outgrowths. The remaining 53 embryos showed normal trophoblast outgrowths, and their ICM cells proliferated well (Fig. 2). These embryos included not only the wild type and heterozygotes, but also homozygous mutants, although in a smaller number than expected from the Mendelian ratio (Table 1). These results strongly suggest that the growth of the Trsp (−/−) embryos became defective by the time shortly after implantation. It is also conceivable that most of the above six preimplantation embryos that did not grow in culture were Trsp (−/−).
Table 1.
Genotype analysis of Trsp (+/−) intercross progeny
| Age | Genotype
|
Resorption | ||
|---|---|---|---|---|
| +/+ | +/− | −/− | ||
| 3 weeks | 72 | 111 | 0 | NA |
| Male | 37 | 58 | 0 | |
| Female | 35 | 53 | 0 | |
| 12.5 dpc | 5 | 9 | 0 | 7 |
| 8.5 dpc | 3 | 8 | 0 | 6 |
| 6.5* dpc | 2 | 6 | 0 | 2 |
| Subtotal | 10 | 23 | 0 | 15 |
| 3.5 dpc† | 15 | 32 | 6 | NA |
NA, not applicable.
Embryonic tissues were recovered from sectioned decidua (see text and Materials and Methods).
Only those embryos that showed trophoblast outgrowth and propagation of the ICM cells in the following culture were genotyped. Of 59 blastocysts examined, 6 did not show outgrowth, and could not be genotyped, accordingly.
Figure 2.
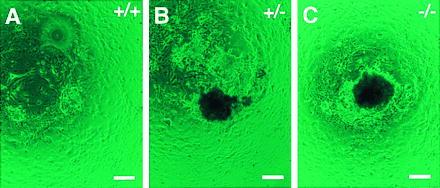
Culture of the Trsp (+/+; A), (+/−; B), and (−/−; C) preimplantation embryos in vitro. Trophoblast outgrowths with ICM cell proliferation after 10 days in culture. The genotypes were determined by PCR using the cultured cells. (Bars = 100 μm.)
Analysis of Heterozygous Knockout Mice.
The heterozygous mutant mice appeared normal in their development, gross anatomy, and behavior, and they were fertile. To investigate this phenotype of the Trsp (+/−) mice further, we determined the levels of tRNASec relative to those in the (+/+) mice in various organs, and compared them with those of tRNASer. As shown in Table 2, the levels of tRNASec in Trsp (+/−) mice were at ≈50–80% of those in the (+/+) mice in most organs. It is interesting that the level of tRNASec in the brain was similar between the Trsp (+/−) and (+/+) mice, and that the tRNASer level in the Trsp (+/−) brain was twice as high as in the (+/+) brain. Accordingly, the relative ratio of tRNASec to tRNASer was about one-half that in the Trsp (+/−) brain like in other organs. These results suggest that the levels of tRNASec in most organs reflect the gene dosage, whereas that in the brain is controlled by an additional mechanism. To investigate whether the amount of tRNASec plays a rate-limiting effect on the biosynthesis of various selenoproteins, we determined the activity of glutathione peroxidase (GPx) in the heterozygous mutants and compared it with that of the wild type. As shown in Table 3, the enzyme activity per milligram of liver protein was essentially at the same level as in the wild type. This result is consistent with the similar intensities of the GPx bands between (+/−) and (+/+) livers in the western analysis (Fig. 3). Not surprisingly, the mRNA levels determined by Northern blot analysis were also similar to those in the wild-type liver and kidney, respectively (Table 3). Accordingly, the approximately half level tRNASec in the Trsp (+/−) mice does not appear to be rate-limiting at least for GPx biosynthesis in the liver (see Discussion).
Table 2.
Relative levels of tRNASec in Trsp (+/−) mouse organs
| Organ | tRNASec*, % | tRNASer*, % | Ratio Sec/Ser†, % |
|---|---|---|---|
| Liver | 87 | 100 | 87 |
| Heart | 67 | 96 | 69 |
| Lung | 68 | 93 | 73 |
| Spleen | 73 | 140 | 52 |
| Thymus | 62 | 104 | 60 |
| Stomach | 54 | 109 | 49 |
| Small intestine | 74 | 158 | 47 |
| Brain | 101 | 206 | 49 |
| Testis | 81 | 116 | 70 |
| Kidney | 42 | 120 | 35 |
Relative levels of tRNASec and tRNASer in the Trsp (+/−) mouse organs are shown as percentages to those in the Trsp (+/+) mice.
Relative ratios of tRNASec to tRNASer in the Trsp (+/−) mouse organs are shown as percentages to those in the Trsp (+/+) mice.
Table 3.
Relative amounts of GPx in Trsp (+/−) mice
| Organ | mRNA, % | Protein, % | Enzyme activity, % |
|---|---|---|---|
| Liver | 84 | 104 | 97 |
| Kidney | 131 | ND | ND |
Relative amounts of GPx mRNA in the Trsp (+/−) mice are shown as percentages to the Trsp (+/+) mice, both calibrated to the amounts of G3PDH mRNA, whereas those of GPx protein are shown as the mean ratio of the GPx western band intensities shown in Fig. 3. The GPx enzyme activity is shown as the ratio of the Trsp (+/−) mouse liver S-100 fraction activity per milligram of protein compared with that in the Trsp (+/+) liver. The mean GPx activity ±SE was 79 ± 4 units per mg for the Trsp (+/+) liver and 71 ± 4 units per mg for the Trsp (+/−) liver. ND, not determined.
Figure 3.
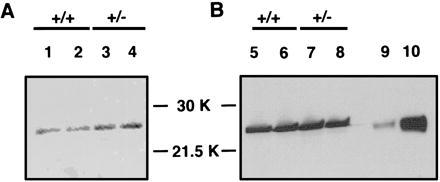
Immunoblot quantitation of GPx in Trsp (+/−) mice. The band for GPx of the relative molecular mass ≈26 kDa is seen between the molecular weight markers of 30 kDa and 21.5 kDa. Two independent antibodies against GPx were used. (A) Sheep anti-bovine GPx. (B) Rabbit anti-bovine GPx. Lanes 1, 2, 5, and 6 show liver extracts from the Trsp (+/+) mice, whereas lanes 3, 4, 7, and 8 show those from Trsp (+/−) mice. On lanes 9 and 10, purified human GPx was loaded at 1 μg and 10 μg, respectively. The results of the densitometric determination of the bands are summarized in Table 3.
DISCUSSION
Some mutants have been reported to die around implantation (reviewed in ref. 32), when the decidual reaction takes place in the uterus in response to the trophoblasts (33). Such lethalities as degeneration at the early egg cylinder stage are usually due to failures of the embryos to establish the normal connection between the trophoblasts and the decidua. Because many selenoproteins participate in scavenging reactive oxygen species (5), either directly or indirectly, it is possible that the failure to elaborate selenoproteins in Trsp (−/−) embryos makes the ICM cells more vulnerable to reactive oxygen species generated by physiological levels of metabolites. It is likely that reactive oxygen species production increases significantly upon implantation, when mammalian embryonic cells accelerate their growth rates dramatically (33). One of the mutants that cause peri-implantation lethality is evx1 knockout (34). Presumptive evx1 (−/−) embryos elicit a decidual response and invade the uterine wall but fail to differentiate extraembryonic tissues or to form the egg cylinder; a similar phenotype to that of the Trsp (−/−) embryos. Moreover, the evx1(−/−) blastocysts appear normal and, when placed in culture, the trophoblast outgrowth and ICM cell proliferation take place almost normally, like the Trsp (−/−) embryos in culture. In this connection, it is interesting that the ICM cells of homozygous embryos of the thioredoxin gene (Txn) knockout mutation do not proliferate in culture, although the blastocysts proceed to trophoblast outgrowth (30). It is worth noting that human T-cell thioredoxin reductase, which is essential for the thioredoxin function, is another selenoprotein (7). In addition to selenoproteins and thioredoxins, mammalian cells elaborate many anti-oxidative systems such as superoxide dismutase, catalase, glutaredoxin (35), and thiol-specific antioxidant (36). However, deficiency in catalase (37) or extracellular superoxide dismutase (38) does not affect mammalian development. These findings, taken together, suggest that each reducing system carries out a particular function essential for ontogeny in a specific spatio-temporal manner, although some of them appear to be redundant mechanisms to ensure the optimal redox states for various biological reactions in a fail-safe manner.
Hill et al. (39) reported that selenium deficiency causes a fall in the concentrations of selenoproteins. The GPx concentration is affected more severely than that of selenoprotein P, and this regulation is at both transcription and translation levels. As shown in Tables 2 and 3, and Fig. 3, however, neither the encoding mRNA, the protein amount, or the enzyme activity of GPx was decreased significantly in the heterozygous Trsp knockout mice. Accordingly, it is unlikely that the amount of tRNASec itself is the rate-limiting factor in selenoprotein biosynthesis. It is rather likely that the availability of selenium itself plays a key role. For example, seryl-tRNASec or aminoacrylyl-tRNASec accumulating without conversion to selenocysteyl-tRNASec in the absence of reactive selenium may causes early termination of the nascent peptides, followed by a concomitant rapid degradation of both the protein and mRNA. In fact, several circumstances have been identified in which the eukaryotic UGA codon that encodes selenocysteine can alternatively function as a stop codon, under conditions such as limiting selenium (24, 40). In this connection, it would be interesting to investigate the phenotype of the Trsp(+/−) mice under conditions of limited selenium intake.
In conclusion, the Trsp(+/−) mice we have generated should provide an animal model suitable for various investigations of selenoproteins.
Acknowledgments
We thank Dr. Y. Tamai for the PGK-HSV-tk-bpA cassette, T. Doetschman for ES cell line D3a2, P. Soriano for the PGKneo cassette, and R. Mizutani for the anti-GPx antibody. We also thank D. Hatfield and M. Matsui for fruitful discussions.
ABBREVIATIONS
- dpc
days post coitum
- GPx
glutathione peroxidase
- ICM
inner cell mass
- tRNASec
selenocysteine tRNA
- ES
embryonic stem
- PGKneo
phosphoglycerate kinase I promoter/neo
References
- 1.Stadtman T C. Annu Rev Biochem. 1990;59:111–127. doi: 10.1146/annurev.bi.59.070190.000551. [DOI] [PubMed] [Google Scholar]
- 2.Böck A. Trends Biochem Sci. 1991;16:463–467. doi: 10.1016/0968-0004(91)90180-4. [DOI] [PubMed] [Google Scholar]
- 3.Stadtman T C. J Biol Chem. 1991;266:16257–16260. [PubMed] [Google Scholar]
- 4.Böck A, Forchhammer K, Heider J, Leinfelder W, Sawers G, Veprek B, Zinoni F. Mol Microbiol. 1991;5:515–520. doi: 10.1111/j.1365-2958.1991.tb00722.x. [DOI] [PubMed] [Google Scholar]
- 5.Stadtman T C. Annu Rev Biochem. 1996;65:83–100. doi: 10.1146/annurev.bi.65.070196.000503. [DOI] [PubMed] [Google Scholar]
- 6.Low S C, Berry M J. Trends Biochem Sci. 1996;21:203–208. [PubMed] [Google Scholar]
- 7.Gladyshec V N, Jeang K-T, Stadtman T C. Proc Natl Acad Sci USA. 1996;93:6146–6151. doi: 10.1073/pnas.93.12.6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatfield D. Trends Biochem Sci. 1985;10:201–204. [Google Scholar]
- 9.Martin G W, Harney J W, Berry M J. RNA. 1996;2:171–182. [PMC free article] [PubMed] [Google Scholar]
- 10.Leinfelder W, Sehelin E, Mandrand-Berthelot M-A, Böck A. Nature (London) 1988;331:723–725. doi: 10.1038/331723a0. [DOI] [PubMed] [Google Scholar]
- 11.Lee B J, Worland P J, Davis J N, Stadtman T C, Hatfield D L. J Biol Chem. 1989;264:9724–9727. [PubMed] [Google Scholar]
- 12.Hatfield D, Lee B J, Hampton L, Diamond A M. Nucleic Acids Res. 1991;19:939–943. doi: 10.1093/nar/19.4.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hatfield D, Choi I S, Ohama T, Jung J-E, Diamond A M. In: Selenium in Biology and Human Health. Burk R F, editor. New York: Springer; 1994. pp. 25–44. [Google Scholar]
- 14.McBride O W, Rajagopalan M, Hatfield D. J Biol Chem. 1987;262:11163–11166. [PubMed] [Google Scholar]
- 15.Mitchell A, Bale A E, Lee B J, Hatfield D, Harley J, Rundle S A, Fan Y S, Fukushima Y, Shows T B, McBride O W. Cytogenet Cell Genet. 1992;61:117–120. doi: 10.1159/000133385. [DOI] [PubMed] [Google Scholar]
- 16.Ohama T, Choi I S, Hatfield D L, Johnson K R. Genomics. 1994;19:595–596. doi: 10.1006/geno.1994.1116. [DOI] [PubMed] [Google Scholar]
- 17.Bösl M R, Seldin M F, Nishimura S, Taketo M. Mol Gen Genet. 1995;248:247–252. doi: 10.1007/BF02191590. [DOI] [PubMed] [Google Scholar]
- 18.Baron C, Heider J, Böck A. Proc Natl Acad Sci USA. 1993;90:4181–4185. doi: 10.1073/pnas.90.9.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sturchler C, Westhof E, Carbon P, Krol A. Nucleic Acids Res. 1993;21:1073–1079. doi: 10.1093/nar/21.5.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu X-Q, Gross H J. Nucleic Acids Res. 1993;21:5589–5594. doi: 10.1093/nar/21.24.5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diamond A M, Choi I S, Crain P F, Hashizume T, Pomerantz S C, Cruz R, Steer C J, Hill K E, Burk R F, McCloskey J A, Hatfield D L. J Biol Chem. 1993;268:14215–14223. [PubMed] [Google Scholar]
- 22.Myslinski E, Krol A, Carbon P. Nucleic Acids Res. 1993;20:203–209. doi: 10.1093/nar/20.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berry M J, Maia A L, Kieffer J D, Harney J W, Larsen P R. Endocrinology. 1992;131:1848–1852. doi: 10.1210/endo.131.4.1396330. [DOI] [PubMed] [Google Scholar]
- 24.Berry M J, Harney J W, Ohama T, Hatfield D L. Nucleic Acids Res. 1994;22:3753–3759. doi: 10.1093/nar/22.18.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gelpi G, Sontheimer E J, Rodrigues-Sanchez J L. Proc Natl Acad Sci USA. 1992;89:9739–9743. doi: 10.1073/pnas.89.20.9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soriano P, Montgomery C, Geske R, Bradley A. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 27.Munir K M, French D C, Dube D K, Loeb L A. J Biol Chem. 1992;267:6584–6589. [PubMed] [Google Scholar]
- 28.Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M M. Proc Natl Acad Sci USA. 1995;92:4482–4486. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oshima M, Oshima H, Kobayashi M, Tsutsumi M, Taketo M M. Cancer Res. 1995;55:2719–2722. [PubMed] [Google Scholar]
- 30.Matsui M, Oshima M, Oshima H, Takaku K, Maruyama T, Yodoi J, Taketo M M. Dev Biol. 1996;178:179–185. doi: 10.1006/dbio.1996.0208. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto Y, Takahashi K. Arch Biochem Biophys. 1993;305:541–545. doi: 10.1006/abbi.1993.1458. [DOI] [PubMed] [Google Scholar]
- 32.Copp A J. Trends Genet. 1995;11:87–93. doi: 10.1016/S0168-9525(00)89008-3. [DOI] [PubMed] [Google Scholar]
- 33.Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the Mouse Embryo. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. [Google Scholar]
- 34.Spyropoulos D D, Capecchi M R. Genes Dev. 1994;8:1949–1961. doi: 10.1101/gad.8.16.1949. [DOI] [PubMed] [Google Scholar]
- 35.Luthman M, Ericksson S, Holmgren A, Thelander L. J Embryol Exp Morphol. 1979;36:363–371. [Google Scholar]
- 36.Chae H A, Robinson K, Poole L B, Church G, Storz G, Rhee S G. Proc Natl Acad Sci USA. 1994;91:7017–7021. doi: 10.1073/pnas.91.15.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahara S. Lancet. 1952;263:1101–1106. doi: 10.1016/s0140-6736(52)90939-2. [DOI] [PubMed] [Google Scholar]
- 38.Carlson L M, Honsson J, Edlund T, Markland S L. Proc Natl Acad Sci USA. 1995;92:6264–6268. doi: 10.1073/pnas.92.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hill K E, Lyons P R, Burk R F. Biochem Biophys Res Commun. 1992;185:260–263. doi: 10.1016/s0006-291x(05)80984-2. [DOI] [PubMed] [Google Scholar]
- 40.DePalo D, Kinlaw W B, Zhao C, Engelberg-Kulka H, St. Germain D L. J Biol Chem. 1994;269:16223–16228. [PubMed] [Google Scholar]