

Abstract
The experiments investigated the applicability of two established criteria for arrhythmogenicity in Scn5a+/Δ and Scn5a+/− murine hearts modelling the congenital long QT syndrome type 3 (LQT3) and the Brugada syndrome (BrS). Monophasic action potentials (APs) recorded during extrasystolic stimulation procedures from Langendorff-perfused control hearts and hearts treated with flecainide (1 μM) or quinidine (1 or 10 μM) demonstrated that both agents were pro-arrhythmic in wild-type (WT) hearts, quinidine was pro-arrhythmic in Scn5a+/Δ hearts, and that flecainide was pro-arrhythmic whereas quinidine was anti-arrhythmic in Scn5a+/− hearts, confirming clinical findings. Statistical analysis confirmed a quadratic relationship between epicardial and endocardial AP durations (APDs) in WT control hearts. However, comparisons between plots of epicardial against endocardial APDs and this reference curve failed to correlate with arrhythmogenicity. Restitution curves, relating APD to diastolic interval (DI), were then constructed for the first time in a murine system and mono-exponential growth functions fitted to these curves. Significant (P < 0.05) alterations in the DI at which slopes equalled unity, an established indicator of arrhythmogenicity, now successfully predicted the presence or absence of arrhythmogenicity in all cases. We thus associate changes in the slopes of restitution curves with arrhythmogenicity in models of LQT3 and BrS.
Keywords: Action potential, Arrhythmia, Heart, Heart rate, Heart excitation, Venticular muscle, Ventricle
Introduction
Mutations in the SCN5a gene encoding the α-subunit of the cardiac voltage-gated Na+ channel are well established to underlie hereditary arrhythmic syndromes which may result in sudden cardiac death. On the one hand, gain-of-function mutations which increase inward depolarising currents and consequently prolong the ventricular action potential are associated with arrhythmogenicity in the congenital long QT syndrome type 3 (LQT3) [23]. On the other hand, loss-of-function mutations which decrease such currents are associated with arrhythmogenicity in the Brugada syndrome (BrS) [1]. In this case, spatial heterogeneities in the degree of shortening of the ventricular action potential result in characteristic electrocardiographic changes, namely a partial right bundle branch block pattern with ST segment elevation in the right precordial leads [2].
Despite the similar clinical outcomes of LQT3 and BrS, class 1A and 1C, anti-arrhythmic agents, exemplified by quinidine and flecainide respectively, exert contrasting effects on arrhythmogenicity in these two syndromes [31]. Thus in LQT3, quinidine is pro-arrhythmic [33] and flecainide is anti-arrhythmic [22]. In contrast, in BrS quinidine is anti-arrhythmic [24], while flecainide is pro-arrhythmic [7] and used to unmask the arrhythmic phenotype in asymptomatic patients. These contrasts are precisely reflected in results from our murine models of LQT3 [39] and BrS [38], emphasizing the translatability of results obtained from such systems. Furthermore, both quinidine and flecainide are known to have the potential for pro-arrhythmic effects [12, 30].
These similarities and differences have parallels in the physiological changes thought to underlie arrhythmogenicity in LQT3 and BrS. Thus circus-type re-entry [2], requiring both an initiating trigger and a re-entrant substrate, has been implicated in arrhythmogenicity in both these conditions. Of these requirements, both conditions are associated with triggering extrasystoles [3]. In LQT3, these are initiated by early after-depolarizations attributable to action potential prolongation [42], while in BrS they are initiated by phase 2 re-entry [20, 46]. Furthermore, in both LQT3 and BrS, arrhythmia occurs against a re-entrant substrate [3]. In LQT3 this is provided by increased transmural dispersion of repolarization [13, 27, 42]. While increased transmural dispersion of repolarization has also been demonstrated in some studies on BrS [3], increased epicardial dispersion of repolarization [2] and slowed conduction [29] are also implicated. Thus studies in murine hearts have associated alterations in transmural dispersion of repolarization, reflected in relationships between epicardial and endocardial action potential durations, with arrhythmogenicity in models of LQT3 but failed to do so in models of BrS [21, 33, 38, 39, 42].
Arrhythmogenicity has often been associated with increases in the slopes of restitution curves, plotting action potential duration against the preceding diastolic interval, to greater than unity [14, 17–19, 28, 40]. Possible mechanisms that might underlie this association have been discussed in detail on a number of occasions [11, 25, 43, 44]. Furthermore, decreases in the slopes of such curves are associated with the anti-arrhythmic effects of several clinically important agents including amiodarone, bretylium and procainamide [11, 26, 32]. Such associations have previously been demonstrated in a pharmacological model of LQT2 [45] but has not been assessed in LQT3 or in BrS.
The present study compares an analysis examining transmural dispersion of repolarization, assessed by comparing epicardial and endocardial action potential durations obtained over a range of heart rates and previously established to predict arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts [34], with another examining effects on restitution curve parameters. This represents the first occasion on which restitution curves have been constructed in a murine system, in relation to BrS or LQT3, or indeed in any genetic model of an hereditary arrhythmic syndrome. To this end, the monophasic action potential (MAP) technique and an established Langendorff-perfused system [15, 34–36] are used to study genetically modified mice modelling LQT3 (Scn5a+/Δ) [13] and BrS (Scn5a+/−) [29] before and after exposure to quinidine and flecainide. We proceed to demonstrate that while the former criterion fails, the latter produces predictions in full agreement with the arrhythmogenic findings in every case and thus suggest that alterations in the slopes of restitution curves are associated with arrhythmogenicity in both LQT3 and BrS.
Materials and methods
Experimental animals
Wild-type (WT), Scn5a+/Δ and Scn5a+/− 129 Sv mice aged 3–6 months were housed at 21 ± 1°C with 12 h light/dark cycles. They were fed sterile chow (RM3 Maintenance Diet, SDS, Witham, Essex, UK) and had free access to water. All procedures complied with the UK Animals (Scientific Procedures) Act 1986.
Solutions
Solutions were based on bicarbonate-buffered Krebs-Henseleit solution (119 mM NaCl, 25 mM NaHCO3, 4 mM KCl, 1.2 mM KH2PO4, 1 mM MgCl2, 1.8 mM CaCl2, 10 mM glucose and 2 mM Na-pyruvate; pH adjusted to 7.4) and were bubbled with 95% O2/5% CO2 (British Oxygen Company, Manchester, U.K). Drug-containing solutions were prepared by adding flecainide (Sigma-Aldrich, Poole, UK) to final concentration of 1 μM and quinidine (Sigma-Aldrich, Poole, UK) to final concentrations of 1 and 10 μM: these concentrations were chosen to reflect clinically therapeutic values (effective free and total plasma concentrations of 0.79 and 1.48 μM, respectively, for flecainide and 0.74 and 7.9 μM, respectively, for quinidine) [9].
Preparation
A Langendorff-perfusion protocol previously adapted for murine hearts [4] was used. Briefly, mice were killed by cervical dislocation [Schedule 1: UK Animals (Scientific Procedures) Act 1986] before hearts were quickly excised and placed in ice-cold bicarbonate-buffered Krebs-Henseleit solution. Short sections of aorta were cannulated under the surface of the solution and attached to a custom-made 21-gauge cannula filled using an aneurysm clip (Harvard Apparatus, Edenbridge, Kent, UK), within 90 s of excision. Fresh Krebs-Henseleit solution was passed through 200- and 5-μm filters (Millipore, Watford, UK) and warmed to 37°C using a water jacket and circulator (Techne model C-85A, Cambridge, UK). This solution was then used for constant-flow retrograde perfusion, thereby circumventing any changes in coronary vascular resistance and therefore flow that might otherwise result from the effects of the drugs studied [10], at 2–2.5 ml/min, driven by a peristaltic pump (Watson-Marlow Bredel model 505S, Falmouth, Cornwall, UK). Hearts were regarded as suitable for experimentation if they regained a healthy pink colour and began to contract spontaneously on re-warming.
Electrophysiological measurements
An epicardial MAP electrode (Hugo Sachs, Harvard Apparatus, UK) was placed against the basal left ventricular epicardium. A custom-made endocardial MAP electrode consisting of two twisted strands of high-purity Teflon-coated silver wire of 0.25 mm diameter (Advent Research Materials, UK) was manufactured. A small access window was created in the interventricular septum to allow access to the left ventricular endocardium [8]. The Teflon coating was removed from the distal 1 mm of the electrode; this was then galvanically chlorided to eliminate DC offset, inserted and placed against the septal endocardial surface. MAPs were amplified, band-pass filtered (0.5 Hz to 1 kHz: Gould 2400S, Gould-Nicolet Technologies, Ilford, Essex, UK) and digitised at a sampling frequency of 5 kHz (micro1401, Cambridge Electronic Design, Cambridge, UK). Analysis of MAPs was performed using Spike II (Cambridge Electronic Design, Cambridge, UK).
Experimental protocol
A bipolar platinum stimulating electrode (1 mm inter-pole spacing) was placed on the basal surface of the right ventricular epicardium. Square-wave stimuli (Grass S48 stimulator, Grass-Telefactor, Slough, UK) of 2 ms duration and amplitudes of twice the excitation threshold were initially applied to hearts at a constant baseline cycle length (BCL) of 125 ms until MAPs showed stable baselines, rapid upstroke phases that reached consistent amplitudes and smooth repolarization phases [16] and for at least 10 min. Before subsequent recordings were made, hearts were then exposed to test solutions for 20 min while stimulation was continued.
In initial experiments, MAPs were recorded during regular stimulation at a BCL of 125 ms. Arrhythmogenicity was then assessed both during regular stimulation and using an extrasystolic stimulation protocol [37] previously adapted for the study of arrhythmogenesis in murine hearts incorporating the Scn5a+/Δ and Scn5a+/− mutations [13, 38] and described below. Hearts were then subjected to an adapted dynamic pacing protocol [17] explained in detail below, and the data obtained used to construct restitution curves.
Data analysis
All data are presented as means ± standard errors of the means and include the number of hearts studied. Comparisons between data sets used analysis of variance (significance threshold set at P ≤ 0.05). Curve fitting of particular functions to data sets used a Levenberg-Marquardt algorithm (OriginPro 7.5, OriginLab, MA, USA).
Results
The experiments made direct measurements of MAPs from intact Langendorff-perfused murine hearts. They compared the extents to which relationships between epicardial and endocardial action potential durations and the slopes of restitution curves are associated with arrhythmogenicity in genetic models of the congenital LQT3 (Scn5a+/Δ) and of the BrS (Scn5a+/−), and in wild-type (WT) controls. They began by successfully demonstrating that these experimental models recapitulated arrhythmic phenotypes observed clinically as well as confirming the anticipated effects of treatment with clinically-relevant [6] concentrations of flecainide (1 μM) and quinidine (1 or 10 μM). Hearts were exposed to solutions for 20 min while stimulated at a constant BCL of 125 ms before recording was commenced.
Arrhythmogenicity in WT, Scn5a+/Δ and Scn5a+/− hearts subject to both regular and extrasystolic stimulation recapitulates clinical observations
Figures 1 and 2 show results obtained from WT (Figs. 1a, 2a), Scn5a+/Δ (Figs. 1b, 2b) and Scn5a+/− (Figs. 1c, 2c) hearts treated with control solution (A) and with solutions containing 1 μM flecainide (B), 1 μM quinidine (C) and 10 μM quinidine (D). Figure 1 illustrates MAP waveforms recorded from the epicardia of hearts during the extrasystolic stimulation procedure showing arrhythmic waveforms in situations where arrhythmic activity was observed in >50% of cases; Fig. 2 quantifies the incidence of such arrhythmic activity both during regular stimulation (i) and during the extrasystolic stimulation procedure (ii).
Fig. 1.
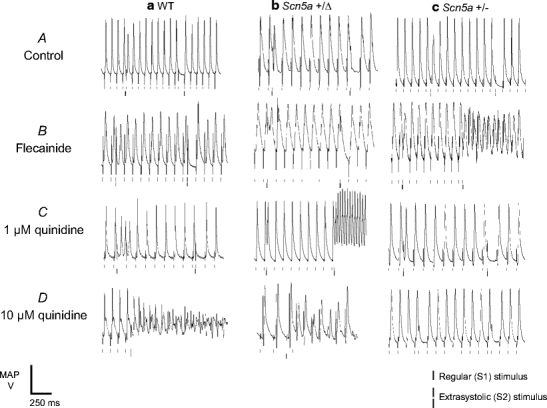
Arrhythmic activity resulting from extrasystolic stimulation. Epicardial monophasic action potential (MAP) recordings illustrating the result of the application of extrasystolic stimuli to WT (a), Scn5a+/Δ (b) and Scn5a+/− (c) hearts treated with control solution (A) and with solutions containing 1 μM flecainide (B), 1 μM quinidine (C) and 10 μM quinidine (D). Traces show arrhythmic activity where this was observed in ≥50% of cases
Fig. 2.
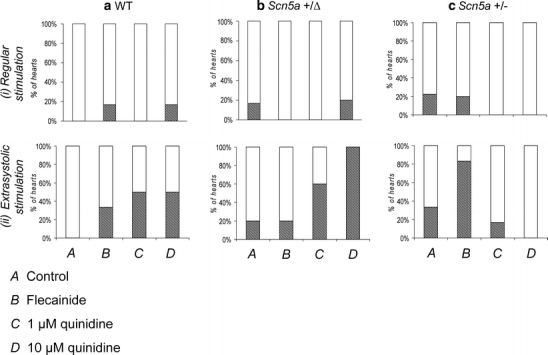
Incidences of arrhythmic activity in WT, Scn5a+/Δ and Scn5a+/− hearts subjected to the extrasystolic stimulation procedure. Percentage of WT (a), Scn5a+/Δ (b) and Scn5a+/− (c) hearts demonstrating arrhythmic activity during regular stimulation (i) and during the extrasystolic stimulation procedure (ii). In both these cases, MAPs were obtained during treatment with control solution (A) and with solutions containing 1 μM flecainide (B), 1 μM quinidine (C) and 10 μM quinidine (D). Hashing indicates hearts demonstrating arrhythmic activity, and dots indicate hearts not demonstrating such activity
The frequencies of arrhythmic phenomena in hearts subjected to regular stimulation at a BCL of 125 ms were relatively low irrespective of the genetic or pharmacological conditions but were nevertheless consistent with clinical expectations. First, zero out of seven WT hearts (~0%, Fig. 2a, i) exposed to control solution (A) demonstrated arrhythmic activity, while treatment with flecainide (B) led to arrhythmic activity in only one out of six (~17%) cases. Furthermore, such hearts never showed arrhythmic activity during treatment with 1 μM quinidine (C, zero out of six hearts, ~0%) and again did so in only one out of six cases (~17%) with 10 μM quinidine (D). Secondly, arrhythmic activity did occur in one out of six (~17%) Scn5a+/Δ hearts (Fig. 2b) treated with control solution (A) and was absent in all cases (zero out of six hearts, ~0%) during treatment with flecainide (B). Arrhythmic activity was not observed (zero out of four hearts, ~0%) during treatment with 1 μM quinidine (C) but did occur in one out of five hearts (~20%) exposed to 10 μM quinidine (D). Thirdly, two out of nine Scn5a+/− hearts (~22%, Fig. 2c) exhibited arrhythmic activity during exposure to control solution (A). Such arrhythmic activity was also observed in one out of five hearts (~20%) treated with flecainide (B). Arrhythmic activity was then absent after treatment with either 1 μM (C) or 10 μM (D) quinidine (zero out of five, 0%, and zero out of six, 0%, hearts, respectively).
In contrast, extrasystolic stimulation (Figs. 1 and 2, ii) revealed clearer trends in arrhythmogenicity in full agreement with clinical expectations. First, while zero out of seven WT hearts (0%, Fig. 2a, ii) exposed to control solution (A) exhibited arrhythmic activity, treatment with flecainide (B) increased the incidence of such events to two out of six hearts (~33%). Furthermore, exposure to 1 μM quinidine (C) resulted in arrhythmic activity in four out of eight hearts (~50%), while exposure to 10 μM quinidine (D) resulted in arrhythmic activity in three out of six hearts (~50%). Secondly, one out of five Scn5a+/Δ hearts (~20%, Fig. 2b, ii) demonstrated arrhythmic activity during exposure to either control solution (A) or flecainide (B). However, exposure to quinidine resulted in arrhythmic activity in three out of five hearts (~60%) at 1 μM (C) and four out of four hearts (~100%) at 10 μM (D). Thirdly, Scn5a+/− hearts (Fig. 2c, ii) demonstrated such arrhythmic activity in three out of nine cases (~33%) during treatment with control solution (A) but in five out of six cases (~83%) during treatment with flecainide (B). Furthermore, arrhythmic activity was observed in only one out of six such hearts (~17%) during treatment with 1 μM quinidine (C) and in zero out of six hearts during exposure to 10 μM quinidine (D).
Graphical analyses of arrhythmogenic tendency
In an effort to explore the possible basis for these arrhythmic phenomena, subsequent experiments made systematic measurements of epicardial and endocardial action potential durations (at 90% repolarization, APD90s) during a dynamic pacing protocol [17]. This consisted of cycles each comprising 100 stimuli, initially delivered at a BCL of 175 ms. BCL was then decremented in 50-ms steps with each subsequent cycle until a reproducible sequence of consistently shaped MAP waveforms was no longer obtained. Steady states were consistently reached within the first 50 responses, and therefore mean values of APD90 were obtained from the final 50 action potentials in each cycle. This approach provided data that could be used to evaluate over the full range of BCLs the relationship between arrhythmogenicity and two possible electrophysiological properties. Thus it was possible first to evaluate transmural repolarization gradients and secondly to evaluate slopes of restitution curves relating APD90 to the preceding DI, given by BCL minus APD90 [17, 25, 28, 43].
Relationships between epicardial and endocardial action potential durations are not consistently associated with either the presence or absence of arrhythmogenicity
Alterations in relationship between epicardial and endocardial APD90 determined at a single BCL has previously been associated with arrhythmogenicity in hearts obtained from our Scn5a+/Δ mice [42]. The present study now extends this analysis from a single BCL to a range of BCLs and applies it to Scn5a+/− hearts for the first time, plotting endocardial APD90 on the abscissa and epicardial APD90 on the ordinate over the full range of BCLs investigated (triangles, Fig. 3). Such plots have previously been used to assess arrhythmogenicity in hypokalaemic WT murine hearts [34] and provided possible criteria for arrhythmogenicity. The control plot obtained from WT hearts perfused with control solution (six hearts, Fig. 3a, A) gave a quadratic relationship between endocardial and epicardial APD90 (solid line), in agreement with previous work [34]. Thus the curve y = ax2 + bx + c, where a = 0.0097 ± 0.0028, b = 1.46 ± 0.27 and c = −5.06 ± 6.34, gave a reduced χ2 of 0.49. Repeating the regression analysis this time fitting the line y = mx + c gave a reduced χ2 of 0.86, and permitted the derivation of an F statistic for the appropriate, N + 1 = 17 data points, given by 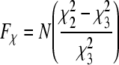 in which the subscripts on the right-hand side of the equation denote the number of parameters involved [5]. This yielded a probability of exceeding F of <0.05, suggesting that inclusion of the quadratic term was justified, again in full agreement with the previous study [34].
in which the subscripts on the right-hand side of the equation denote the number of parameters involved [5]. This yielded a probability of exceeding F of <0.05, suggesting that inclusion of the quadratic term was justified, again in full agreement with the previous study [34].
Fig. 3.
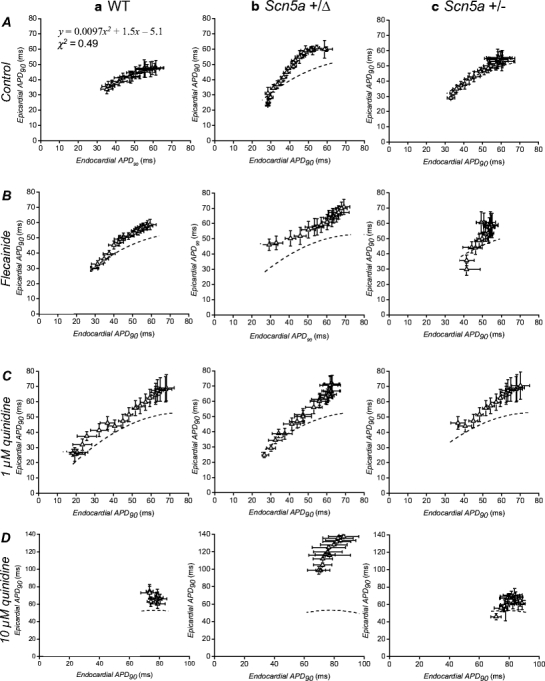
Relationships between epicardial and endocardial action potential durations (at 90% repolarization, APD90s) obtained over a range of baseline cycle lengths. WT (a), Scn5a+/Δ (b) and Scn5a+/− (c) hearts were exposed to control solution (A) and to solutions containing 1 μM flecainide (B), 1 μM quinidine (C) and 10 μM quinidine (D) and stimulated at baseline cycle lengths of between 80 ms and 175 ms. Broken lines indicate reference curves constructed by least-squares fitting of the equation y = ax2 + bx + c to data points obtained from wild-type hearts treated with control solution
Data points obtained under the range of genetic and pharmacological conditions studied could then be compared with this reference curve (broken lines, Fig. 3) with the expectation that the departure of points to the left of this curve might be associated with arrhythmogenicity [34]. However, data sets obtained before and after the pharmacological manoeuvres satisfied this expectation in WT but clearly failed to do so in the remaining genetically modified hearts.
First, treatment of WT hearts (Fig. 3a) with flecainide (five hearts, B), 1 μM quinidine (six hearts, C) and 10 μM quinidine (six hearts, D) all resulted in points falling to the left of the reference curve, in parallel with the occurrence of arrhythmic activity under these conditions (Fig. 2a, B, C and D). Secondly, data points obtained from Scn5a+/Δ hearts (Fig. 3b) treated with control solution (five hearts, A) again fell to the left of the reference curve in contrast with the low incidence of arrhythmic activity in this setting (Fig. 2b, A). This pattern was maintained when such hearts were exposed to flecainide (five hearts, Fig. 3b, B), once again in contrast with the low frequency which arrhythmic activity was observed under these conditions (Fig. 2b, B). Exposure to 1 μM quinidine (five hearts, Fig. 3b, C) and 10 μM quinidine (six hearts, D) again both resulted in points falling to the left of the control curve, this time in parallel with the high incidence of arrhythmic activity in these settings (Figs. 2b, C and 3b, D). Thirdly, data points obtained from Scn5a+/− hearts (Fig. 3c) treated with control solution (six hearts, A) consistently fell on the reference curve, in contrast with the occurrence of arrhythmic activity in this setting (Fig. 2c, A). Furthermore, treatment with flecainide (six hearts, Fig. 3c, B) failed to result in a left-shift of data points, in contrast with the large increase in the incidence of arrhythmic activity (Fig. 2c, B). In addition, points obtained during treatment with either 1 μM quinidine (five hearts, Fig. 3c, C) or 10 μM quinidine (seven hearts, D) again fell to the left of the reference curve, in contrast with the anti-arrhythmic effect of this agent demonstrated in Fig. 2c, C and D.
Alterations in the slopes of restitution curves correlate with the presence and absence of arrhythmic activity
Restitution curves (data points, left ordinate) were then plotted using the APD90 (left ordinate) and DI data obtained during the dynamic pacing protocol from the epicardia (circles, Fig. 4) and endocardia (squares, Fig. 5) of WT (Figs. 4a, 5a), Scn5a+/Δ (Figs. 4b, 5b) and Scn5a+/− (Figs. 4c, 5c) hearts for the first time. Hearts were studied during treatment with control solution (A) and with solutions containing flecainide (B), 1 μM quinidine (C) or 10 μM quinidine (D). Under all conditions, epicardial and endocardial APD90 both decreased as DI decreased. Increases or decreases in the slopes of such relationships were in full agreement with the presence or absence of arrhythmic activity under all conditions studied.
Fig. 4.
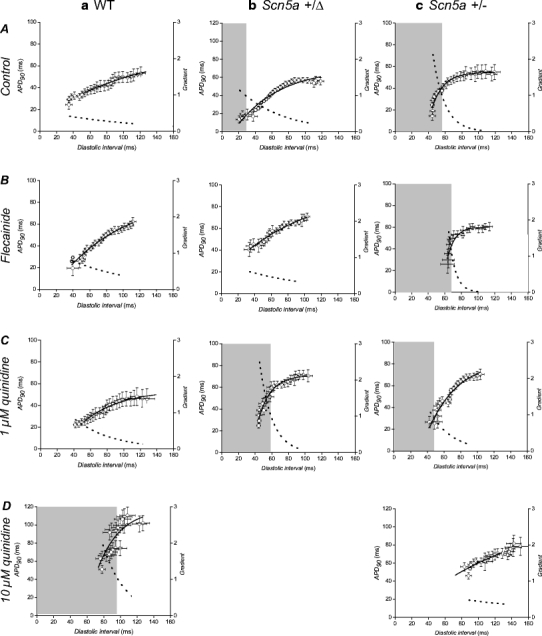
Epicardial restitution curves plotting action potential duration (at 90% repolarization, APD90) against preceding diastolic interval (DI). Data were obtained from the epicardia (circles) of WT (a), Scn5a+/Δ (b) and Scn5a+/− (c) hearts exposed to control solution (A) and to solutions containing 1 μM flecainide (B), 1 μM quinidine (C) and 10 μM quinidine (D). Curves are fitted with mono-exponential growth functions obtained by least squares fitting to the experimental values of APD90 and DI (solid lines, left ordinates). Gradients were obtained by differentiation of the fitted functions (broken lines, right axes). Shaded boxes indicate ranges of DI values at which such gradients exceed unity
Fig. 5.
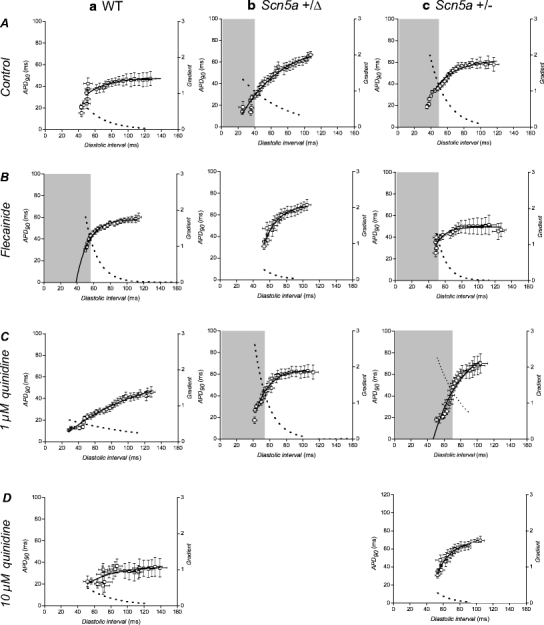
Endocardial restitution curves plotting action potential duration (at 90% repolarization, APD90) against preceding diastolic interval (DI). Data were obtained from the endocardia (squares) of WT (a), Scn5a+/Δ (b) and Scn5a+/− (c) hearts exposed to control solution (A) and to solutions containing 1 μM flecainide (B), 1 μM quinidine (C) and 10 μM quinidine (D). Curves are fitted with mono-exponential growth functions obtained by least squares fitting to the experimental values of APD90 and DI (solid lines, left ordinates). Gradients were obtained by differentiation of the fitted functions (broken lines, right axes). Shaded boxes indicate ranges of DI values at which such gradients exceed unity
Restitution curves yield parameters predictive of arrhythmogenicity
To permit quantitative analysis, simple mono-exponential growth functions, as used previously [28], were fitted to the experimental data. These took the form:
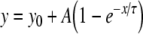 |
1 |
where y represents APD90, x represents DI, and y0, A and τ are constants obtained by least squares fitting to the experimental values of APD90 and DI in each case. These fitted parameters, along with corresponding reduced χ2 values, are shown in Tables 1 (epicardium) and 2 (endocardium). Curve fits performed on data sets obtained by separating successive odd- and even-numbered action potentials yielded statistically indistinguishable (P > 0.05) parameters in all cases (Tables 3 and 4) thus eliminating the possibility of error arising as a result of averaging. The corresponding gradients were then given by:
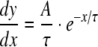 |
2 |
and take their largest values at the shortest BCLs studied.
Table 1.
Fit parameters for epicardial restitution curves
| Condition | y0 (ms) | A (ms) | τ (ms) | χ2 |
|---|---|---|---|---|
| WT (6 hearts) | 1.6 ± 4.4 | 56.2 ± 2.4 | 58.4 ± 9.8 | 1.4 |
| WT + flecainide (5 hearts) | −16.3 ± 9.4 | 104.7 ± 7.1 | 82.8 ± 28.0 | 5.9 |
| WT + 1 μM quinidine (6 hearts) | −38.3 ± 17.4 | 92.4 ± 14.7 | 45.1 ± 9.4 | 2.5 |
| WT + 10 μM quinidine (6 hearts) | −863.0 ± 1,387.1 | 981.6 ± 1,369.1 | 27.1 ± 15.9 | 113.3 |
| Scn5a+/Δ (5 hearts) | −19.6 ± 7.2 | 90.0 ± 4.4 | 53.1 ± 10.7 | 14.0 |
| Scn5a+/Δ + flecainide (5 hearts) | −16.1 ± 6.5 | 82.2 ± 12.3 | 95.3 ± 42.1 | 3.5 |
| Scn5a+/Δ + 1 μM quinidine (5 hearts) | −677.1 ± 306.1 | 748.0 ± 304.7 | 15.2 ± 2.2 | 10.0 |
| Scn5a+/− (6 hearts) | −674.8 ± 329.9 | 729.4 ± 329.1 | 14.2 ± 2.1 | 8.4 |
| Scn5a+/− + flecainide (6 hearts) | −56,359.8 ± 2,559.7 | 52,677.3 ± 2.2 | 7.8 ± 2.5 | 14.8 |
| Scn5a+/− + 1 μM quinidine (5 hearts) | −118.3 ± 57.4 | 202.5 ± 49.4 | 36.4 ± 10.5 | 16.7 |
| Scn5a+/− + 10 μM quinidine (7 hearts) | −1.7 ± 44.7 | 131.9 ± 62.3 | 158.4 ± 262.2 | 10.8 |
Results of least-squares fits to the function 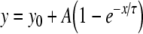 under the conditions indicated
under the conditions indicated
Table 2.
Fit parameters for endocardial restitution curves
| Condition | y0 (ms) | A (ms) | τ (ms) | χ2 |
|---|---|---|---|---|
| WT (6 hearts) | −11.8 ± 3.7 | 73.8 ± 1.7 | 65.2 ± 8.4 | 1.2 |
| WT + flecainide (5 hearts) | −769.0 ± 244.1 | 827.3 ± 243.7 | 14.5 ± 1.3 | 1.4 |
| WT + 1 μM quinidine (6 hearts) | −65.8 ± 100.4 | 113.5 ± 98.3 | 24.9 ± 12.0 | 11.7 |
| WT + 10 μM quinidine (6 hearts) | −41.9 ± 71.5 | 78.2 ± 68.7 | 32.8 ± 19.3 | 13.6 |
| Scn5a+/Δ (5 hearts) | −31.9 ± 9.0 | 109.9 ± 5.1 | 49.6 ± 9.0 | 10.3 |
| Scn5a+/Δ + flecainide (5 hearts) | −561.1 ± 1.2 | 631.6 ± 15.1 | 18.7 ± 0.3 | 6.7 |
| Scn5a+/Δ + 1 μM quinidine (5 hearts) | −499.7 ± 143.8 | 563.0 ± 142.9 | 16.3 ± 1.6 | 56.0 |
| Scn5a+/− (6 hearts) | −281.7 ± 9.8 | 43.5 ± 6.4 | 48.5 ± 19.0 | 4.3 |
| Scn5a+/− + flecainide (6 hearts) | −1,050.6 ± 1,558.5 | 1,100.1 ± 1,557.8 | 11.7 ± 4.0 | 10.0 |
| Scn5a+/− + 1 μM quinidine (5 hearts) | −261.2 ± 117.9 | 350.5 ± 107.8 | 34.2 ± 9.3 | 21.7 |
| Scn5a+/− + 10 μM quinidine (7 hearts) | −220.1 ± 322.0 | 319.1 ± 316.8 | 26.2 ± 12.7 | 45.5 |
Results of least-squares fits to the function 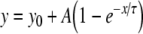 under the conditions indicated
under the conditions indicated
Table 3.
Fit parameters for endocardial restitution curves separating successive odd- and even-numbered action potentials
| Condition | Odd | Even | ||||||
|---|---|---|---|---|---|---|---|---|
| y0 (ms) | A (ms) | τ (ms) | χ2 | y0 (ms) | A (ms) | τ (ms) | χ2 | |
| WT (6 hearts) | 13.4 ± 7.6 | 40.5 ± 12.9 | 50.3 ± 21.4 | 6.3 | 8.1 ± 11.2 | 58.3 ± 25.4 | 48.2 ± 32.3 | 4.6 |
| WT + flecainide (5 hearts) | −14.7 ± 8.9 | 105.2 ± 9.0 | 88.0 ± 31.3 | 5.8 | −44.6 ± 18.5 | 117.4 ± 14.2 | 49.0 ± 9.9 | 1.5 |
| WT + 1 μM quinidine (6 hearts) | −20.6 ± 9.9 | 75.8 ± 6.2 | 56.4 ± 13.2 | 3.8 | −34.2 ± 11.7 | 89.2 ± 9.3 | 45.9 ± 7.8 | 3.3 |
| WT + 10 μM quinidine (6 hearts) | −1,391.7 ± 3,195.9 | 1,496.0 ± 3,182.0 | 21.1 ± 14.3 | 117.9 | −393.1 ± 5.3.0 | 532.8 ± 481.7 | 41.1 ± 27.6 | 115.7 |
| Scn5a+/Δ (5 hearts) | −23.9 ± 8.3 | 92.0 ± 5.3 | 47.8 ± 9.0 | 12.4 | −24.4 ± 8.6 | 92.6 ± 5.6 | 47.6 ± 8.9 | 11.8 |
| Scn5a+/Δ + flecainide (5 hearts) | 14.6 ± 6.3 | 77.1 ± 8.4 | 84.1 ± 32.2 | 3.4 | −33.2 ± 46.4 | 110.1 ± 41.9 | 36.4 ± 13.1 | 2.0 |
| Scn5a+/Δ + 1 μM quinidine (5 hearts) | −250.2 ± 12.8 | 483.0 ± 24.5 | 18.8 ± 7.8 | 12.4 | −254.7 ± 321.2 | 328.1 ± 318.3 | 20.0 ± 7.7 | 5.3 |
| Scn5a+/− (6 hearts) | −850.4 ± 282.7 | 904.2 ± 282.1 | 13.5 ± 1.3 | 4.3 | −913.3 ± 283.3 | 968.6 ± 282.7 | 13.7 ± 1.2 | 3.7 |
| Scn5a+/− + flecainide (6 hearts) | −1,473.8 ± 3,144.5 | 1,534.3 ± 2,141.2 | 14.7 ± 7.6 | 34.3 | −2,108.8 ± 4,489.9 | 2,171.3 ± 4,486.8 | 14.0 ± 6.7 | 35.0 |
| Scn5a+/− + 1 μM quinidine (5 hearts) | −46.8 ± 51.2 | 146.8 ± 21.3 | 59.5 ± 43.5 | 9.4 | −46.7 ± 50.6 | 149.2 ± 20.2 | 61.0 ± 44.2 | 9.3 |
| Scn5a+/− + 10 μM quinidine (7 hearts) | 7.3 ± 34.2 | 167.7 ± 264.8 | 272.8 ± 78.3 | 10.8 | 2.8 ± 44.6 | 143.1 ± 121.3 | 192.2 ± 405.8 | 12.0 |
Results of least-squares fits to the function 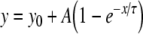 under the conditions indicated
under the conditions indicated
Table 4.
Fit parameters for endocardial restitution curves separating successive odd- and even-numbered action potentials
| Condition | Odd | Even | ||||||
|---|---|---|---|---|---|---|---|---|
| y0 (ms) | A (ms) | τ (ms) | χ2 | y0 (ms) | A (ms) | τ (ms) | χ2 | |
| WT (6 hearts) | −17.4 ± 5.3 | 73.8 ± 2.8 | 67.3 ± 13.5 | 4.7 | −38.9 ± 15.4 | 91.6 ± 13.0 | 46.5 ± 8.5 | 1.8 |
| WT + flecainide (5 hearts) | −49.2 ± 26.2 | 113.8 ± 23.0 | 35.7 ± 8.2 | 7.6 | −367.3 ± 171.3 | 426.9 ± 170.6 | 17.7 ± 2.4 | 1.3 |
| WT + 1 μM quinidine (6 hearts) | −393.2 ± 368.6 | 437.7 ± 368.6 | 14.9 ± 4.3 | 18.1 | −1,337.3 ± 1,206.4 | 1,384.5 ± 1,205.7 | 11.5 ± 2.5 | 14.7 |
| WT + 10 μM quinidine (6 hearts) | −17.7 + −39.0 | 53.2 ± 35.0 | 42.6 ± 28.9 | 11.2 | −28.0 ± 62.7 | 66.6 ± 58.2 | 39.6 ± 29.7 | 17.3 |
| Scn5a+/Δ (5 hearts) | −149.6 ± 131.4 | 221.0 ± 128.6 | 26.4 ± 7.6 | 0.9 | −42.3 ± 10.5 | 115.3 ± 8.2 | 40.8 ± 5.0 | 2.6 |
| Scn5a+/Δ + flecainide (5 hearts) | −1,133.8 ± 784.2 | 1,201.4 ± 783.1 | 15.2 ± 2.6 | 2.0 | −2,164.0 ± 1,195.7 | 2,233.3 ± 1,195.0 | 13.6 ± 1.6 | 1.4 |
| Scn5a+/Δ + 1 μM quinidine (5 hearts) | −2,720.4 ± 3,917.8 | 2,779.7 ± 3,917.2 | 10.4 ± 2.8 | 3.4 | −3,527.2 ± 4,674.78 | 3,603.0 ± 4,674.1 | 11.1 ± 2.7 | 3.9 |
| Scn5a+/− (6 hearts) | −134.6 ± 28.9 | 195.3 ± 28.0 | 22.8 ± 2.1 | 3.3 | −149.1 ± 33.5 | 212.0 ± 32.6 | 22.9 ± 2.2 | 3.6 |
| Scn5a+/− + flecainide (6 hearts) | −13.8 ± 29.2 | 66.2 ± 27.1 | 32.5 ± 11.1 | 1.8 | −55.4 ± 55.5 | 108.6 ± 54.6 | 23.6 ± 5.9 | 1.2 |
| Scn5a+/− + 1 μM quinidine (5 hearts) | −1,979.9 ± 1,709.0 | 2,051.5 ± 1,704.6 | 15.9 ± 2.8 | 13.4 | −108.4 ± 15,101.8 | 10,911.4 ± 15,098.3 | 11.8 ± 3.0 | 14.8 |
| Scn5a+/− + 10 μM quinidine (7 hearts) | −332.1 ± 447.2 | 431.4 ± 439.8 | 25.2 ± 12.7 | 116.0 | −160.0 ± 289.5 | 262.2 ± 281.9 | 20.1 ± 18.1 | 48.1 |
Results of least-squares fits to the function 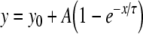 under the conditions indicated
under the conditions indicated
Figures 4 and 5 thus superimpose optimisations of Eq. 1 to the data shown in Tables 1 and 2 (solid lines, left-hand ordinates). Furthermore, they show the corresponding gradients calculated using Eq. 2 (broken lines, right-hand axes). Shaded regions where present indicate DIs less than the critical DI at which these gradients exceed unity: the existence of such a region is an established criterion for arrhythmogenesis [25]. Critical DI was calculated as:
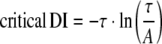 |
3 |
Figure 6 proceeds to show these values of critical DI derived from the fitted values of A and τ for the epicardia (filled bars) and endocardia (open bars) under each of the above conditions. In contrast to the findings from the analysis of the relationships between epicardial and endocardial APD90s described above, the present analysis of epicardial and endocardial restitution properties together provided datasets in complete agreement with the observed incidences of arrhythmic activity.
Fig. 6.
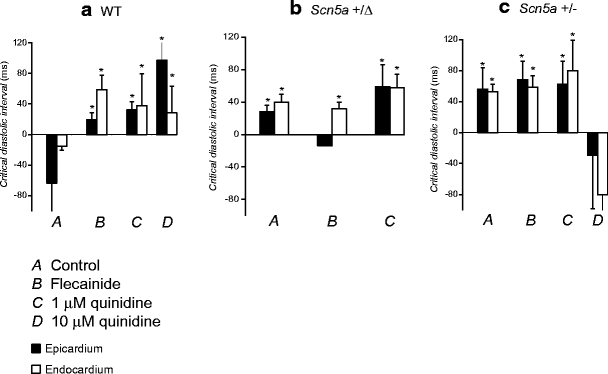
Critical diastolic intervals obtained from restitution curves. Critical diastolic intervals at which gradients equalled unity obtained from epicardial (filled bars) and endocardial (open bars) restitution curves shown in Figs. 4 and 5 in WT (a), Scn5a+/Δ (b) and Scn5a+/− (c) hearts exposed to control solution (A) and to solutions containing 1 μM flecainide (B), 1 μM quinidine (C) and 10 μM quinidine (D). Asterisks indicate values that are significantly (P < 0.05) larger than those recorded in WT hearts exposed to control solution
First, in WT hearts (Fig. 6a) exposed to control solution (A) the critical DI took a value of −63.63 ± 14.41 ms in the epicardium and −15.43 ± 4.97 ms in the endocardium. Treatment with flecainide (B) significantly (P < 0.05) increased these values to 19.45 ± 9.39 ms in the epicardium and 58.75 ± 18.71 ms in the endocardium. This critical DI was attained in the endocardium (shaded region in Fig. 5a, B), in parallel with the increased incidence of arrhythmogenesis observed during the extrasystolic stimulation procedure under these conditions (Fig. 2, ii). Treatment with 1 μM quinidine (Fig. 6a, C) also significantly (P < 0.05) increased the critical DI in both the epicardium and endocardium (32.30 ± 7.80 ms and 27.80 ± 14.53 ms respectively) in parallel with the observed arrhythmogenicity on extrasystolic stimulation under these conditions (Fig. 2, ii), although such values were not attained during the dynamic pacing protocol. Furthermore, treatment with 10 μM quinidine (Fig. 6a, D) again significantly (P < 0.05) increased these values to 97.20 ± 57.90 ms in the epicardium and to 28.50 ± 34.52 ms in the endocardium. This DI was attained in the epicardium in parallel with the observed arrhythmogenicity during the extrasystolic stimulation procedure (Fig. 2, ii). Thus, increases in the critical DI are precisely paralleled with arrhythmogenicity in WT hearts treated with flecainide and quinidine.
Secondly, critical DIs obtained from Scn5a+/Δ hearts (Fig. 6b) were always significantly greater than those in WT controls (P < 0.05 in all cases) in parallel with the increased incidence of arrhythmic activity observed during the extrasystolic stimulation procedure (Fig. 2, ii). Thus, in hearts treated with control solution (Fig. 6b, A) the critical DI increased to 28.04 ± 8.08 ms in the epicardium and 39.38 ± 10.28 ms in the endocardium: These values were attained in both the epicardium and endocardium in association with the increased incidence of arrhythmic activity on extrasystolic stimulation observed (Fig. 2, ii) and in full agreement with clinical expectations [23]. Treatment with flecainide (Fig. 6b, B) decreased the epicardial value (−14.09 ± 9.05 ms, P < 0.05 as compared to hearts treated with control solution) but did not significantly affect the endocardial value (21.41 ± 8.58 ms, P > 0.05 as compared to hearts treated with control solution), having no effect on the observed incidence of arrhythmic activity on extrasystolic stimulation (Fig. 2, ii). However, treatment with 1 μM quinidine (Fig. 6b, C) significantly (P < 0.05) increased both epicardial and endocardial values (59.09 ± 26.91 ms and 57.64 ± 16.68 ms, respectively) as compared to those obtained from such hearts treated with control solution, in association with the observed increases in the incidence of arrhythmic activity resulting from extrasystolic stimulation (Fig. 2, ii) and again in agreement with expectations from clinical work [33]. It was not possible to obtain MAPs over a sufficiently wide range of BCLs to permit the construction of such curves in Scn5a+/Δ hearts treated with 10 μM quinidine.
Thirdly, critical DI values were also increased in Scn5a+/− hearts (Fig. 6c) in association with the increased incidence of arrhythmic activity observed. Thus, in hearts treated with control solution (A), the critical DIs were significantly (P < 0.05) increased as compared to WT controls in both the epicardium and endocardium to 56.00 ± 27.79 ms and 52.59 ± 9.50 ms, respectively. These values were attained in both the epicardium and endocardium, fulfilling clinical expectations [1]. Treatment with flecainide (B) did not significantly alter these values (68.14 ± 24.09 ms in the epicardium and 58.56 ± 14.75 ms in the endocardium). These were again attained in both epicardium and endocardium in association with the arrhythmic activity observed on extrasystolic stimulation (Fig. 2, ii). Furthermore, treatment with quinidine at a concentration of 1 μM (Fig. 6c, C) had no significant effect on critical DIs in either the epicardium or endocardium (62.45 ± 29.74 ms and 79.63 ± 39.31 ms, respectively). These values were again attained in both epicardium and endocardium. However, treatment with quinidine at 10 μM significantly decreased both epicardial and endocardial critical DIs, such that they became statistically indistinguishable from WT controls (29.01 ± 9.27 ms and −80 ± 3.58 ms, respectively). This was associated with the complete absence of arrhythmogenesis during the extrasystolic stimulation procedure (Fig. 2, ii).
Thus, while relationships between epicardial and endocardial action potential durations obtained over a range of cycle lengths are poorly correlated with arrhythmogenicity, restitution curves are, in general, predictive of arrhythmogenicity under the genetic and pharmacological conditions studied.
Discussion
The congenital LQT3 and the BrS constitute two important examples of genetic causes for ventricular arrhythmia and sudden cardiac death [3]. These conditions result from similarly targeted mutations in the SCN5a gene coding for the cardiac Na+ channel: gain-of-function mutations are characteristic of LQT3, while loss-of-function mutations are characteristic of BrS [1, 23].
The present study began by using the MAP technique to verify that Langendorff-perfused murine hearts modelling LQT3 (Scn5a+/Δ) [13] and BrS (Scn5a+/−) [29] recapitulated features of the incidence of arrhythmic activity reported clinically. These results were in full agreement with previous physiological and pharmacological findings from our LQT3 [39] and BrS [38] models. Epicardial and endocardial MAP recordings were then used to evaluate the applicability of two established criteria in predicting arrhythmogenicity in these models over the full physiological range of heart rates (quantified as baseline cycle lengths, BCLs).
Transmural dispersion of repolarization was studied over a range of BCLs by comparing epicardial and endocardial action potential durations (at 90% repolarization, APD90s) in the two genetic models. Such an analysis had previously been demonstrated to reveal arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts [34]. Furthermore, changes in this relationship investigated at a single BCL had previously been associated with arrhythmogenicity in our Scn5a+/Δ hearts [42]. Plots of epicardial against endocardial APD90 in WT hearts treated with control solution gave reference relationships best fitted by a quadratic function, in full agreement with previous work [34]. Treatment of such WT hearts with either quinidine or flecainide resulted in the data points falling to the left of the reference curves, reflecting increased transmural repolarization gradients, in association with increased incidences of arrhythmic activity and again in full agreement with a previous study [34]. However, such plots failed to consistently predict the presence or absence of arrhythmogenicity in the Scn5a+/Δ and Scn5a+/− hearts. Such left-shifting of points was observed in Scn5a+/Δ hearts under all pharmacological conditions studied, despite the established anti-arrhythmic effect of flecainide in this setting. Furthermore, data points obtained from Scn5a+/− hearts treated with control solution fell on the reference curve despite the increased incidence of arrhythmic activity observed. Treatment with flecainide failed to result in a left-shift of points, despite its established pro-arrhythmic effect; treatment with quinidine did result in such a shift, despite its anti-arrhythmic effect.
These discrepancies prompted an investigation for alternative criteria for arrhythmogenicity. We therefore explored the applicability of a second approach involving restitution curves plotting APD90 against the preceding DI. Arrhythmic activity occurring in a range of settings has previously been associated with increases in the slopes of such curves to greater than unity [14, 17–19, 28, 40]. Such an association has been demonstrated in a canine pharmacological model of the congenital long QT syndrome type 2 [45] but has not previously been studied in any models of LQT3 or BrS in any species. Furthermore, decreases in the slopes of such curves have been associated with the effects of clinically important anti-arrhythmic drugs, including the class 1A agent procainamide in a canine model [32]. However, such slopes have not been examined in relation to either quinidine or flecainide, nor have any such studies previously been attempted in murine models. This study accordingly proceeded to construct epicardial and endocardial restitution curves for the first time in a murine system and in relation to LQT3 or BrS in any species.
In contrast to the previous criterion, epicardial and endocardial restitution curves taken together yielded parameters in precise agreement with the presence or absence of arrhythmic phenomena demonstrated. In WT hearts, treatment with either quinidine or flecainide significantly increased the critical DI at which the slopes of the restitution curves exceeded unity, in full agreement with the increased incidences of arrhythmic activity observed. Not only did the Scn5a+/Δ and Scn5a+/− mutations result in significant increases in critical DI in association with increased rates of arrhythmic activity, but quinidine and flecainide exerted contrasting effects on this critical DI in agreement with clinical expectations in both cases. Thus, in Scn5a+/Δ hearts, treatment with quinidine increased critical DIs in association with its pro-arrhythmic effect. Furthermore, in Scn5a+/− hearts, treatment with quinidine decreased critical DIs in association with the decreased incidence of arrhythmic activity observed, while treatment with flecainide had the opposite effect. Thus class 1A agents may result in either increases (quinidine in WT and Scn5a+/Δ murine hearts) or decreases (quinidine in Scn5a+/− murine hearts and procainamide in canine hearts) [32] in the critical DI: Such changes are in full agreement with effects on arrhythmogenicity in all cases.
Taken together, these results demonstrate that criteria considering effects on the slopes of restitution curves successfully account for arrhythmogenicity in murine models of arrhythmic syndromes resulting from mutations in the Scn5a Na+ channel, as well as for the effects of quinidine and flecainide on such arrhythmogenicity. These observations might form the basis for future clinical explorations of the utility of restitution curves obtained using the MAP technique [28, 40] in the assessment of arrhythmic propensity in hereditary arrhythmic syndromes. Such a robust means of assessing arrhythmogenicity might be particularly useful given the known variable expressivity of the arrhythmic phenotype in patients harbouring such mutations [41].
Acknowledgements
We thank the James Baird Fund, the Frank Elmore Fund, the Medical Research Council, the Wellcome Trust, the British Heart Foundation and Downing College, Cambridge for their generous support. Ian N. Sabir and Lucia M. Li have contributed equally to this work.
References
- 1.Antzelevitch C, Brugada P, Brugada J, Brugada R, Towbin JA, Nademanee K (2003) Brugada syndrome: 1992–2002: A historical perspective. J Am Coll Cardiol 41:1665–1671 [DOI] [PubMed]
- 2.Antzelevitch C, Burashnikov A, Di Diego JM (2003) Cellular and ionic mechanisms underlying arrhythmogenesis. In: Cardiac repolarization: bridging basic and clinical science. Humana Press, Totowa, N.J
- 3.Antzelevitch C, Yan GX, Shimizu W (1999) Transmural dispersion of repolarization and arrhythmogenicity: the Brugada syndrome versus the long QT syndrome. J Electrocardiol 32(Suppl):158–165 [DOI] [PubMed]
- 4.Balasubramaniam R, Grace AA, Saumarez RC, Vandenberg JI, Huang CL (2003) Electrogram prolongation and nifedipine-suppressible ventricular arrhythmias in mice following targeted disruption of KCNE1. J Physiol 552:535–546 [DOI] [PMC free article] [PubMed]
- 5.Bevington PR (1969) Data reduction and error analysis for the physical sciences. McGraw-Hill, New York
- 6.Birkett DJ (1997) Therapeutic drug monitoring. Aust Prescr 20:9–11
- 7.Brugada R (2000) Use of intravenous antiarrhythmics to identify concealed Brugada syndrome. Curr Control Trials Cardiovasc Med 1(1):45–47 [DOI] [PMC free article] [PubMed]
- 8.Casimiro MC, Knollmann BC, Ebert SN, Vary JC Jr, Greene AE, Franz MR, Grinberg A, Huang SP, Pfeifer K (2001) Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange-Nielsen Syndrome. Proc Natl Acad Sci USA 98:2526–2531 [DOI] [PMC free article] [PubMed]
- 9.Farkas A, Curtis MJ (2002) Limited antifibrillatory effectiveness of clinically relevant concentrations of class I antiarrhythmics in isolated perfused rat hearts. J Cardiovasc Pharmacol 39:412–424 [DOI] [PubMed]
- 10.Fernandez del Pozo B, Perez-Vizcaino F, Fernandez C, Zaragoza F, Tamargo J (1997) Effects of several class I antiarrhythmic drugs on isolated rat aortic vascular smooth muscle. Gen Pharmacol 29:539–543 [DOI] [PubMed]
- 11.Garfinkel A, Kim YH, Voroshilovsky O, Qu Z, Kil JR, Lee MH, Karagueuzian HS, Weiss JN, Chen PS (2000) Preventing ventricular fibrillation by flattening cardiac restitution. Proc Natl Acad Sci USA 97:6061–6066 [DOI] [PMC free article] [PubMed]
- 12.Grace AA, Camm AJ (1998) Quinidine. N Engl J Med 338:35–45 [DOI] [PubMed]
- 13.Head CE, Balasubramaniam R, Thomas G, Goddard CA, Lei M, Colledge WH, Grace AA, Huang CL (2005) Paced electrogram fractionation analysis of arrhythmogenic tendency in DeltaKPQ Scn5a mice. J Cardiovasc Electrophysiol 16:1329–1340 [DOI] [PubMed]
- 14.Karagueuzian HS, Khan SS, Hong K, Kobayashi Y, Denton T, Mandel WJ, Diamond GA (1993) Action potential alternans and irregular dynamics in quinidine-intoxicated ventricular muscle cells. Implications for ventricular proarrhythmia. Circulation 87:1661–1672 [DOI] [PubMed]
- 15.Killeen M, Thomas G, Gurung I, Goddard C, Fraser J, Mahaut-Smith M, Colledge H, Grace A, Huang C (2007) Arrhythmogenic mechanisms in the isolated perfused hypokalemic murine heart. Acta Physiol 189:33–46 [DOI] [PMC free article] [PubMed]
- 16.Knollmann BC, Katchman AN, Franz MR (2001) Monophasic action potential recordings from intact mouse heart: validation, regional heterogeneity, and relation to refractoriness. J Cardiovasc Electrophysiol 12:1286–1294 [DOI] [PubMed]
- 17.Koller ML, Riccio ML, Gilmour RF Jr (1998) Dynamic restitution of action potential duration during electrical alternans and ventricular fibrillation. Am J Physiol 275:H1635–H1642 [DOI] [PubMed]
- 18.Koller ML, Riccio ML, Gilmour RF Jr (2000) Effects of [K+]o on electrical restitution and activation dynamics during ventricular fibrillation. Am J Physiol Heart Circ Physiol 279:H2665–H2672 [DOI] [PubMed]
- 19.Lee JJ, Kamjoo K, Hough D, Hwang C, Fan W, Fishbein MC, Bonometti C, Ikeda T, Karagueuzian HS, Chen PS (1996) Reentrant wave fronts in Wiggers’ stage II ventricular fibrillation. Characteristics and mechanisms of termination and spontaneous regeneration. Circ Res 78:660–675 [DOI] [PubMed]
- 20.Lukas A, Antzelevitch C (1996) Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia. Cardiovasc Res 32:593–603 [DOI] [PubMed]
- 21.Milberg P, Reinsch N, Wasmer K, Monnig G, Stypmann J, Osada N, Breithardt G, Haverkamp W, Eckardt L (2005) Transmural dispersion of repolarization as a key factor of arrhythmogenicity in a novel intact heart model of LQT3. Cardiovasc Res 65:397–404 [DOI] [PubMed]
- 22.Moss A, Windle J, Hall W, Zareba W, Robinson J, McNitt S, Severski P, Rosero S, Daubert J, Qi M, Cieciorka M, Manalan A (2005) Safety and efficacy of flecainide in subjects with Long QT-3 syndrome (KPQ mutation): a randomized, double-blind, placebo-controlled clinical trial. Annals of Non-invasive Electrocardiology 10(suppl):59–66 [DOI] [PMC free article] [PubMed]
- 23.Moss AJ, Kass RS (2005) Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest 115:2018–2024 [DOI] [PMC free article] [PubMed]
- 24.Napolitano C, Priori S (2006) Brugada Syndrome. Orphanet J Rare Dis 1:35–40 [DOI] [PMC free article] [PubMed]
- 25.Nolasco JB, Dahlen RW (1968) A graphic method for the study of alternation in cardiac action potentials. J Appl Physiol 25:191–196 [DOI] [PubMed]
- 26.Omichi C, Zhou S, Lee MH, Naik A, Chang CM, Garfinkel A, Weiss JN, Lin SF, Karagueuzian HS, Chen PS (2002) Effects of amiodarone on wave front dynamics during ventricular fibrillation in isolated swine right ventricle. Am J Physiol Heart Circ Physiol 282:H1063–H1070 [DOI] [PubMed]
- 27.Opthof T, Coronel R (2005) Transmural dispersion in LQT3 and arrhythmogenesis. Cardiovasc Res 68:336–337, author reply 338–339 [DOI] [PubMed]
- 28.Pak HN, Hong SJ, Hwang GS, Lee HS, Park SW, Ahn JC, Moo Ro Y, Kim YH (2004) Spatial dispersion of action potential duration restitution kinetics is associated with induction of ventricular tachycardia/fibrillation in humans. J Cardiovasc Electrophysiol 15:1357–1363 [DOI] [PubMed]
- 29.Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI, Colledge WH, Grace AA (2002) Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci USA 99:6210–6215 [DOI] [PMC free article] [PubMed]
- 30.Pratt CM, Moye LA (1995) The cardiac arrhythmia suppression trial. Casting suppression in a different light. Circulation 91:245–247 [DOI] [PubMed]
- 31.Priori SG, Napolitano C, Schwartz PJ, Bloise R, Crotti L, Ronchetti E (2000) The elusive link between LQT3 and Brugada syndrome: The role of flecainide challenge. Circulation 102:945–947 [DOI] [PubMed]
- 32.Riccio ML, Koller ML, Gilmour RF Jr (1999) Electrical restitution and spatiotemporal organization during ventricular fibrillation. Circ Res 84:955–963 [DOI] [PubMed]
- 33.Roden D, Anderson M (2006) Proarrhythmia. Handb Exp Pharmacol 171:73–97 [DOI] [PubMed]
- 34.Sabir IN, Fraser JA, Cass TR, Grace AA, Huang CL (2007a) A quantitative analysis of the effect of cycle length on arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. Pflugers Arch PMID: 17437126 [DOI] [PMC free article] [PubMed]
- 35.Sabir IN, Fraser JA, Killeen MJ, Grace AA, Huang CL (2007b) The contribution of refractoriness to arrhythmic substrate in hypokalaemic Langendorff-perfused murine hearts. Pflugers Arch 454:209–222 [DOI] [PMC free article] [PubMed]
- 36.Sabir IN, Killeen MJ, Goddard CA, Thomas G, Gray S, Grace AA, Huang CL (2007c) Transient alterations in transmural repolarization gradients following premature ventricular depolarizations in arrhythmogenic hypokalaemic Langendorff-perfused murine hearts. J Physiol 581:277–289 [DOI] [PMC free article] [PubMed]
- 37.Saumarez RC, Grace AA (2000) Paced ventricular electrogram fractionation and sudden death in hypertrophic cardiomyopathy and other non-coronary heart diseases. Cardiovasc Res 47:11–22 [DOI] [PubMed]
- 38.Stokoe KS, Balasubramaniam R, Goddard CA, Colledge WH, Grace AA, Huang CL (2007) Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/− murine hearts. J Physiol 578:69–84 [DOI] [PMC free article] [PubMed]
- 39.Stokoe KS, Thomas G, Goddard CA, Colledge WH, Grace AA, Huang CL (2007) Effects of flecainide and quinidine on arrhythmogenic properties of Scn5a+/Δ murine hearts modelling long QT syndrome 3. J Physiol 578:69–84 [DOI] [PMC free article] [PubMed]
- 40.Taggart P, Sutton P, Chalabi Z, Boyett MR, Simon R, Elliott D, Gill JS (2003) Effect of adrenergic stimulation on action potential duration restitution in humans. Circulation 107:285–289 [DOI] [PubMed]
- 41.Tan HL, Hofman N, van Langen IM, van der Wal AC, Wilde AA (2005) Sudden unexplained death: heritability and diagnostic yield of cardiological and genetic examination in surviving relatives. Circulation 112:207–213 [DOI] [PubMed]
- 42.Thomas G, Gurung IS, Killeen MJ, Hakim P, Goddard CA, Mahaut-Smith MP, Colledge WH, Grace AA, Huang CL (2007a) Effects of L-type Ca2+ channel antagonism on ventricular arrhythmogenesis in murine hearts containing a modification in the Scn5a gene modelling human long QT syndrome 3. J Physiol 578:85–97 [DOI] [PMC free article] [PubMed]
- 43.Weiss JN, Chen PS, Qu Z, Karagueuzian HS, Garfinkel A (2000) Ventricular fibrillation: how do we stop the waves from breaking? Circ Res 87:1103–1107 [DOI] [PubMed]
- 44.Weiss JN, Karma A, Shiferaw Y, Chen PS, Garfinkel A, Qu Z (2006) From pulsus to pulseless: the saga of cardiac alternans. Circ Res 98:1244–1253 [DOI] [PubMed]
- 45.Yamauchi S, Yamaki M, Watanabe T, Yuuki K, Kubota I, Tomoike H (2002) Restitution properties and occurrence of ventricular arrhythmia in LQT2 type of long QT syndrome. J Cardiovasc Electrophysiol 13:910–914 [DOI] [PubMed]
- 46.Yan G-X, Antzelevitch C (1999) Cellular Basis for the Brugada Syndrome and Other Mechanisms of Arrhythmogenesis Associated With ST-Segment Elevation. Circulation 100:1660–1666 [DOI] [PubMed]