

Abstract
The Cdc6 protein is essential for the assembly of pre-replicative complexes (pre-RCs) at origins of DNA replication in the budding yeast Saccharomyces cerevisiae. This reaction is blocked in vivo by the cyclin-dependent kinase Cdc28p, together with its regulatory subunits, the B type cyclins that are present throughout S, G2, and M phases. Because the destruction of B type cyclins and the consequent inactivation of the kinase are essential for exit from mitosis, pre-RC formation can only occur after passage through mitosis. Therefore, pre-RC formation has been proposed to be essential for coupling S phase and mitosis and for limiting DNA replication to once per cell cycle. The Mcm2–7 family of proteins has been implicated in limiting replication to once per cell cycle from experiments with Xenopus egg extracts. Here we show that the Mcm proteins of budding yeast are abundant and are quantitatively found in a chromatin-enriched fraction specifically during the G1 phase of the cell cycle. This chromatin binding depends on the de novo synthesis of Cdc6p, providing evidence that a conserved biochemical pathway plays a critical role in coordinating DNA replication with mitosis in both yeast and higher eukaryotes. Cdc6p and the origin recognition complex can be selectively removed from this chromatin-enriched fraction without removing the Mcm proteins. From these results, we propose that Cdc6p (and the origin recognition complex) nucleates the binding of Mcm proteins to chromatin, but once bound, the Mcm proteins appear to interact tightly with some other component of chromatin.
Understanding how DNA replication is regulated is an important goal in cell cycle research. Classic cell fusion experiments have defined two levels to this regulation. First, S phase cells produce a diffusible factor that triggers DNA replication in G1 nuclei. Second, G2 nuclei do not respond to the S phase activity; therefore, passage through mitosis is required to make nuclei competent for another round of DNA replication (1, 2).
Experiments in a wide variety of organisms have indicated that the cyclin-dependent kinases (cdk) play two key roles in regulating DNA replication. First, a cdk and a companion cyclin are required to trigger DNA replication at the start of S phase. Second, the cdk–cyclin pairs that are active from the beginning of the S phase until the end of mitosis act to block re-replication (reviewed in refs. 3–6). Thus, the destruction of cyclins at the end of mitosis is required both for exiting mitosis and removing the block to re-replication.
In budding yeast, the six subunit origin recognition complex (ORC) (7) appears to remain bound at origins during most or all of the cell cycle (8, 9). Pre-replicative complexes (pre-RCs) assemble at replication origins at the end of mitosis (9) in a reaction that requires Cdc6p (10), a protein that is essential for initiating DNA replication and that interacts with ORC (11). Cdc6p can only promote the formation of pre-RCs before a “point of no return” that occurs in late G1 (12). This point of no return coincides with the activation of two B type cyclins (Clbs), Clb 5 and Clb 6, which are also required for triggering S phase. The fact that the point of no return can be delayed by deleting these cyclins provides evidence that Clbs are required for establishing the block to pre-RC formation. In addition, inactivation of Clb kinase in G2 by overexpression of the Clb kinase inhibitor p40SIC1 is sufficient to drive the formation of pre-RCs (13). Previous experiments in fission yeast, Schizosaccharomyces pombe, have indicated that the mitotic cdk–cyclin pair cdc2+–cdc13+ functions to inhibit re-replication (14–16). Together, these experiments argue that one mechanism by which Clb kinase blocks re-replication is by blocking the formation of new pre-RCs. The mechanism of this inhibition is currently unknown.
From experiments in Xenopus egg extracts, a “licensing” reaction, essential for initiating DNA replication, has been described; this reaction is blocked by the presence of the nuclear envelope and, therefore, can only occur after nuclear envelope breakdown in mitosis (17). This reaction leads to the loading of the Mcm2–7 family of proteins onto chromatin (18–21). The Mcm proteins were first identified in yeast in genetic screens for minichromosome maintenance mutants (22–24) and cell division cycle mutants (25–29). In budding yeast, five related Mcm proteins have been characterized: Mcm2p, Mcm3p (23), Cdc46p (30), Cdc47p (31), and Cdc54p (32). A sixth protein, most closely related to the S. pombe mis5+ (24), has been identified in the yeast genome project. Additional factors are required for the binding of the Mcm proteins to chromatin, including homologues of ORC (33–35) and Cdc6p (36).
In this article, we show that the budding yeast Mcm proteins, like their Xenopus counterparts, are loaded onto chromatin during G1 in a Cdc6p-dependent reaction. This suggests that DNA replication is limited to once per cell cycle by a biochemical pathway widely conserved among eukaryotes. We also show that the Mcm proteins are bound to chromatin during G1 at levels that far exceed the number of active replication origins. Furthermore, the Mcm proteins remain bound to chromatin in vitro after selective extraction of ORC and Cdc6p. From these experiments we propose that Cdc6p and ORC are essential for the loading of the Mcm proteins onto chromatin; however, once loaded, maintenance of Mcm chromatin binding does not appear to require ORC or Cdc6p.
MATERIALS AND METHODS
Strain Construction.
The cdc6–1 strain is congenic with A364A (MATa ura1 ade1 ade2 tyr1 his7 lys2 gal1–1). The cdc47–1 strain is congenic with W303–1a (MATa ade2–1 ura3–1 his3–11, 15 trp1–1 leu2–3, 112 can1–100). Cdc46p, Cdc47p, and Mcm3p were tagged using the strategy previously described (37) in the strain PY26 (MATa ura3–52 trp1 prb1–1122 prc1–407 pep4–3 leu2–3, 112 NUC1::LEU2) or in the above strains. The URA3 gene in 4142 (MATa cdc15–2, cdc6::hisGURA3hisG, trp1::TRP1 MET-CDC6, leu2; ref. 10) was removed after selection on 5-fluoroorotic acid to generate YLD1, and Cdc46p or Cdc47p were tagged in this strain as described above. In all of the resulting strains, the tagged gene is expressed from its own promoter to minimize the possibility of altering protein levels. Because the tagged gene is the only gene present and because each of these genes is essential, the tag does not interfere with the essential function of these proteins. We found during the course of our analysis that the Mcm3p strain was rendered temperature-sensitive by the tag and was, therefore, not pursued. The tag consists of a single c-myc epitope (37) and nine histidine residues fused in frame to the C terminus of each protein. Cdc46p was purified to near homogeneity using Ni-NTA agarose chromatography and immunoaffinity chromatography using immobilized 9E10 (unpublished data). For the experiments shown in Fig. 2, extracts were prepared by bead beating essentially as described (9).
Figure 2.
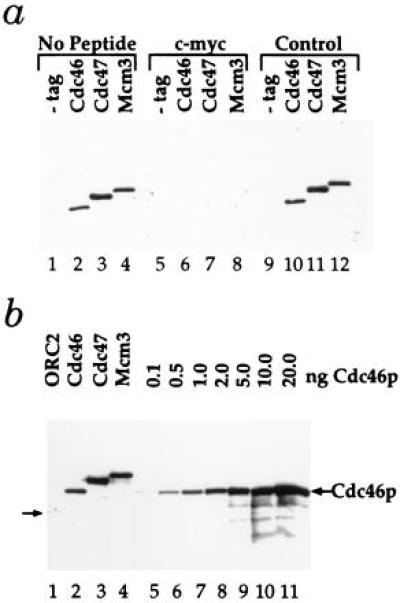
Detection and abundance of Mcm proteins. (a) Whole-cell extracts from either the untagged parental strain PY26 (−tag) or strains containing the indicated tagged genes were subjected to immunoblot analysis. Blots were probed with the 9E10 monoclonal antibody specific for the c-myc epitope either alone (No Peptide) or after preincubation with the c-myc peptide (c-myc) or an unrelated control peptide (Control). (b) Whole-cell extracts from strains containing the indicated tagged genes (lanes 1–4) were subjected to immunoblot analysis alongside equal amounts of an extract from an untagged strain to which the indicated amounts of purified, tagged Cdc46p were added (lanes 5–11).
Chromatin Purification.
To examine chromatin binding, extracts were prepared from “semi-intact” cells as described (38) with the following modifications. After spheroplasting and regrowth, cells were washed three times with lysis buffer (0.4M Sorbitol/150 mM potassium acetate/2 mM magnesium acetate/20 mM Pipes/KOH, pH 6.8/1 mM phenylmethylsulfonyl fluoride/10 μg/ml leupeptin/1 μg/ml pepstatin A/10 mM benzamidine). Cells were resuspended in lysis buffer at no more than 8 × 108 cells per ml and lysed by addition of Triton X-100 to a final concentration of 1%. The chromatin-enriched fraction was isolated after centrifugation for 15 min at 15,800 × g, and the supernatant was carefully removed. Nocodazole and α factor blocks were performed as described (9). During all blocks, morphology was monitored to ensure that cells remained blocked. For Deoxyribonuclease1 (DNase1) treatment, extracts were incubated with 3 Kunitz units of DNase1. In all cases, lysates were separated into a supernatant and a pellet as described above after a 5-min incubation on ice. For the immunoblot analysis described in this paper, proteins were transferred to nitrocellulose and blocked with 5% dry milk in Tris-buffered saline containing 0.1% Tween 20. Purified 9E10 was used at 12.5 μg/ml. Purified 9H85 (anti-Cdc6p monoclonal) was used at 5 μg/ml. JAB12 is a polyclonal antibody raised to recombinant Orc2p protein and was used at a 1:500 dilution. Horseradish peroxidase-coupled anti-mouse was used with the monoclonal antibodies, horseradish peroxidase-coupled protein A was used with JAB12 (Sigma), and immunoreactive bands were visualized with enhanced chemiluminescence (ECL, Amersham) according to the manufacturer’s instructions.
RESULTS AND DISCUSSION
Chromatin Binding Assay.
Genomic footprinting experiments have provided considerable detail about the regulation of protein–DNA complexes at budding yeast origins. However, with this approach it is difficult to determine which proteins are actually bound to replication origins in chromatin. Consequently, we have developed a procedure to partially purify chromatin from budding yeast cell lysates. In this procedure, spheroplasts are lysed by addition of non-ionic detergent, and chromatin is collected by centrifugation through sorbitol-containing buffer. As shown in Fig. 1a, when fractions from the purification are analyzed for protein content by SDS/PAGE, less than 5% of the total cellular protein is found in the chromatin-enriched fraction. Conversely, nearly all of the total cellular DNA is found in this fraction (Fig. 1b). Therefore, this procedure results in a considerable purification of chromatin.
Figure 1.
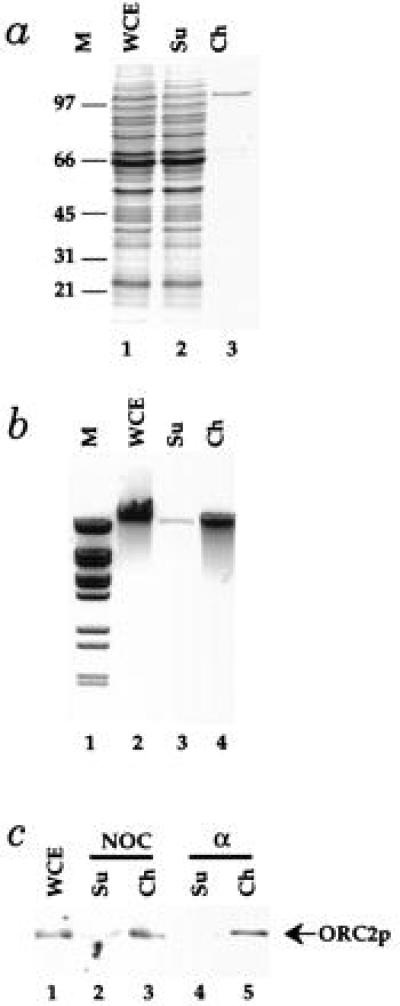
A chromatin isolation procedure. The chromatin isolation procedure described in Materials and Methods was applied to cell lysates from PY26. (a) The protein content of fractions from the chromatin purification were examined by SDS/PAGE in 10% acrylamide gels followed by staining with Coomassie brilliant blue. Protein from the starting whole cell extract (WCE), the supernatant (Su), and the purified chromatin fraction (Ch) from equal cell equivalents was loaded. The positions of molecular weight markers (M) are shown on the left. (b) DNA was purified by phenol extraction and ethanol precipitation from the same fractions and subjected to electrophoresis in a 0.8% agarose gel containing ethidium bromide. (c) The amounts of ORC2p in fractions from this procedure was determined by immunoblotting as described in Materials and Methods for cells blocked in G2 with nocodazole or G1 with α factor.
Genomic footprints of replication origins in G2 are very similar to the footprints produced in vitro with purified ORC (9). Furthermore, this postreplicative footprint is thermolabile in an orc2 temperature-sensitive mutant (39). Because Orc2p, the 72-kDa subunit of ORC, is not present in large excess over the number of replication origins in G2 blocked cells (40), it represents a useful example of a chromatin-bound protein that is not in excess over its binding sites in chromatin. As shown in Fig. 1c, although less than 5% of the total cellular protein is in the chromatin fraction, virtually all of the Orc2p is found in this fraction from cells blocked in G2. Thus, the chromatin purification protocol does not appear to disrupt initiation complexes.
Because the pre-RC causes significant extension of the ORC footprint in G1, resulting in protection of the highly diagnostic ORC-induced DNase1 hypersensitive sites as well as other sites in domain B (9, 39), our evidence that ORC is actually a component of pre-RCs has been circumstantial (see ref. 10 for discussion). Therefore, it was of interest to examine whether ORC was bound to chromatin during G1. As shown in Fig. 1c, virtually all of the Orc2p is also found in the chromatin fraction from G1 blocked cells. This is consistent with the idea that ORC is a component of pre-RCs.
Cdc46p, Cdc47p, and Mcm3p Are Abundant Cellular Proteins.
For biochemical analysis, we have constructed strains in which individual Mcm proteins, expressed from their own promoters, have been tagged at their C termini with an epitope from the c-myc gene (41) to allow their detection by immunoblotting. Fig. 2a demonstrates that c-myc tagged Cdc46p, Cdc47p, and Mcm3p can be detected in crude whole-cell extracts. Single polypeptides between 100 and 120 kDa are specifically detected in extracts of each of the indicated tagged strains but not the untagged parental strain (Fig. 2, lanes 1–4). This immunoreactivity is blocked by the c-myc peptide (Fig. 2, lanes 5–8) but not by an unrelated control peptide (lanes 9–12). Therefore, we can unambiguously identify these three Mcm proteins in crude whole-cell extracts.
We have previously described strains in which Orc2p was also tagged with the c-myc epitope (ref. 37; Fig. 2b, lane 1). Because the strategy we have used ensures that each of the proteins is tagged with the same epitope and is expressed from its own promoter, we can directly compare the levels of these proteins, assuming that the tag does not alter the protein copy number. We have previously shown that this tag does not affect the levels of Orc2p (40). Comparison of lane 1 with lanes 2–4 in Fig. 2b indicates that each of the Mcm proteins examined is present at considerably higher levels than Orc2p. To determine the amount of each Mcm protein per cell, levels of the individual Mcm proteins in whole-cell extracts from 1.1 × 106 cells of the tagged strains (Fig. 2b, lanes 2–4) was compared with the indicated amounts of purified Cdc46p added to a whole-cell extract from 1.1 × 106 cells of the parental untagged strain (lanes 5–11). This analysis indicated that there are approximately 2 ng of Cdc46p, 5 ng of Cdc47p, and 3 ng of Mcm3p, corresponding to 12,000, 30,000, and 18,000 copies per cell, respectively. Thus, each of these Mcm proteins is at least 20–50 times more abundant than ORC. Tye and coworkers (42) have recently examined the abundance of Mcm proteins estimating the intracellular levels of Mcm2 and Mcm3 to be 40,000 and 200,000 copies per cell, respectively. Although our estimate of the amount of Mcm3 per cell is lower, it is still in great excess over the amount of ORC.
Cdc46p and Cdc47p Are Bound to Chromatin During G1.
We next examined the possibility that Mcm proteins might be in the chromatin-enriched fraction. Consistent with previous analysis (30), the overall levels of Cdc46p are very similar in G1 and G2/M blocked cells (Fig. 3a, lanes 1 and 4). However, less than 5% of the total Cdc46p is found in chromatin fractions from cells blocked in G2/M with nocodazole (Fig. 3a, lanes 1–3), whereas approximately half of the Cdc46p is found in the chromatin fraction from cells arrested in G1 with the α factor mating pheromone (lanes 4–6). To further investigate the temporal regulation of Mcm chromatin binding, we have examined the chromatin binding status of Cdc47p in cells blocked at various points in the cell cycle. As is the case with Cdc46p, very little Cdc47p is in the chromatin fraction from nocodazole-blocked cells (Fig. 3b, lanes 1–3). cdc15 mutants block at the end of anaphase, after sister chromatids have separated but before cyclin B destruction (43). Fig. 3b (lanes 4–6) shows that Cdc47p is absent from the chromatin fraction in cdc15-blocked cells. Again, as is the case with Cdc46p, approximately half of the Cdc47p is in the chromatin fraction in cells blocked in G1 with the α factor mating pheromone (Fig. 3b, lanes 7–9). Furthermore, Cdc47p remains in the chromatin fraction in cells blocked at the G1/S transition using a cdc7 temperature-sensitive mutant (Fig. 3b, lanes 10–12). Cells blocked in S phase with the ribonucleotide reductase inhibitor hydroxyurea show a significant reduction but not a complete loss of Cdc47p in the chromatin fraction (Fig. 3b, lanes 13–15). Taken together, these results indicate that Cdc46p and Cdc47p bind to chromatin after mitosis but before Start and are displaced from chromatin during S phase.
Figure 3.

Cell-cycle-regulated chromatin binding of Cdc46p and Cdc47p. (a) Whole-cell extracts (WCE), supernatants (Su), and chromatin fractions (Ch) were prepared as described in Materials and Methods. SDS/PAGE was performed as described in Fig. 1, and immunoblots were performed as described in Materials and Methods. (a) Cdc46p is chromatin-bound in G1 but not G2/M. PY26 containing tagged Cdc46p was blocked with either nocodazole (NOC) or alpha factor (α) before chromatin isolation. (b) Cdc47p is chromatin-bound throughout G1 but not in the S, G2/M, or late M phases. W303–1a containing tagged Cdc47p and no additional mutation (lanes 1–3, 7–9, and 13–15), the cdc15–2 mutation (lanes 4–6), or the cdc7–1 mutation (lanes 10–12) were blocked either with the indicated inhibitors or by raising cultures of mutant strains to the nonpermissive temperature until a uniform arrest was achieved before chromatin isolation.
To provide further evidence that the G1-specific appearance of these Mcm proteins in the chromatin fraction actually represents chromatin binding, we have performed additional experiments. As shown in Fig. 4a, the appearance of Cdc46p in the chromatin fraction from G1 blocked cells is insensitive to the addition of 250 mM NaCl. However, as shown in Fig. 4b, the presence of Cdc46p in this chromatin fraction at 250 mM NaCl is completely abolished by treatment with DNase1. The requirement for NaCl in this experiment is because moderate salt concentrations are required to solubilize the digested chromatin (44). Therefore, DNA integrity is essential for the appearance of the Mcm proteins in the chromatin fraction. Yan et al. (45) have also found a fraction of Mcm2 and Mcm3 proteins in a DNase1 sensitive pellet; however, under the conditions they employed, this was a much smaller fraction of the total Mcm protein than seen here. Experiments using Xenopus egg extracts have shown that the appearance of Mcm proteins in a similar pellet fraction depends on addition of sperm chromatin (18–21, 36), suggesting that the Mcm proteins are interacting with chromatin or some other component of the nucleus that forms after chromatin addition. We note that neither the Xenopus nor yeast experiments unambiguously demonstrate direct binding of Mcm proteins to chromatin; however, in both cases, intact genomic DNA is required for the appearance of Mcm proteins in a chromatin enriched fraction.
Figure 4.
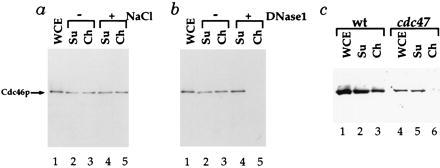
Characterization of Cdc46p chromatin binding. (a) Cdc46p chromatin binding is insensitive to low salt concentrations. Supernatant and chromatin fractions from the Cdc46p tagged strain blocked in G1 with α factor were prepared as above and divided into aliquots. One aliquot (−NaCl, lanes 2 and 3) was untreated. Another was treated with 0.25M NaCl before chromatin isolation. (b) Cdc46p chromatin binding is lost after DNase1 treatment. Supernatant and chromatin fractions from the Cdc46p tagged strain blocked in G1 with α factor were prepared as above in 0.25 M NaCl and divided into aliquots. One aliquot of this extract was untreated (−DNase1), whereas the other (+DNase1) was treated with 3 Kunitz units of DNase1 before chromatin isolation. (c) Cdc46p chromatin binding is lost in a cdc47 mutant. Chromatin fractions were prepared as described above for either a wild-type (wt) or congenic cdc47–1 mutant strain (see Materials and Methods) from cells first blocked with α factor at 25°C and then shifted to 37°C for 1.5 h.
A great deal of evidence from yeast and from higher eukaryotes indicates that the Mcm proteins functionally interact with each other (46, 47). Therefore, if the G1-specific binding of Cdc46p to chromatin has biological relevance, it might be affected by mutations in other Mcm proteins. That this is the case is shown in Fig. 4c. A wild-type and a congenic cdc47 mutant strain were first blocked in G1 at the permissive temperature (25°C) and subsequently shifted to the restrictive temperature (37°C). Under these conditions, not only is the absolute level of Cdc46p reduced in the cdc47 mutant, but little or no Cdc46p is now found in the chromatin fraction. Therefore, the appearance of Cdc46p in an enriched chromatin fraction occurs only in G1 and depends on the integrity of the DNA and upon the activity of at least one other Mcm protein. Consequently, we conclude that Cdc46p is chromatin-bound specifically during G1.
Cdc6p Is Required for Mcm Protein Chromatin Binding.
We next examined the role of Cdc6p in the G1-specific Mcm chromatin binding. We have previously used strains in which the CDC6 gene was under the control of the MET3 promoter, which can be repressed by methionine, to demonstrate that Cdc6p is essential for pre-RC formation (10). Fig. 5a shows that Cdc6p is also required for Mcm chromatin binding in G1. In this experiment, cells were first synchronized in late anaphase using the cdc15 temperature-sensitive mutation. None of the Cdc46p or Cdc47p is bound to chromatin in these cells (Fig. 5a, lanes 1–3). Cells were then released into G1 (α factor) either with or without de novo Cdc6p synthesis. In the presence of Cdc6p, approximately half of the Cdc46p and Cdc47p is in the chromatin fraction (Fig. 5a, lanes 4–6); however, in the absence of Cdc6p, none of these proteins is found in the chromatin fraction (Fig. 5a, lanes 7–9). We also found no Cdc46p in the chromatin fraction during G1 in a cdc6 temperature-sensitive mutant (Fig. 5b, lanes 4–6). Chromatin binding of Cdc46p is severely compromised even at the permissive temperature in this mutant preventing us from genetically addressing whether Cdc6p action is continuously required during G1 for Mcm chromatin binding (data not shown).
Figure 5.
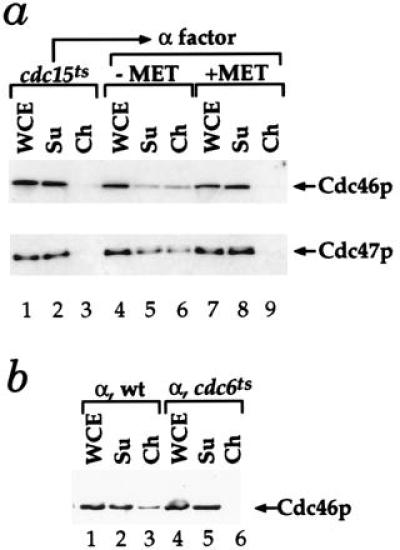
Cdc46p and Cdc47p chromatin binding in G1 depends on Cdc6p. (a) De novo Cdc6p synthesis in G1 is essential for Cdc46p and Cdc47p chromatin binding. The levels of Cdc46p or Cdc47p bound to chromatin were examined in cells harboring a single CDC6 gene under the control of the MET3 promoter either in late anaphase (cdc15ts) or after release from late anaphase into G1 either with (−MET) or without (+MET) Cdc6p expression (10). (b) Cdc46p chromatin binding is defective in a cdc6 mutant during G1. Cdc46p chromatin binding was examined with a wild-type and congenic cdc6–1 mutant after α factor arrest at 24°C.
Evidence that ORC and Cdc6p Are Not Required to Maintain Mcm Chromatin Binding.
In Fig. 6, we examined the effect of NaCl on chromatin binding more closely. As shown in Fig. 4a, intermediate levels of NaCl have little effect on the binding of the Mcm proteins to chromatin. At a slightly higher NaCl concentration (300 mM), a small fraction of the Cdc46p is removed from chromatin (Fig. 6, top bands, compare lanes 2 and 3 with lanes 4 and 5); however, much of the Cdc46p remains chromatin-bound. As shown in Fig. 1c, Orc2p is quantitatively bound to chromatin in G1. However, 300 mM NaCl is sufficient to completely remove Orc2p from G1 chromatin (Fig. 6, lanes 4 and 5, middle bands). In these G1 blocked cells, we estimate that there are approximately 2,000 copies of Cdc6p per cell (data not shown) and that approximately one-third of these molecules are bound to chromatin (Fig. 6, lanes 2 and 3, bottom bands). Whether it is significant that this corresponds roughly to the number of replication origins per cell is not known. NaCl (300 mM), however, removes essentially all of the Cdc6p from G1 chromatin (Fig. 6, lanes 4 and 5, bottom bands). Because the Cdc6 protein is only present at detectable levels during G1 (48), these experiments do not address whether Cdc6p can bind to chromatin after DNA replication. These experiments do indicate, however, that Orc2p and Cdc6p can be quantitatively removed without quantitatively removing Cdc46p from chromatin.
Figure 6.
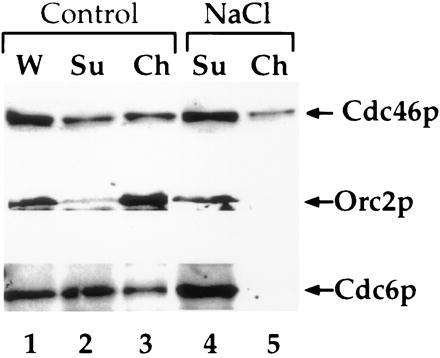
Removal of ORC and Cdc6p from chromatin does not remove Cdc46p. Lysates were either untreated (Control) or treated with 0.3 M NaCl (NaCl) for 5 min on ice before chromatin isolation. Immunoblots were performed on these fractions with 9E10 to detect the tagged Cdc46p, JAB12 to detect ORC2, or 9H85 to detect Cdc6 as described in Materials and Methods.
From this and previous work, we propose the model shown in Fig. 7. ORC remains bound at replication origins during the entire cell cycle (8–10). The demonstration that ORC is chromatin-bound during G1 and G2 (Fig. 1c) provides additional support for this statement. Cdc6p is essential for the loading of the Mcm proteins onto pre-replicative chromatin. At present we do not know whether Cdc6p acts as a loading factor for Mcm proteins or is actually a component of pre-RCs. That some Cdc6p appears to be chromatin-bound in G1 may support this idea; however, further experiments are required to establish this possibility. Cdc28/Clb kinase plays a dual role in regulating this process by activating the firing of pre-replicative origins (ref. 49; together with the Cdc7p/Dbf4p protein kinase) and by preventing the Cdc6p-dependent pre-RC assembly at post-replicative origins (12, 13). We do not know whether the direct target of Cdc28/Clb inhibition is Cdc6p itself or some other component in the pathway such as ORC or the Mcm proteins; however, Cdc6p has been reported to interact with Cdc28/Clb kinase (12, 50). Cdc6p-dependent pre-RC formation is not limited to mitosis but can occur during G1 in budding yeast cells (9, 12). In this regard, we note that the Xenopus licensing factor system is inactive in mitotic extracts (51), indicating that re-replication may not be blocked solely by nuclear envelope permeability. We suggest that the pathway described in Fig. 7 may be involved in regulating the initiation of DNA replication and limiting it to once per cell cycle in all eukaryotes.
Figure 7.
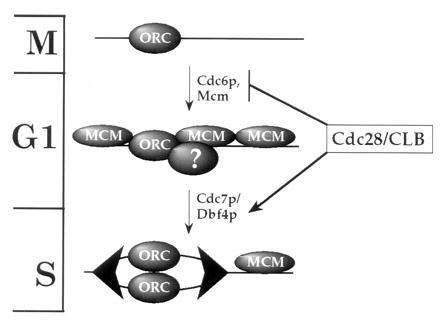
A model for the regulation of eukaryotic DNA replication. Large arrowheads represent replication complexes at forks. Cell cycle phases are indicated on the left. Details of the model are discussed in the text.
Acknowledgments
We thank I. Goldsmith for oligonucleotide synthesis, G. Evan for peptide synthesis, and J. Steel for antibody production. We also thank Tim Hunt for critical reading of the manuscript.
ABBREVIATIONS
- ORC
origin recognition complex
- pre-RC
pre-replicative complex
References
- 1.Guttes S, Guttes E. J Cell Biol. 1968;37:761–772. doi: 10.1083/jcb.37.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rao P N, Johnson R T. Nature (London) 1970;225:159–164. doi: 10.1038/225159a0. [DOI] [PubMed] [Google Scholar]
- 3.Nasmyth K. Science. 1996;274:1643–1645. doi: 10.1126/science.274.5293.1643. [DOI] [PubMed] [Google Scholar]
- 4.Diffley J F X. Genes Dev. 1996;10:2819–2830. doi: 10.1101/gad.10.22.2819. [DOI] [PubMed] [Google Scholar]
- 5.Stillman B. Science. 1996;274:1659–1664. doi: 10.1126/science.274.5293.1659. [DOI] [PubMed] [Google Scholar]
- 6.Stern B, Nurse P. Trends Genet. 1996;12:345–350. [PubMed] [Google Scholar]
- 7.Bell S P, Stillman B. Nature (London) 1992;357:128–134. doi: 10.1038/357128a0. [DOI] [PubMed] [Google Scholar]
- 8.Diffley J F X, Cocker J H. Nature (London) 1992;357:169–172. doi: 10.1038/357169a0. [DOI] [PubMed] [Google Scholar]
- 9.Diffley J F X, Cocker J H, Dowell S J, Rowley A. Cell. 1994;78:303–316. doi: 10.1016/0092-8674(94)90299-2. [DOI] [PubMed] [Google Scholar]
- 10.Cocker J H, Piatti S, Santocanale C, Nasmyth K, Diffley J F X. Nature (London) 1996;379:180–182. doi: 10.1038/379180a0. [DOI] [PubMed] [Google Scholar]
- 11.Liang C, Weinreich M, Stillman B. Cell. 1995;81:667–676. doi: 10.1016/0092-8674(95)90528-6. [DOI] [PubMed] [Google Scholar]
- 12.Piatti S, Bohm T, Cocker J H, Diffley J F X, Nasmyth K. Genes Dev. 1996;10:1516–1531. doi: 10.1101/gad.10.12.1516. [DOI] [PubMed] [Google Scholar]
- 13.Dahmann C, Diffley J F X, Nasmyth K A. Curr Biol. 1995;5:1257–1269. doi: 10.1016/s0960-9822(95)00252-1. [DOI] [PubMed] [Google Scholar]
- 14.Broek D, Bartlett R, Crawford K, Nurse P. Nature (London) 1991;349:388–393. doi: 10.1038/349388a0. [DOI] [PubMed] [Google Scholar]
- 15.Moreno S, Nurse P. Nature (London) 1994;367:236–242. doi: 10.1038/367236a0. [DOI] [PubMed] [Google Scholar]
- 16.Hayles J, Fisher D, Woollard A, Nurse P. Cell. 1994;78:813–822. doi: 10.1016/s0092-8674(94)90542-8. [DOI] [PubMed] [Google Scholar]
- 17.Blow J J, Laskey R A. Nature (London) 1988;332:546–548. doi: 10.1038/332546a0. [DOI] [PubMed] [Google Scholar]
- 18.Kubota Y, Mimura S, Nishimoto S-I, Takisawa H, Nojima H. Cell. 1995;81:601–609. doi: 10.1016/0092-8674(95)90081-0. [DOI] [PubMed] [Google Scholar]
- 19.Chong J P J, Mahbubani H M, Khoo C-Y, Blow J J. Nature (London) 1995;375:418–421. doi: 10.1038/375418a0. [DOI] [PubMed] [Google Scholar]
- 20.Madine M A, Khoo C-Y, Mills A D, Laskey R A. Nature (London) 1995;375:421–424. doi: 10.1038/375421a0. [DOI] [PubMed] [Google Scholar]
- 21.Madine M A, Khoo C Y, Mills A D, Mushal C, Laskey R A. Curr Biol. 1995;5:1270–1279. doi: 10.1016/s0960-9822(95)00253-3. [DOI] [PubMed] [Google Scholar]
- 22.Maine G T, Sinha P, Tye B-K. Genetics. 1984;106:365–385. doi: 10.1093/genetics/106.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan H, Gibson S, Tye B-K. Genes Dev. 1991;5:944–957. doi: 10.1101/gad.5.6.944. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi K, Yamada H, Yanagida M. Mol Biol Cell. 1994;5:1145–1158. doi: 10.1091/mbc.5.10.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moir D, Stewart S E, Osmond B C, Botstein D. Genetics. 1982;100:547–563. doi: 10.1093/genetics/100.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hennessy K M, Lee A, Chen E, Botstein D. Genes Dev. 1991;5:958–969. doi: 10.1101/gad.5.6.958. [DOI] [PubMed] [Google Scholar]
- 27.Miyake S, Okishio N, Samejima I, Hiraoka Y, Toda T, Saitoh I, Yanagida M. Mol Biol Cell. 1993;4:1003–1015. doi: 10.1091/mbc.4.10.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forsburg S L, Nurse P. J Cell Sci. 1994;107:2779–2788. doi: 10.1242/jcs.107.10.2779. [DOI] [PubMed] [Google Scholar]
- 29.Coxon A, Maundrell K, Kearsey S E. Nucleic Acids Res. 1992;20:5571–5577. doi: 10.1093/nar/20.21.5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hennessy K M, Clark C D, Botstein D. Genes Dev. 1990;4:2252–2263. doi: 10.1101/gad.4.12b.2252. [DOI] [PubMed] [Google Scholar]
- 31.Dalton S, Whitbread L. Proc Natl Acad Sci USA. 1995;92:2514–2518. doi: 10.1073/pnas.92.7.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitbread L A, Dalton S. Gene. 1995;155:113–117. doi: 10.1016/0378-1119(94)00925-i. [DOI] [PubMed] [Google Scholar]
- 33.Carpenter P B, Mueller P R, Dunphy W G. Nature (London) 1996;379:357–360. doi: 10.1038/379357a0. [DOI] [PubMed] [Google Scholar]
- 34.Rowles A, Chong J P J, Brown L, Howell M, Evan G I, Blow J J. Cell. 1996;87:287–296. doi: 10.1016/s0092-8674(00)81346-x. [DOI] [PubMed] [Google Scholar]
- 35.Romanowski P, Madine M A, Rowles A, Blow J J, Laskey R A. Curr Biol. 1996;6:1416–1425. doi: 10.1016/s0960-9822(96)00746-4. [DOI] [PubMed] [Google Scholar]
- 36.Coleman T R, Carpenter P B, Dunphy W G. Cell. 1996;87:53–63. doi: 10.1016/s0092-8674(00)81322-7. [DOI] [PubMed] [Google Scholar]
- 37.Micklem G, Rowley A, Harwood J, Nasmyth K, Diffley J F X. Nature (London) 1993;366:87–89. doi: 10.1038/366087a0. [DOI] [PubMed] [Google Scholar]
- 38.Conradt B, Shaw J, Vida T, Emr S, Wickner W. J Cell Biol. 1992;119:1469–1479. doi: 10.1083/jcb.119.6.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santocanale C, Diffley J F X. EMBO J. 1996;15:6671–6679. [PMC free article] [PubMed] [Google Scholar]
- 40.Rowley A, Cocker J H, Harwood J, Diffley J F X. EMBO J. 1995;14:2631–2641. doi: 10.1002/j.1460-2075.1995.tb07261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evan G, Lewis G, Ramsay G, Bishop J M. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lei M, Kawasaki Y, Tye B-K. Mol Cell Biol. 1996;16:5081–5090. doi: 10.1128/mcb.16.9.5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Surana U, Amon A, Dowzer C, McGrew J, Byers B, Nasmyth K. EMBO J. 1993;12:1969–1978. doi: 10.1002/j.1460-2075.1993.tb05846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu L-C, Fisher P A, Broach J R. J Biol Chem. 1986;262:883–891. [PubMed] [Google Scholar]
- 45.Yan H, Merchant A M, Tye B-K. Genes Dev. 1993;7:2149–2160. doi: 10.1101/gad.7.11.2149. [DOI] [PubMed] [Google Scholar]
- 46.Chong J P J, Thömmes P, Blow J J. Trends Biochem Sci. 1996;21:102–106. [PubMed] [Google Scholar]
- 47.Kearsey S E, Labib K, Maiorano D. Curr Opin Genet Dev. 1996;6:208–214. doi: 10.1016/s0959-437x(96)80052-9. [DOI] [PubMed] [Google Scholar]
- 48.Piatti S, Lengauer C, Nasmyth K. EMBO J. 1995;14:3788–3799. doi: 10.1002/j.1460-2075.1995.tb00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwob E, Bohm T, Mendenhall M D, Nasmyth K. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 50.Elsasser S, Lou F, Wang B, Campbell J L, Jong A. Mol Biol Cell. 1996;7:1723–1735. doi: 10.1091/mbc.7.11.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blow J J. J Cell Biol. 1993;122:993–1002. doi: 10.1083/jcb.122.5.993. [DOI] [PMC free article] [PubMed] [Google Scholar]