

Abstract
In the insulinoma cell line INS-1, a model system for glucose-regulated insulin secretion, the mitogen-activating protein (MAP) kinases/extracellular signal-regulated protein kinases, ERK1 and ERK2 are activated up to 15-fold by physiological concentrations of glucose, in the range of 3–12 mM. The related MAP kinase family members, the c-Jun–N-terminal kinases/stress-activated protein kinases are insensitive to glucose, while the p38 MAP kinase is slightly glucose responsive (1.5-fold). ERK activation is dependent on glucose metabolism and the subsequent increase in calcium influx. Inhibiting activation of ERK1 and ERK2 with the MEK1/2 inhibitor PD98059 has no effect on insulin secretion, indicating that ERK activity is not necessary for secretion under these conditions. Glucose activates ERK1 and ERK2 in cytosolic and purified nuclear fractions of INS-1 cells and more of each is found in nuclei from glucose-treated cells. These findings suggest that some of the glucose-dependent actions of ERKs will be exerted in the nucleus.
Mitogen-activated protein (MAP) kinases (MAPKs) are activated by many extracellular factors and are involved in numerous regulatory processes (1, 2). The best studied MAPKs, extracellular signal-regulated protein kinases 1 and 2 (ERK1 and ERK2), are the terminal enzymes in a ubiquitous and pleiotropic three-kinase cascade, consisting of Raf isoforms that activate the MAP/ERK kinases, MEK1 and MEK2, which in turn activate ERK1 and ERK2. The best documented actions of ERK1 and ERK2 are on fibroblast proliferation and differentiation of PC12 cells. In addition, they have been implicated in many other cellular events, including secretion. ERKs are activated in neurons, adrenal chromaffin cells and islet model systems by secretory stimuli (3–5). Evidence demonstrating a requirement for their activities in secretion, however, has not yet been obtained.
Insulin biosynthesis and secretion from the beta cells of the islets of Langerhans is triggered by increasing concentrations of circulating glucose, in the range of 3–20 mM (6, 7). Both transcription and translation are regulated, although the short term effects of glucose are believed to be predominantly on translation (8). Nutrients including amino acids and fatty acids potentiate effects of glucose on insulin secretion. Neural inputs and hormones influence secretion and biosynthesis in a glucose-dependent manner (9). In particular, hormones that enhance production of cAMP (10) potentiate glucose-mediated insulin release.
The stimulation of insulin secretion by glucose depends on its metabolism (11–13), as indicated by studies with nonmetabolizable sugars and with mannoheptulose, which blocks glucose phosphorylation. Secretion is initiated following closure of an ATP-dependent potassium channel and the subsequent entry of calcium through voltage-sensitive calcium channels (14). The potassium channel is a target, indirectly, for sulfonylurea oral hypoglycemic drugs (15). Elevated intracellular calcium then aids in docking secretory vesicles at the plasma membrane for fusion and release.
Van Obberghen and coworkers (5) reported that glucose led to activation of ERK1 in the rat insulinoma cell line INS-1, and that increased ERK1 activity correlated with secretion, although ERKs were activated by agents that did not promote secretion, indicating that ERK activation in another context is not sufficient for secretion. Thus, we used the INS-1 system to examine the potential requirement for ERK activity in insulin secretion. We determined that insulin secretion occurs normally even if ERK activity is blocked. We also found a marked translocation of ERKs to the nucleus elicited by glucose. Thus, it is likely that ERK activity, although not required for insulin vesicle fusion and release, is a significant regulator of glucose-dependent nuclear events involved in β-cell function.
MATERIALS AND METHODS
Cell Culture and Preparation of Cell Extracts.
The rat insulinoma cell line INS-1 was maintained as described (16). INS-1 cells were preincubated in Krebs–Ringer-bicarbonate-Hepes (KRBH) buffer at 37°C and then exposed to test agents in KRBH without serum at 37°C as indicated. Cells were lysed as described (5). The supernatants were stored at −80°C or assayed immediately.
Immunoprecipitation and Western Blot Analysis.
Anti-ERK1 antiserum Y691 that recognizes both ERK1 and ERK2 and antiserum X837 that immunoprecipitates ERK1 were as described (17). Antibodies to MEK1 (A2227) and MEK2 (A2228) selectively immunoblotted and immunoprecipitated the indicated MEK isoform (18). Antibodies that recognize the active forms of ERK1 and ERK2 were from Promega; their selective recognition of the high activity forms was confirmed as described elsewhere (A. Khokhlatchev, S. Xu, J. English, P. Wu, E. Schaefer, and M.H.C., unpublished). Antibodies were raised against recombinant stress-activated protein kinase β (SAPKβ) (O977) and recombinant p38 (P287) using methods described previously (19), and were used to immunoprecipitate c-Jun–N-terminal kinase (JNK)/SAPK and p38, respectively. Immune complex assays were performed as described (18). Immunoblots were developed with the Amersham enhanced chemiluminescence kit.
Kinase Assays.
Activity of ERK1, MEK1, MEK2, JNK/SAPK, or p38 was measured in 30 μl of 10 mM Hepes (pH 8.0), 10 mM MgCl2, 1 mM dithiothreitol, 1 mM benzamidine, and 50 μM ATP ([γ-32P]ATP, 5–15 cpm/fmol) containing 0.3 mg/ml substrate at 30°C for 15 min (ERK1) or 30 min (MEK1, MEK2, JNK/SAPK, and p38) using immunoprecipitated kinase associated with 10 μl of protein A-Sepharose beads. The substrates of ERK1, MEK1/2, JNK/SAPK, and p38 were myelin basic protein, recombinant ERK2 K52R, glutathione S-transferase (GST)-c-Jun(1–221) and GST-ATF2(1–254), respectively. Recombinant His6-ERK2 K52R, GST-ATF2(1–254), and GST-c-Jun(1–221) were expressed and purified as described (20, 21). The reactions were analyzed by electrophoresis in polyacrylamide gels (18).
Materials and Miscellaneous Methods.
Tolbutamide, glyburide and forskolin were obtained from Sigma. PD98059 was from New England Biolabs. All were dissolved in dimethyl sulfoxide for use. Bacterial expression vectors encoding GST-c-Jun(1–221) and GST-ATF2(1–254) were kindly provided by Michael Karin (University of California, San Diego, La Jolla). Tumor necrosis factor α (TNF-α) and interleukin 1β (IL-1β) were from Promega. Protein concentrations were assayed using the Lowry method with BSA as standard.
RESULTS
Glucose Activates ERK1 and ERK2 in the Insulinoma Cell Line INS-1.
As a prelude to a study of the role of ERKs in secretion, we verified an earlier report (5) by characterizing effects of glucose on ERK activities in INS-1 cells. Fig. 1A Upper shows an immunoblot of equal amounts of lysate protein from INS-1 cells probed with an antibody that selectively recognizes active, phosphorylated forms of ERK1 and ERK2. A comparable immunoblot, probed with an antibody that recognizes all forms of ERK1 and ERK2 (Fig. 1A Lower), reveals equal amounts of the two proteins in all lanes. ERK1 and ERK2 are activated in INS-1 cells in standard growth medium (11 mM glucose). When glucose is withdrawn for 1 h, activity of both enzymes drops to nearly undetectable levels. When 15 mM glucose is restored for 30 min, both kinases are activated. Comparable ERK activation is also observed when the cells are preincubated in 3 mM glucose, close to the in vivo basal glucose concentration, before stimulation by 15 mM glucose. Forskolin potentiates the effect of glucose, having a modest effect alone (Fig. 1 A and B). In agreement, immune complex kinase assays (Fig. 1B) indicated a 3-fold activation of ERK1 by 30 min of exposure to 15 mM glucose on average and a 10-fold effect of glucose plus forskolin. Glucose has no effect on ERK activity in 293 cells (Fig. 1E) or in RIN m5F cells, which have lost the capacity for glucose-responsive insulin secretion (data not shown).
Figure 1.
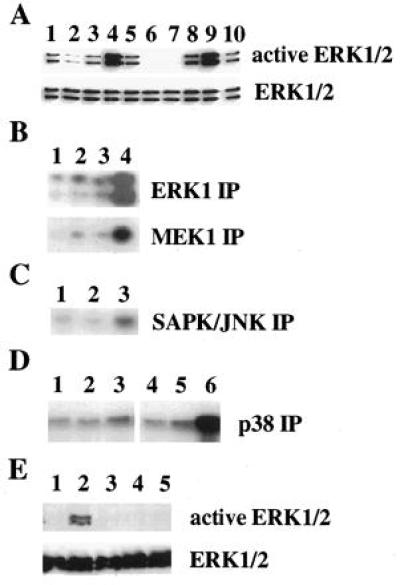
Effect of glucose on protein kinase activities in INS-1 and 293 cells. (A) Activation of ERK1 and ERK2 detected by immunoblotting of INS-1 cell lysates with active ERK antibody (Upper) and Y691 (Lower). Lane 1, INS-1 cells were maintained in growth medium containing 11 mM glucose. Cells pretreated in either 0 (lanes 6–10) or 3 (lanes 2–5) mM glucose for 1 h and then treated in KRBH for 30 min as follow: 0 mM glucose (lane 6), 3 mM glucose (lanes 2 and 7), 15 mM glucose (lanes 3 and 8), 15 mM glucose and 10 μM forskolin (lanes 4 and 9), and 10 μM forskolin (lanes 5 and 10). (B) Immune complex kinase assays of ERK1 and MEK1. Cells were preincubated in 1 mM glucose for 1 h and then incubated in KRBH without glucose (lane 1), with 15 mM glucose (lane 2), with 10 μM forskolin (lane 3), or with 15 mM glucose plus 10 μM forskolin (lane 4) for 30 min. The activities of ERK1 and MEK1 were assayed [X837, ERK-1 (Upper) or A2227, MEK1 (Lower)] using myelin basic protein or ERK2 K52R as substrates. (C) Immune complex kinase assays of JNK/SAPK. Cells were preincubated as in B and incubated in KRBH alone (lane 1), with 15 mM glucose plus 10 μM forskolin (lane 2) for 30 min, or with 10 ng/ml TNF-α and 10 ng/ml IL-1β (lane 3) for 20 min. JNK/SAPK was immunoprecipitated with O977 and assayed with GST-c-Jun(1–221). (D) Immune complex kinase assays of p38. Cells were pretreated as in B and incubated in KRBH alone (lane 1), with 15 mM glucose (lane 2) or with 15 mM glucose plus forskolin (lane 3) for 30 min; in KRBH alone (lane 4), with 10 ng/ml TNF-α (lane 5) or with 10 ng/ml IL-1β (lane 6). p38 was immunoprecipitated with P287 and assayed with GST-ATF2(1–254). In B–D, autoradiograms are shown. (E) 293 cells were serum-starved overnight and glucose was removed for the final 1 h. The cells were incubated in KRBH alone for 5 min (lane 1), KRBH with 10% fetal bovine serum for 5 min (lane 2), KRBH for 30 min (lane 3), KRBH with 15 mM glucose (lane 4), and with 15 mM glucose plus forskolin (lane 5) for 30 min. Immunoblots with active ERK antibody (Upper) and Y691 (Lower). Experiments in A and B were performed five times and in C–E two to three times.
The related p38 MAPK and 46- and 54-kDa forms of JNK/SAPK are present in INS-1 cells, as determined by immunoblotting with anti-protein antibodies. JNK/SAPK is unresponsive to glucose (Fig. 1C), while the p38 MAPK is increased marginally by glucose plus forskolin (1.5-fold, Fig. 1D). These two cascades are activated by inflammatory cytokines and have been associated with cellular stress responses (22–25); thus, it is possible that they may have a modulatory effect on secretion. Therefore, we compared the effects of cytokines on these kinases in INS-1 cells. All three are activated to different extents by interleukin-1β. ERK1 activity is increased by 2-fold (data not shown); JNK/SAPK activity is increased by 3.5-fold; and p38 activity is increased by 6-fold.
The time course of ERK1 activation by glucose with or without forskolin has been examined and compared with effects of serum, an activator of ERK in most cell types. In contrast to the expectation from a fibroblast, serum has only a small effect on activation of ERK1 or ERK2 in INS-1 cells (Fig. 2A and data not shown). On the other hand, at short times of exposure, glucose activates ERK1 by more than 5-fold in the presence or absence of forskolin. The effect of glucose alone decreases by 30 min, at which time the potentiating effect of forskolin is apparent. The activation of ERK1 by glucose plus forskolin exceeds an additive response by 30 min and reaches 15-fold by 60 min. In the presence or absence of forskolin, an effect on ERK1 and ERK2 activity could be detected at a glucose concentration of 3 mM (2.5-fold in the presence of forskolin) or lower (data not shown), at or below the threshold concentration for insulin secretion (Fig. 2C, also Figs. 4 A and C, and 5 A and D). Increasing glucose to 12 mM maximally increased activation of ERK1; no further increase was detected at 20 mM glucose in the presence of forskolin.
Figure 2.
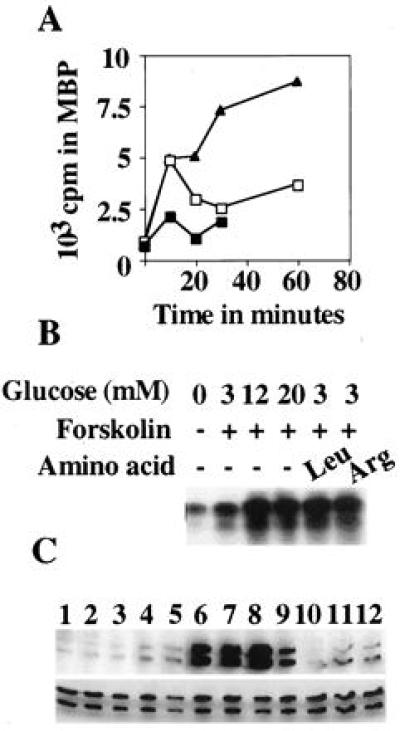
Effect of glucose and amino acids on activation of ERK1 in INS-1 cells. (A) Cells preincubated as described in legend to Fig. 1 were treated with serum (▪) and glucose with (▴) or without (□) forskolin. (B) ERK1 was immunoprecipitated from cells treated with the indicated concentrations of glucose and amino acids, leucine, and arginine and assayed in immune complexes. The myelin basic protein bands were excised and 32P-incorporation was quantitated (A), or an autoradiogram (B) is shown. (C) Cells were preincubated without glucose for 1 h and then treated for 30 min as follows: 0 mM glucose (lane 1), 1 mM glucose (lane 2), 2 mM glucose (lane 3), 3 mM glucose (lane 4), 5 mM glucose (lane 5), 10 mM glucose (lane 6), 15 mM glucose without (lanes 7 and 10), or with forskolin (lanes 8 and 11) and forskolin alone (lanes 9 and 12). 5 mM EGTA was included in lanes 10–12. Immunoblots with active ERK antibody (Upper) and Y691 (Lower). Experiments were performed at least twice.
Figure 4.
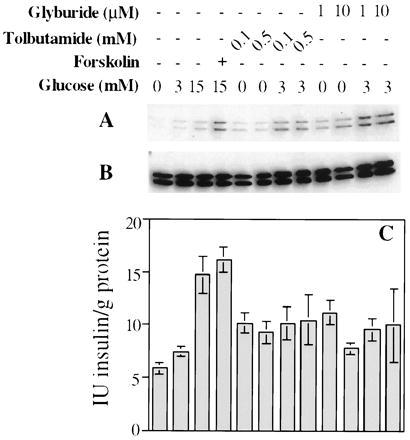
Effect of insulin secretagogues on insulin secretion and ERK1/2 activation in INS-1 cells. (A–C) Cells were deprived of glucose for 2 h and then treated with the agents indicated for 2 h at 37°C. Immunoblots using active ERK antibody (A) and Y691 (B) are shown. Matching insulin secretion profiles are shown in (C). Insulin was measured by radioimmunoassay with the Coat-a-Count insulin kit (Diagnostic Products, Los Angeles). A representative of three experiments is shown. The apparent enhancement in intensity of the signals from active ERKs in cells treated with glyburide plus glucose is probably due to increased protein loaded from glyburide-treated samples.
Potentiation of ERK Activation in INS-1 Cells Mirrors Potentiation of Insulin Secretion.
To test the ability of other agents that enhance insulin secretion to activate ERKs, we examined the capacities of leucine and arginine to act alone or to potentiate glucose-dependent activation of ERKs (Fig. 2B). Although without significant effects alone below mM concentrations, either leucine or arginine at 10 μM increases the effect of forskolin plus 3 mM glucose on ERK1/ERK2 activity. The activity attained was similar to that caused by 12–20 mM glucose plus forskolin.
Activation of ERK1/ERK2 Requires Glucose Metabolism and Calcium Entry.
We wished to determine if ERK activation occurred through the same upstream mechanisms as glucose effects on insulin biosynthesis and secretion. To determine if ERK activation by glucose requires glucose metabolism, a requisite event in stimulation of insulin secretion, effects of 2-deoxyglucose, 3-O-methylglucose, and mannoheptulose were investigated (Fig. 3). None of these agents activates ERKs (Fig. 3A). Further, the addition of 20 mM mannoheptulose, a competitive substrate of glucokinase, completely blocks the effect of glucose under all conditions (Fig. 3B) in INS-1 cells. The activities of the upstream kinases MEK1 and MEK2 have also been examined under these conditions. Mannoheptulose did not block serum activation of ERKs in 293 fibroblasts. MEK1 is activated by the same stimuli as ERK1 (Fig. 3C), but MEK2 is unresponsive (S. Xu and M.H.C., personal communication).
Figure 3.
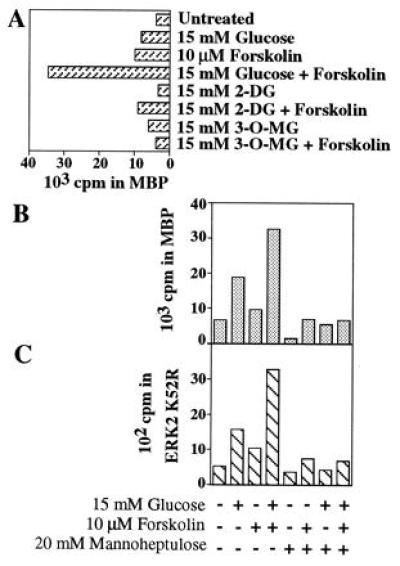
Effect of glucose analogs on ERK1 and MEK1 activities. Cells were exposed to 2-deoxyglucose (2-DG) or 3-O-methylglucose (3-OMG) (A) and ERK1 activity was measured in immune complexes. Cells were exposed to mannoheptulose and ERK1 (B) or MEK1 (C) activity was measured in immune complexes as in Fig. 1. Cells were treated with indicated agents for 30 min. 32P-incorporation into substrates is plotted. Experiments were performed at least twice.
To determine if ERKs are activated at a step following glucose metabolism, effects of the oral hypoglycemic drugs glyburide and tolbutamide were examined. Treatment of INS-1 cells with glyburide or tolbutamide for 2 h increased immunoreactive insulin in the medium by ≈2-fold (Fig. 4C) and also increased ERK activity. Addition of EGTA to INS-1 cells stimulated with glucose alone, glucose with forskolin (Fig. 2C) or KCl (data not shown) blocks ERK activation. This is consistent with the findings of Frödin et al. that Ca2+ influx causes ERK activation (5).
Inhibition of MEK Activity Blocks ERK Activation Without Influencing Insulin Secretion.
To determine if ERK activity is required for secretion, activation of ERK1 and ERK2 was blocked with PD98059, an inhibitor of MEK1 and MEK2 developed by Parke–Davis. This inhibitor blocks ERK activation in several cell types and blocks processes such as neurite outgrowth in nerve growth factor-treated PC12 cells (26–28). In INS-1 cells exposed to 100 μM PD98059, the effect of glucose plus forskolin on ERK activity was reduced to control levels (Fig. 5 A and C). On the other hand, the amount of insulin released into the medium by treatment of cells with glucose plus forskolin was the same whether or not cells were exposed to PD98059 (Fig. 5B). PD98059 did not block secretion even if cells were prestimulated and exposed to the inhibitor for 24 h prior to the second challenge with glucose (data not shown). These findings indicate that ERK activation is not required for insulin secretion.
Figure 5.
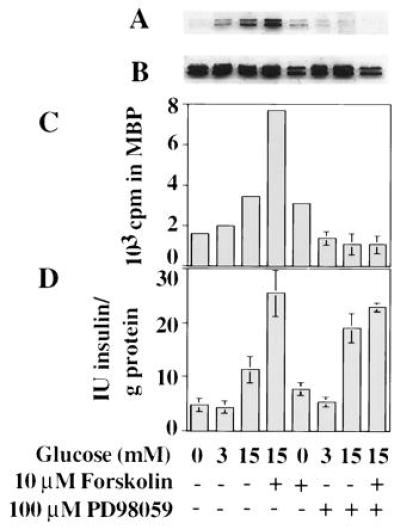
Effect of MEK inhibitor PD98059 on insulin secretion and ERK1/2 activation in INS-1 cells. Cells were preincubated in 0 mM glucose for 1 h and then treated with agents as indicated for 2 h. PD98059 (100 μM) was included in all stages of the experiment from the 1 h preincubation to the 2 h treatment for the relevant samples. Immunoblots using active ERK antibody (A) and Y691 (B) and a corresponding immune complex kinase assay with X837 antibody using myelin basic protein as substrate (C) are shown. Matching insulin secretion profiles are also shown (D). One of three experiments is shown.
Glucose Increases the Amount and the Activity of ERK1 and ERK2 in Nuclei of INS-1 Cells.
To gain insight into other possible functions of ERKs, we examined their distribution in subcellular fractions of cells before and after exposure to glucose. In the absence of glucose there was a small amount of immunoreactive ERK1 and ERK2 associated with the nuclear fraction. Treatment of cells with 15 mM glucose plus forskolin caused a marked increase in the amount and activity of ERK1 and ERK2 in the nuclear fraction (Fig. 6 A and B). Similar effects were observed with 15 mM glucose alone (data not shown). In five experiments, differences in both amount and activity of nuclear ERK1 and ERK2 were induced by glucose; however, the absolute amount of ERK1 and ERK2 in the nuclear fractions from cells not exposed to glucose varied from just barely detectable to substantial. This may be a consequence of the time of glucose deprivation, contamination of nuclei with cytosolic protein, or loss of ERKs from nuclei during preparation.
Figure 6.
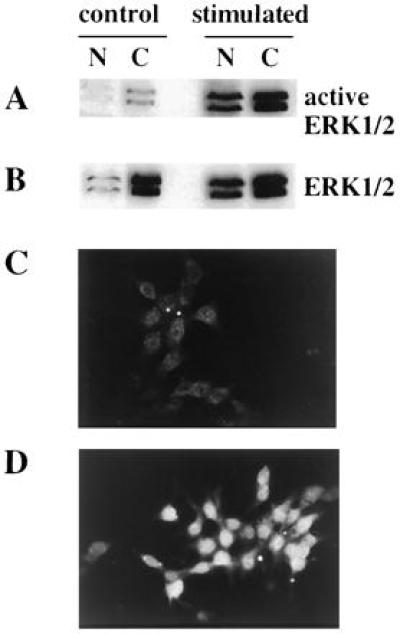
Presence of active ERK1/2 in subcellular fractions of INS-1 cells exposed to glucose and forskolin. (A and B) Cells were deprived of glucose for 2 h and then incubated in KRBH alone (control) or with 15 mM glucose plus forskolin (stimulated) for 30 min. Nuclear (N) and cytosol (C) fractions were prepared as described in (31, 34). Briefly, the cells were incubated in 10 mM Hepes (pH 7.9), 10 mM KCl, 100 mM NaF, 2 mM Na3VO4, 1 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 0.1 mM EDTA, and 0.1 mM EGTA for 20 min on ice. The cells were then lysed with 0.5% Nonidet P-40 and layered on top of 1 ml of 1 M sucrose in the hypotonic buffer and centrifuged at 1,600 × g for 10 min. The nuclei were pelleted and the cytosolic fraction remained on top of the sucrose layer. The nuclear pellet was resuspended in 1 M sucrose in buffer and centrifuged again as described above. The nuclear proteins were extracted from the pellet with 20 mM Hepes (pH 7.9), 0.42 M NaCl, 100 mM NaF, 2 mM Na3VO4, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 mM EDTA, and 1 mM EGTA. Equal amounts of protein were loaded in each lane. Immunoblots with active ERK antibody (A) and Y691 (B). (C and D) Immunofluorescence studies of active ERK1/2 in INS-1 cells. INS-1 cells were cultured for 2 days on coverslips coated with poly-l-lysine (Sigma; Mr > 300,000). The cells were deprived of glucose for 4 h and then exposed to KRBH (C) or KRBH (D) with 15 mM glucose plus forskolin for 30 min. The cells were fixed in 3.7% paraformaldehyde in PBS for 10 min and permeabilized in 0.1% Triton X-100 in PBS for 10 min. The cells were then blocked with 10 mg/ml bovine serum albumin and 0.1% Triton X-100 in PBS for 30 min. The cells were incubated in affinity-purified active ERK antibody (1:50) in PBS containing 1% Triton X-100 and 10 mg/ml albumin overnight at 4°C in a humidified chamber. After three washes with the blocking solution, the cells were incubated in tetramethylrhodamine B isothiocyanate-conjugated goat anti-rabbit antibody (1:2,000) (Cappel) for 30 min. The cells were washed three times in the blocking solution and two times in PBS before mounting for analysis by fluorescence microscopy.
Immunolocalization demonstrated faint, diffuse staining throughout the cytoplasm of glucose-deprived cells, with some, occasionally punctate, staining visible in the nucleus (Fig. 6C). In cells exposed to 15 mM glucose plus forskolin, a dramatic increase in intensity was observed in the cytoplasm and, in particular, in the nucleus, which occupies much of the cell volume (Fig. 6D). These data are consistent with results of subcellular fractionation.
DISCUSSION
Several studies have suggested a relationship of ERK activation with secretion. Electroconvulsive shock, which causes massive release of neurotransmitters, activates ERKs in discrete regions of the brain (3). ERK phosphorylation and activation is induced in adrenal chromaffin cells by a variety of secretagogues, and an essential role for it has been proposed (4). Given the complexity of biological processes regulated by signal transducing enzymes, it is unlikely that any one cascade could individually be sufficient to induce a process like secretion. Indeed, activation of ERKs by stimuli that do not elicit secretion supports this idea (5). However, many proteins are undoubtedly required for secretion including components of the secretory machinery, metabolic enzymes and signaling molecules. Thus, we determined if ERK activity is required for insulin secretion. Inhibition of ERK activation by glucose with PD98059 or through the use of dominant negative mutants in the kinase cascade (unpublished data) has no effect on insulin secretion. In spite of the coincidence of ERK activation and secretion, signaling by these enzymes is not required for vesicle fusion and release. Thus, glucose-regulated calcium influx generates two apparently independent signals, one that leads to vesicle fusion and a second that causes activation of ERK that may lead to regulatory events that support glucose-dependent insulin secretion.
ERK1 and ERK2 are activated over a physiological range of glucose concentrations and by other secretagogues. We traced the steps involved in glucose-mediated insulin secretion to determine which step leads to ERK activation. Glucose phosphorylation and calcium uptake are required. Ample evidence from this study and that of Frödin et al. (5) indicates that ERK activation is primarily a consequence of Ca2+ entry, provoked not only by glucose but also by sulfonylureas and by KCl. The molecular mechanisms used by Ca2+ to induce ERK activation are undefined. Preliminary experiments suggest that Ras is required (unpublished data). It has been reported that glucose has no effect on ERK activity in rat islets (29). Nevertheless, in preliminary experiments we find ERKs are activated substantially in intact rat islets by glucose (B. Wicksteed, S.K., M.H.C., and C. Rhodes, unpublished data), much as they are in INS-1 cells. The discrepancy could be attributed to differences in sensitivity of the detection method for ERK activity.
Subcellular localization plays an important part in determining the functions of ERK1 and ERK2. The kinetics of their activation influence the efficiency of their nuclear translocation and access to nuclear substrates in several cell types. In many resting cells ERK1 and ERK2 are in the cytoplasm and associated with microtubules (30). Stimulation of fibroblasts with serum, CCL39 cells with thrombin, and PC12 cells with NGF induces the translocation of a portion of ERK1 and ERK2 from the cytoplasm to the nucleus (31–33). Thus, mitogens and differentiating agents are thought to elicit prolonged activation and nuclear retention of these enzymes, while less potent activators cause transient activation and minimal nuclear accumulation. Surprisingly, exposure of INS-1 cells to glucose, with or without forskolin, causes the phosphorylated forms of ERK1 and ERK2 to accumulate in the nucleus. Glucose activates ERK1 and ERK2 in the cytosol and causes their nuclear translocation, and ERK1 and ERK2, already in nuclei, become activated.
These findings strongly suggest that some of the glucose-dependent actions of ERKs will be exerted in the nucleus. Active ERKs in the nucleus may be involved in maintaining important differentiated properties of these cells, such as insulin biosynthesis or the transcription of other genes that participate in the secretory function of the cells. Many agents activate multiple ERK pathways. In fibroblasts serum, for example, increases the activities not only of the ERK cascade, but also the JNK/SAPK pathway, although to a lesser extent. In contrast, in INS-1 cells glucose has no effect on the JNK/SAPK pathway, activates the ERK pathway substantially, and the p38 MAPK pathway only slightly. Thus, this system offers an opportunity to study the role of ERKs isolated from other related kinase cascades in an important biological context.
Acknowledgments
We thank Joe Albanesi for helpful discussions, Chris Newgard and Pete Antinozzi for advice about maintenance of INS-1 cells and insulin secretion assays, Peiqun Wu for bacterial proteins used as substrates and p38 immune complex kinase assays, Jessie English for critical reading of the manuscript, and Jo Hicks for its preparation. This work was supported by National Institutes of Health Research Grant DK34128.
ABBREVIATIONS
- MAP
mitogen-activated protein
- MAPK
MAP kinase
- ERK1 and ERK2
extracellular signal-regulated protein kinases 1 and 2
- JNK
c-Jun–N-terminal kinase
- SAPK
stress-activated protein kinase
- MEK
MAP/ERK kinase
- KRBH
Krebs–Ringer-bicarbonate-Hepes
- MBP
myelin basic protein
- TNF-α
tumor necrosis factor α
- IL-1β
interleukin 1β
- GST
glutathione S-transferase
References
- 1.Hunter T. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 2.Cobb M H, Goldsmith E. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 3.Stratton K R, Worley P F, Litz J S, Parsons S J, Huganir R L, Baraban J M. J Neurochem. 1991;56:147–152. doi: 10.1111/j.1471-4159.1991.tb02574.x. [DOI] [PubMed] [Google Scholar]
- 4.Ely C M, Oddie K M, Litz J S, Rossomando A J, Kanner S B, Sturgill T W, Parsons S J. J Cell Biol. 1990;110:731–742. doi: 10.1083/jcb.110.3.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frödin M, Sekine N, Roche E, Filloux C, Prentki M, Wolheim C B, Van Obberghen E. J Biol Chem. 1995;270:7882–7889. doi: 10.1074/jbc.270.14.7882. [DOI] [PubMed] [Google Scholar]
- 6.Ashcroft S J, Ashcroft F M, editors. Insulin: Molecular Biology to Pathology. New York: Oxford Univ. Press; 1992. [Google Scholar]
- 7.Efrat S, Tal M, Lodish H F. Trends Biochem Sci. 1994;19:535–538. doi: 10.1016/0968-0004(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 8.Welsh M, Scherberg N, Gilmore R, Steiner D F. Biochem J. 1986;235:459–467. doi: 10.1042/bj2350459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holz G G, Habener J F. Trends Biochem Sci. 1992;17:388–393. doi: 10.1016/0968-0004(92)90006-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holz G G 4, Leech C A, Habener J F. J Biol Chem. 1995;270:17749–17757. [PMC free article] [PubMed] [Google Scholar]
- 11.German M S. Proc Natl Acad Sci USA. 1993;90:1781–1785. doi: 10.1073/pnas.90.5.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newgard C B, McGarry J D. Annu Rev Biochem. 1995;64:689–719. doi: 10.1146/annurev.bi.64.070195.003353. [DOI] [PubMed] [Google Scholar]
- 13.Hughes S D, Quaade C, Johnson J H, Ferber S, Newgard C B. J Biol Chem. 1993;268:15205–15212. [PubMed] [Google Scholar]
- 14.Ashcroft F M, Rorsman P. Prog Biophys Mol Biol. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- 15.Ämmälä C, Moorhouse A, Gribble F, Ashfield R, Proks P, Smith P A, Sakura H, Coles B, Ashcroft S J H, Ashcroft F M. Nature (London) 1996;379:548. doi: 10.1038/379545a0. [DOI] [PubMed] [Google Scholar]
- 16.Asfari M, Janjic D, Meda P, Li G, Halban P A, Wollheim C B. Endocrinology. 1992;130:167–178. doi: 10.1210/endo.130.1.1370150. [DOI] [PubMed] [Google Scholar]
- 17.Boulton T G, Cobb M H. Cell Regul. 1991;2:357–371. doi: 10.1091/mbc.2.5.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu S, Robbins D, Frost J, Dang A, Lange-Carter C, Cobb M H. Proc Natl Acad Sci USA. 1995;92:6808–6812. doi: 10.1073/pnas.92.15.6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng M, Boulton T G, Cobb M H. J Biol Chem. 1996;271:8951–8958. doi: 10.1074/jbc.271.15.8951. [DOI] [PubMed] [Google Scholar]
- 20.Robbins D J, Zhen E, Owaki H, Vanderbilt C, Ebert D, Geppert T D, Cobb M H. J Biol Chem. 1993;268:5097–5106. [PubMed] [Google Scholar]
- 21.Frost J, Xu S, Hutchison M, Marcus S, Cobb M H. Mol Cell Biol. 1996;16:3707–3713. doi: 10.1128/mcb.16.7.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kyriakis J M, Banerjee P, Nikolakaki E, Dai T, Rubie E A, Ahmad M F, Avruch J, Woodgett J R. Nature (London) 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 23.Lee J C, Laydon J T, Mcdonnell P C, Gallagher T F, Kumary S, Green D, McNulty D, Blumenthal M J, Heys J R, Landvatter S W, Strickler J E, McLaughlin M M, Siemens I R, Fisher S M, Livi G P, White J R, Adams J L, Young P R. Nature (London) 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 24.Han J, Lee J-D, Bibbs L, Ulevitch R J. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 25.Dérijard B, Hibi M, Wu I-H, Barrett T, Su B, Deng T, Karin M, Davis R J. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 26.Pang L, Sawada T, Decker S J, Saltiel A R. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 27.Alessi D R, Cuenda A, Cohen P, Dudley D T, Saltiel A R. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 28.Dudley D T, Pang L, Decker S J, Bridges A J, Saltiel A R. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Persaud S J, Wheeler-Jones C P D, Jones P M. Biochem J. 1996;313:119–124. doi: 10.1042/bj3130119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reszka A A, Seger R, Diltz C D, Krebs E G, Fischer E H. Proc Natl Acad Sci USA. 1995;92:8881–8885. doi: 10.1073/pnas.92.19.8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen R-H, Sarnecki C, Blenis J. Mol Cell Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lenormand P, Sardet C, Pages G, L’Allemain G, Brunet A, Pouysségur J. J Cell Biol. 1993;122:1079–1088. doi: 10.1083/jcb.122.5.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez F A, Seth A, Raden D L, Bowman D S, Fay F S, Davis R J. J Cell Biol. 1993;122:1089–1101. doi: 10.1083/jcb.122.5.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dignam J D, Lebovitz R M, Roeder R G. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]