

SUMMARY
Either herpesvirus entry mediator (HVEM, TNFRSF14) or nectin-1 (PVRL1) is sufficient for herpes simplex virus (HSV) infection of cultured cells. The contribution of individual receptors to infection in vivo and to disease is less clear. To assess this, Tnfrsf14 −/− and/or Pvrl1 −/− mice were challenged intravaginally with HSV-2. Infection of the vaginal epithelium occurred in the absence of either HVEM or nectin-1, but was virtually undetectable when both receptors were absent, indicating that either HVEM or nectin-1 was necessary. Absence of nectin-1 (but not HVEM) reduced efficiency of infection of the vaginal epithelium and viral spread to the nervous system, attenuating neurological disease and preventing external lesion development. While nectin-1 proved not to be essential for infection of the nervous system, it is required for the full manifestations of disease. This study illustrates the value of mutant mice for understanding receptor contributions to disease caused by a human virus.
INTRODUCTION
Viral entry can be mediated by multiple alternative receptors that are expressed in many cell types and tissues, making it difficult to assess the roles of each receptor in disease. Mice with gene disruptions have been of limited use for studying this problem with human viruses because mice are often resistant to these viruses. However, mice are susceptible to HSV infection and disease, and mouse entry receptors are encoded by genes orthologous to the genes for the human entry receptors (Spear et al., 2000). Here we describe the use of mutant mice to explore the roles in HSV disease of two cell surface proteins that can mediate the entry of HSV-2 into cells.
Manifestations of disease caused by HSV infection of children or adults range from none to mucocutaneous lesions to meningitis and encephalitis. The virus replicates in cells of the mucosal or cutaneous epithelium, causing blisters or lesions often confined to the epithelium. HSV also enters neurons and is transported to sensory and autonomic ganglia to establish latent infections. Reactivation of HSV in neurons results in transport of virus to the body surface and in recurrent mucocutaneous lesions, permitting seeding of additional neurons. Occasionally, the virus can spread to the CNS to cause life-threatening disease.
HSV is an enveloped DNA virus, the prototype of the neurotropic alphaherpesviruses. Of the dozen or more envelope glycoproteins in the HSV virion, five have a role in viral entry. Glycoproteins gB and/or gC can mediate the binding of virus to cell surface heparan sulfate or related glycosaminoglycans (Shukla and Spear, 2001). This binding is not sufficient for entry, which requires the interaction of envelope glycoprotein gD with any one of several cell surface molecules. These include HVEM (Montgomery et al., 1996), a member of the tumor necrosis factor receptor family; nectin-1 (Cocchi et al., 1998; Geraghty et al., 1998) and nectin-2 (Warner et al., 1998), cell adhesion molecules belonging to the immunoglobulin superfamily; and specific sites in heparan sulfate (3-O-S-HS) generated by particular 3-O-sulfotransferases (Shukla et al., 1999). Interaction of any of these receptors with gD is thought to trigger conformational changes in gD that result in activation of the fusogenic activity of trimeric gB and/or the heterodimer gH-gL (Carfi et al., 2001; Fusco et al., 2005; Heldwein et al., 2006; Krummenacher et al., 2005; Rey, 2006; Spear et al., 2006), resulting in entry by envelope-membrane fusion.
HVEM and nectin-1 are efficient entry and cell fusion receptors for both serotypes of HSV whereas nectin-2 and 3-O-S-HS appear to be less efficient (Lopez et al., 2000; Shukla et al., 1999). Also, nectin-2 preferentially mediates entry of HSV-2 and certain mutants of HSV-1 (Lopez et al., 2000; Martinez and Spear, 2001; Warner et al., 1998) whereas 3-O-S-HS preferentially mediates entry of HSV-1 (Shukla and Spear, 2001). Mouse forms of these receptors resemble the human forms in their entry and fusion activities with both HSV-1 and HSV-2 (Yoon et al., 2003) except that it has been difficult to document entry activity for mouse nectin-2 (Martinez and Spear, 2001; Shukla et al., 1999). Thus, the best candidates for efficient HSV-2 entry receptors in the mouse are HVEM and nectin-1.
Mice in which the gene encoding HVEM (Tnfrsf14) was disrupted displayed no evident developmental abnormalities but did exhibit enhanced lethal responses to the mitogen, concanavalin A, and increased susceptibility to autoimmune encephalomyelitis (Wang et al., 2005). HVEM has two natural ligands. One is a member of the tumor necrosis factor family and its binding to HVEM on T cells, B cells, neutrophils, macrophages, and possibly dendritic cells can enhance activation and/or differentiation of these cells (Croft, 2005; Murphy et al., 2006). The other is B and T lymphocyte attenuator (BTLA) and the binding of HVEM to BTLA on T cells can repress T cell proliferation and activation (Murphy et al., 2006). Although physiological roles of HVEM have been associated mainly with cells of the lymphoid system, HVEM is widely expressed in a number of other cell types, including epithelial, stromal and endothelial cells (Spear, 2004). Its expression in the nervous system has not been studied, in part because initial screens for transcripts revealed little or no expression in the brain (Hsu et al., 1997; Montgomery et al., 1996).
Mice in which the gene encoding nectin-1 (Pvrl1) was disrupted were fertile and developed normally, for the most part, but exhibited delays in attaining adult size and abnormalities such as microphthalmia (Inagaki et al., 2005). There are four nectins and five nectin-like molecules, all of which participate in cell junctions of various kinds, including adherens junctions (Ogita and Takai, 2006). It seems likely that there is redundancy in functionality of the members of this family because disruptions in expression of specific nectins have deleterious effects on only a limited subset of the junctions in which each molecule can be detected. Gene disruptions have shown that nectin-1 and nectin-3 are both required for a specific junction between pigmented and non-pigmented cell layers in the ciliary epithelia and for normal development of the ciliary body of the eye (Inagaki et al., 2005). Moreover, both nectin-1 and nectin-3 contribute to the formation of puncta adherentia junctions in the mouse hippocampus and influence mossy fiber trajectories (Honda et al., 2006; Mizoguchi et al., 2002). Nectin-1 is widely expressed in a variety of cell types, including epithelial, stromal, endothelial and lymphoid cells, and also in many neurons of the mouse peripheral and central nervous systems (PNS and CNS) (Haarr et al., 2001). Moreover, antibodies specific for nectin-1 have been shown to inhibit entry of virus into cultured human and rodent neurons (Richart et al., 2003; Simpson et al., 2005).
Mice can be infected by HSV-1 or HSV-2 and provide useful models for several aspects of HSV disease in humans. Mice are susceptible to infection via the intravaginal route during the diestrous stage of the estrous cycle, when the vaginal epithelium is only a few cell layers thick and not cornified, but not during other stages when there is extensive cornification. Diestrous can be induced artificially by an injection of progesterone, so that mice do not have to be staged prior to inoculation. This model of HSV infection and disease (Parr et al., 1994) is widely used. Virus replicates in the vaginal epithelium, causing lesions, and then spreads via autonomic and sensory nerves to peripheral ganglia, including dorsal root ganglia (DRG), paracervical ganglia and other autonomic ganglia in the bladder and rectal walls (Parr and Parr, 2003). Secondary lesions occur externally, on the perineal skin and surrounding areas. Clinical symptoms resemble, in some respects, what humans may experience during severe primary genital infection with HSV-2. For example, both mice and humans may experience urinary retention, constipation and paraparesis, as well as internal and external lesions. In mice, virus is more likely to spread also to the CNS causing death, particularly at the doses of virus used in experimental studies.
To investigate the roles of HVEM and nectin-1 in HSV-2 infection and acute disease, we inoculated Tnfrsf14−/− and Pvrl1−/− mice, double mutants and wild-type controls with virus via the intravaginal route and monitored both clinical signs of disease and the spread of virus from the portal of entry to the PNS and CNS. The results showed that either nectin-1 (most efficiently) or HVEM could mediate the infection of cells in the vaginal epithelium and that neither receptor was required for spread of virus to the PNS and CNS or for lethal consequences of disease. Nectin-1 was required, however, for the appearance of secondary external lesions, which may result from horozontal cell-to-cell transmission of infection between mucosal and skin epithelia or zosteriform transmission from neurons to skin cells.
RESULTS
Effects of mouse gene disruptions on clinical signs of disease after HSV-2 inoculation
Two virus strains were used for inoculation: virulent HSV-2(333) and a recombinant form of HSV-2(333) in which a lacZ expression cassette was inserted into an intergenic region, so that presence of virus in cells and tissues could be assessed by staining with the β-galactosidase substrate, X-gal. Unless otherwise indicated, the inoculating dose for each virus, designated here HSV-2 and HSV-2/Gal, was 6 × 105 plaque-forming units (PFU) per mouse delivered intravaginally.
The mice were examined daily after inoculation and clinical symptoms recorded, including the appearance of external signs of infection such as hair loss, inflammation and lesions around the vaginal and rectal openings, around the tail base and down the legs. Symptoms also included signs of lethargy, abdominal distention, dehydration and (rarely) hind-limb paralysis, which were indicative of impending death. Mice showing these latter signs in severe form were euthanized. The results are summarized in the Kaplan Meier survival plots shown in Fig. 1. The top panels show days without external signs of disease and the bottom panels show days remaining alive.
Figure 1. Appearance of external lesions and mortality after intravaginal inoculation of wild-type (+/+), Tnfrsf14−/−, Pvrl+/− and Pvrl−/− mice with HSV-2/Gal or HSV-2.
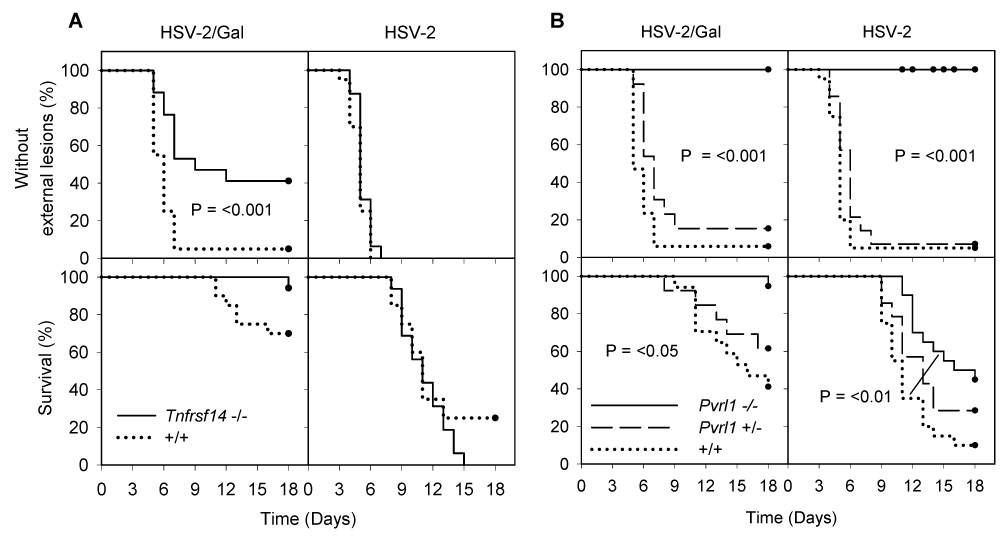
All mice were injected with Depo-Provera 6 days prior to inoculation of virus (6 × 105 PFU per mouse). The mice were examined daily for external lesions. They were also examined for severe morbidity requiring euthanasia. The day on which external lesions were first noted and the day of death or sacrifice were recorded for each animal (n = 16–20 mice for each experimental group). In A, C57BL/6 (+/+) mice are compared with Tnfrsf14−/− mice, in which the gene expressing HVEM was disrupted; in B, littermate controls (both +/+ and Pvrl+/−) are compared with Pvrl−/− mice, in which the gene expressing nectin-1 was disrupted. Kaplan-Meier survival analysis was performed using the Gehan-Breslow test. Statistically significant differences between mice of different genotypes are noted on the relevant panels. In B, the P values shown apply to comparisons of Pvrl−/− mice with either +/+ or Pvrl+/− mice except for the survival curves for mice inoculated with HSV-2. In the latter case, a significant difference was noted only for the comparison between Pvrl−/− and +/+ mice (curves linked by line). The closed circles on each survival curve indicate censored data due to death of mice during the experiment or sacrifice of mice at the conclusion of the experiment. Mice were monitored for at least 21 days but no changes in status were noted between 18 and 21 days.
Absence of HVEM in the Tnfrsf14−/− mice had little or no effect on the course of acute disease, assessed either by the appearance of external lesions or by mortality, after wild-type HSV-2 inoculation. Absence of nectin-1 in the Pvrl−/− mice had significant effects, however. In contrast to the wild-type control mice or Pvrl+/− mice, which were statistically indistinguishable in terms of disease symptoms, none of the Pvrl−/− mice inoculated with HSV-2 showed any signs of external lesions (P <0.001), despite more than half eventually dying from the infection. The survival curve (mortality) for the Pvrl−/− mice also differed significantly from that for the wild-type mice (P <0.01), indicating some attenuation of disease in the absence of nectin-1.
HSV-2/Gal is less virulent than HSV-2 (Fig. 1 and statistical analysis given in the Experimental Procedures). The mutation that abrogates nectin-1 expression (Pvrl−/−) had the expected effect of further attenuating disease caused by HSV-2/Gal. For the mutation abrogating HVEM expression (Tnfrsf14−/−), the somewhat unexpected result was attenuation of disease with HSV-2/Gal but not with HSV-2. There was a statistically significant difference between wild-type and Tnfrsf14−/− mice in the timing of external lesion appearance and number of mice eventually showing such lesions (P <0.001).
Mice expressing nectin-1 but not HVEM (Tnfsfr14−/− Pvrl1+/−), HVEM but not nectin-1 (Pvrl1−/−) and neither (Tnfrsf14−/− Pvrl−/−) were also challenged with HSV-2 at a 10-fold higher dose than that used in Fig. 1 (6 × 106 PFU per mouse instead of 6 × 105). The higher dose of virus did not significantly alter the timing or manifestations of clinical symptoms for the HVEM-expressing and nectin-1-expressing mice (except that one Pvrl1−/− mouse exhibited some inflammation at the vaginal opening without the hair loss and lesions typically observed around the vaginal and rectal openings, tail base and down the legs). None of the double-mutant mice (n=9) showed any signs of disease for more than 21 days (Fig. 2).
Figure 2. Appearance of external lesions and mortality after intravaginal inoculation with HSV-2 of nectin-1-expressing Tnfrsf14−/− Pvrl1+/− mice, HVEM-expressing Pvrl1−/− mice and double-mutant mice.
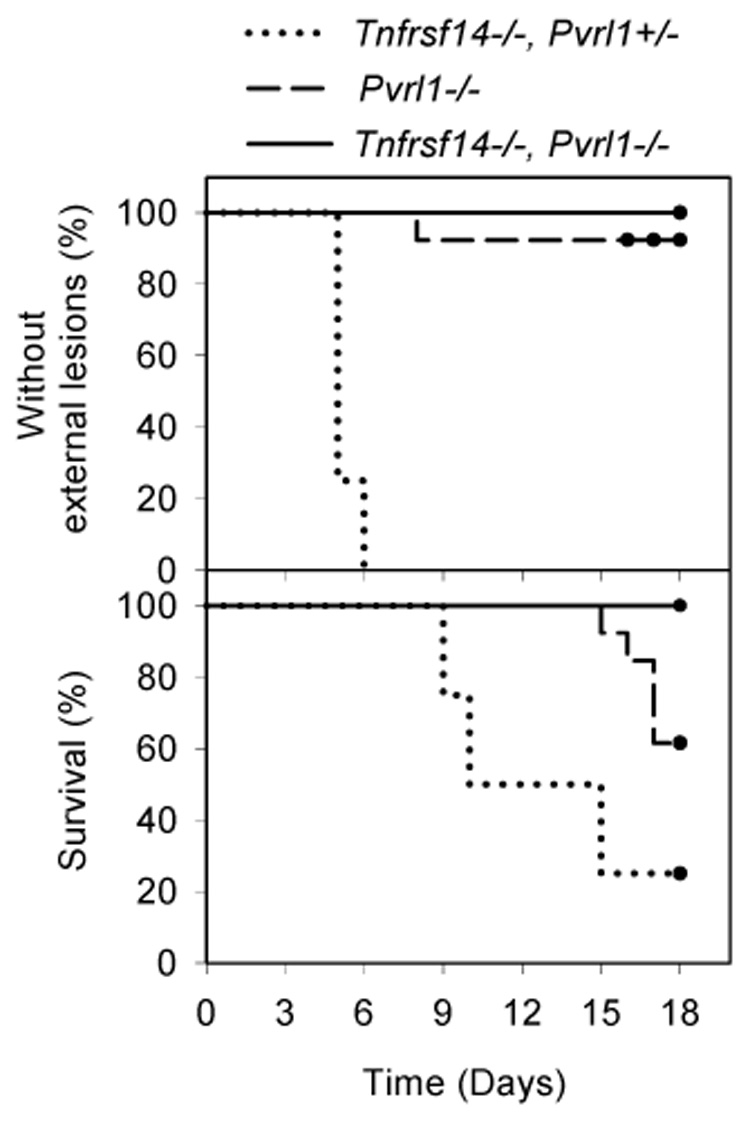
The experiment was done as in Fig. 1 except that the mice were inoculated with 10 times the dose of HSV-2 (6 × 106 PFU/mouse). The single Pvrl1−/− mouse that exhibited an external sign of disease showed some inflammation at the vaginal opening but no hair loss or external lesions around the vagina, rectum or tail or down the legs, as was typical for mice that express nectin-1. Kaplan-Meier survival analysis was performed using the Gehan-Breslow test. Statistically significant differences were noted for all pair-wise comparisons (P <0.02) except for Pvrl1−/− mice vs Tnfrsf14−/− Pvrl1−/− mice in the % remaining free of external lesions (n = 4--Tnfrsf14−/− Pvrl+/−; n=13--Pvrl1−/−; N=9--Tnfrsf14−/− Pvrl1−/−).
The results of these challenge experiments demonstrate that neither nectin-1 nor HVEM is essential for HSV-2 infection of mice via the intravaginal route, as evidenced by symptoms or mortality in 50–100% of the mice inoculated. Expression in mice of at least one of these entry receptors appears to be necessary for disease, based on absence of any clinical symptoms in the double-mutant mice after virus challenge. Absence of nectin-1 alone delayed death and reduced its incidence, but did not prevent it, whereas absence of HVEM alone had little effect on disease caused by HSV-2. Strikingly, absence of nectin-1 completely abrogated external lesions in mice inoculated with either HSV-2/Gal or HSV-2. For mice that expressed nectin-1, mortality was never observed in the absence of external lesions. HVEM-deficient mice inoculated with HSV-2/Gal, but not HSV-2, were partially protected from the development of external lesions.
Tracking of virus infection and spread by monitoring expression of β-galactosidase from HSV-2/Gal
The question arises from Fig. 1 and Fig. 2 as to whether mice failing to show any symptoms of disease had actually become infected by the inoculated virus. As one measure of the efficiency of infection of the vaginal epithelium and spread to the nervous system, wild-type and mutant mice were inoculated with HSV-2/Gal and sacrificed at various times after inoculation for the dissection of vaginas, DRG, spinal cords and perivaginal skin, followed by staining of these tissues with the β-galactosidase substrate, X-gal. The extent of lesion development on the vaginal luminal surface and in nervous system tissue was scored as described in Experimental Procedures.
Fig. 3 shows examples of stained vaginas photographed at 24 hrs after inoculation with two different doses of HSV-2/Gal: 6 × 105 and 6 × 106 PFU/mouse. At the lower dose of virus (Fig. 3A and 3B), the viral lesions were numerous and extensive for wild-type and Tnfrsf14−/− mice but were reduced in number and size for the Pvrl−/− mice. However, mice of all three genotypes invariably had detectable lesions on the vaginal epithelium (no lesion scores of 0). For the double-mutant mice (Tnfrsf14−/− Pvrl−/−), only rare blue spots (<5 per vagina) were observed, if at all, at either dose of inoculated virus (Fig. 3A and 3C) whereas there were significantly increased numbers of lesions for the Pvrl−/− mice at the higher dose. These results show that nectin-1 expression is associated with efficient infection of the vaginal epithelium but, in its absence, HVEM expression permits infection, albeit less efficiently. When neither nectin-1 nor HVEM is expressed, infection of the vaginal epithelium was extremely limited or not detectable even at high doses of inoculated virus. These results also indicate that the failure of some wild-type, Tnfrsf14−/− or Pvrl−/− mice to show symptoms of disease in Fig. 1 was probably not due to failure of the inoculated virus to initiate infection of the vaginal epithelium. On the other hand, failure of double-mutant Tnfrsf14−/− Pvrl−/− mice to show any clinical signs of disease might be accounted for by failure of the virus to establish infection via the intravaginal route of inoculation.
Figure 3. Development of lesions on the vaginal epithelium at 24 hrs after inoculation of C57BL/6 (+/+), Tnfrsf14−/−, Pvrl−/− or double mutant Tnfrsf14−/− Pvrl−/− mice with HSV-2/Gal.
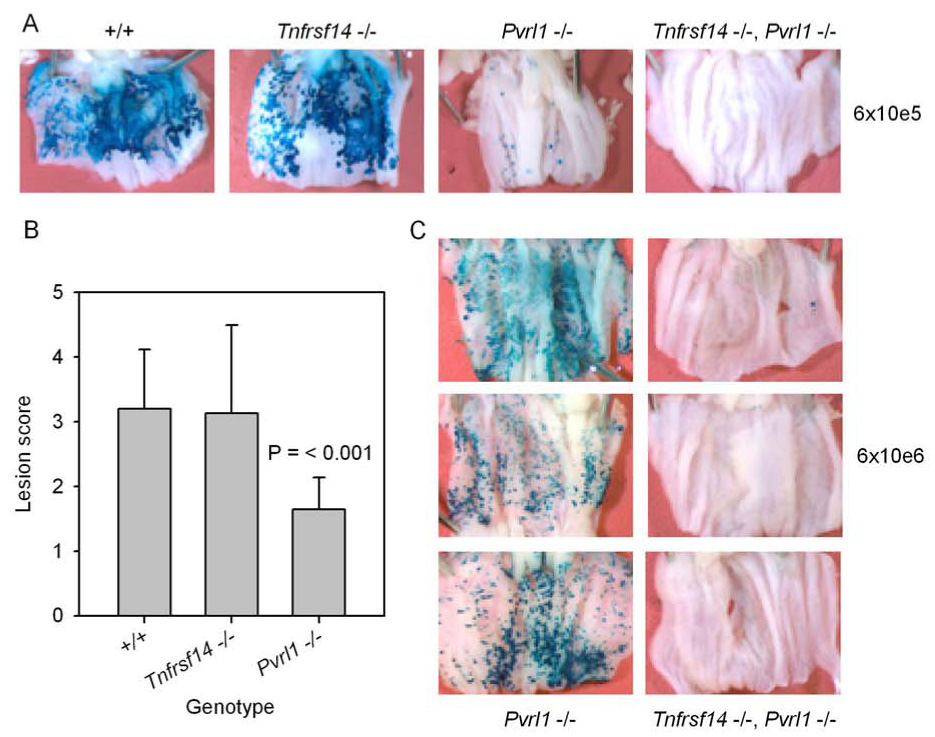
The mice were injected with Depo-Provera as in Fig. 2 and then inoculated with HSV-2/Gal at 6 × 105 PFU per mouse (A and B) or 6 × 106 PFU per mouse (C). At 24 hrs after virus inoculation, the mice were sacrificed. The vaginas were removed and split open longitudinally and then fixed and stained with X-gal to detect β-galactosidase expressed from an insert in the viral genome. The extent of staining and therefore of virus infection was scored on a scale from 0 (no blue lesions noted) to 5 (greater than 80% of vaginal epithelium infected) as described in the text. A - Representative pictures of vaginas from mice of the indicated genotypes inoculated with 6 × 105 PFU per mouse. B - Scores assigned for vaginas from mice of the genotypes noted, after inoculation with HSV-2/Gal at 6 × 105 PFU/mouse. The values shown are means plus standard deviation (n = 8–11). Only the mean for the Pvrl−/− mice was significantly different from that for the wild-type (+/+) mice (t test). C – Pvrl−/− and double-mutant mice (Tnfrsf14−/− Pvrl−/−) were inoculated with HSV-2/Gal at 6 × 106 PFU per mouse and treated as in A. A total of 8 double-mutant mice have been inoculated with either 6 × 105 or 6 × 106 PFU and none have exhibited staining scores other than 0 or 1.
Fig. 4A shows an example of an X-gal-stained spinal column from a wild-type mouse inoculated with HSV-2/Gal. A portion of the image was enlarged to show the staining of DRG. At several timepoints following HSV-2/Gal inoculation, infection of the nervous system (DRG, attached nerves and spinal cord) was scored. There was a significant difference between wild-type littermate controls and Pvrl−/− mice in the lesion scores for days 3, 5, 7 and 9 after virus inoculation (Fig. 4B; P < or = 0.003). Moreover, spread of virus infection to the PNS and CNS was detectable by X-gal staining for only a subset of mice in each Pvrl−/− experimental group (numbers in parentheses in Fig. 4B) whereas this spread of infection was readily detectable in all wild-type mice. Tnfrsf14−/− mice (n = 3 for each time point) had scores indistinguishable from those of the wild-type mice on days 3, 5 and 9 following inoculation (data not shown).
Figure 4. Development of lesions in DRG and the spinal cord after inoculation of Pvrl−/− mice and littermate wild-type (+/+) mice with HSV-2/Gal.
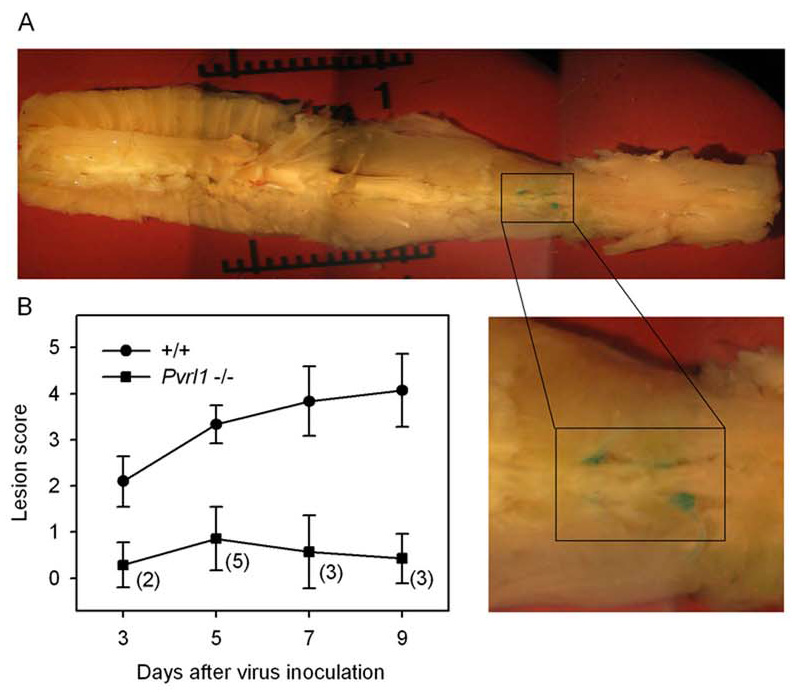
Depo-Provera-treated mice were inoculated with HSV-2/Gal at 6 × 105 PFU per mouse and sacrificed at days 3, 5, 7 and 9 after inoculation. The spinal columns were then dissected out (A), the vertebra cut and the entire tissue stained with X-gal. After staining, the spinal cords with attached ganglia and nerves were teased away from other tissue so that blue foci of infection in these tissues could be scored on a scale from 0 (no blue stain observed) to 5 (most ganglia in sacral and lumbar region positive, attached nerves positive and spinal cord positive in several regions) as described in the text. A - Representative picture of a stained spinal column from a +/+ mouse sacrificed at day 9. The magnified inset shows two heavily stained and two lightly stained ganglia in the lumbar region. B - Scores assigned for infected mice of the genotypes indicated at the days indicated. The values shown are means plus standard deviation (n = 5–7 for the wild-type (+/+) littermate control groups and n = 7 for all Pvrl−/− groups). Shown in parentheses beside each point in the lower curve is the number of Pvrl−/− mice in each group for which lesions could be detected in nervous tissue (score greater than 0). On each day the values for the Pvrl−/− mice were significantly different from those for the +/+ mice (P < or = 0.003, Mann Whitney rank-sum test). Scale bar divisions in A are 0.1 cm.
An example of the typical external lesions caused by HSV-2 is shown in Fig. 5A. Samples of perivaginal skin were dissected from wild-type mice infected with HSV-2/Gal to assess the spread of virus infection and distribution of lesions at the gross (Fig. 5B) and histological (Fig. 5C–F) levels. We failed to observe indications that infection spread directly and contiguously from the vaginal epithelium itself to external perivaginal regions but, instead, noted isolated X-gal stained lesions, some quite distant from the vaginal opening (Fig. 5B). Serial sections of external skin also provided evidence for isolated microscopic viral lesions, often extending into hair follicles (Fig. 5C–F), as previously observed (Parr and Parr, 2003).
Figure 5. Development of external lesions on skin at sites non-contiguous with the vaginal opening and involvement of hair follicles.
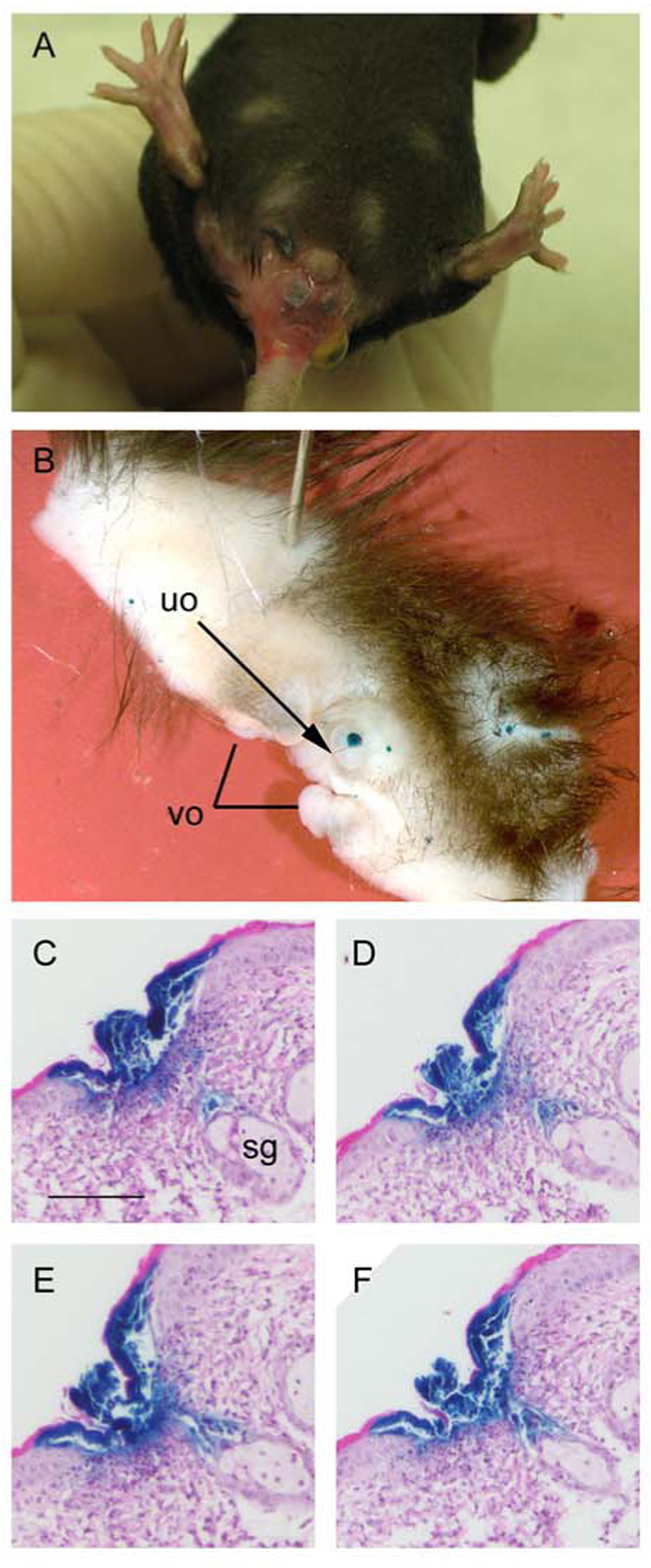
A - A mixed-strain wild-type mouse was treated with Depo-Provera, inoculated with 6 × 105 PFU of HSV-2 and photographed on day 15 to show external hair loss, inflammation and lesions around the vagina and rectum. B–F – A C57BL/6 mouse was treated with Depo-Provera, inoculated with 6 × 105 PFU of HSV-2/Gal and sacrificed 7 days later. Skin was dissected from the area ventral to the vaginal orifice (vo), fixed and stained with X-gal. Areas of skin around the vo suffered hair loss and had been inflamed. B - Isolated lesions (blue foci) at sites distant from the vo. The urethral orifice (uo) is indicated by the arrow. C – F Serial sections, after H&E staining, of paraffin-embedded tissue from the same mouse. The dark blue insoluble product generated by action of β-galactosidase on X-gal identifies infected cells on the epithelial surface of the skin and extending along the hair follicle toward a sebaceous gland (sg). Scale bar in C represents 20 µm for panels C–F.
Tracking of virus infection and spread by titration of infectious HSV-2 in various organs
Groups of wild-type, Tnfrsf14−/− and Pvrl−/− mice were inoculated with HSV-2 at 6 × 105 PFU per mouse and sacrificed at intervals following inoculation for removal of the tissues indicated in Fig. 6. Each organ was weighed, homogenized and serial dilutions prepared for quantification of PFU. On day 1, virus was detected only in the vaginal samples, with significantly lower mean titers for the Pvrl−/− mice than for wild-type controls or Tnfrsf14−/− mice. Viral loads declined in the vaginal samples thereafter. On day 3, maximal mean titers of virus were detected in the DRG of wild-type and Tnfrsf14−/− mice whereas maximal titers were not detected until day 5 for the Pvrl−/− mice. By day 5, maximal titers of virus were detected in the spinal cords of mice of all three genotypes but at significantly lower levels for the Pvrl−/− mice. Finally, virus could be detected in the hindbrains of most wild-type and Tnfrsf14−/− mice by day 7 and 9 but not in any of the Pvrl−/− mice. For the DRG and spinal cords and at timepoints where infectious virus could be detected in these organs for all wild-type and Tnfrsf14−/− mice, there were one or more Pvrl−/− mouse in which virus could not be detected. This study was also done with Pvrl1+/− mice. Mean titers of virus detected in various organs of the Pvrl+/− mice at each timepoint were usually not statistically different from those obtained for the wild-type mice but were nevertheless usually less than those obtained for the wild-type mice, suggesting gene dosage effects (data not shown), as did Fig. 1. Thus, absence of HVEM expression was without effect on the replication of HSV-2 in the organs tested whereas absence of nectin-1 resulted in reduced titers in all organs.
Figure 6. Viral loads in various tissues of wild-type (+/+), Tnfrsf14−/− and Pvrl−/− mice at the days indicated after inoculation with HSV-2.
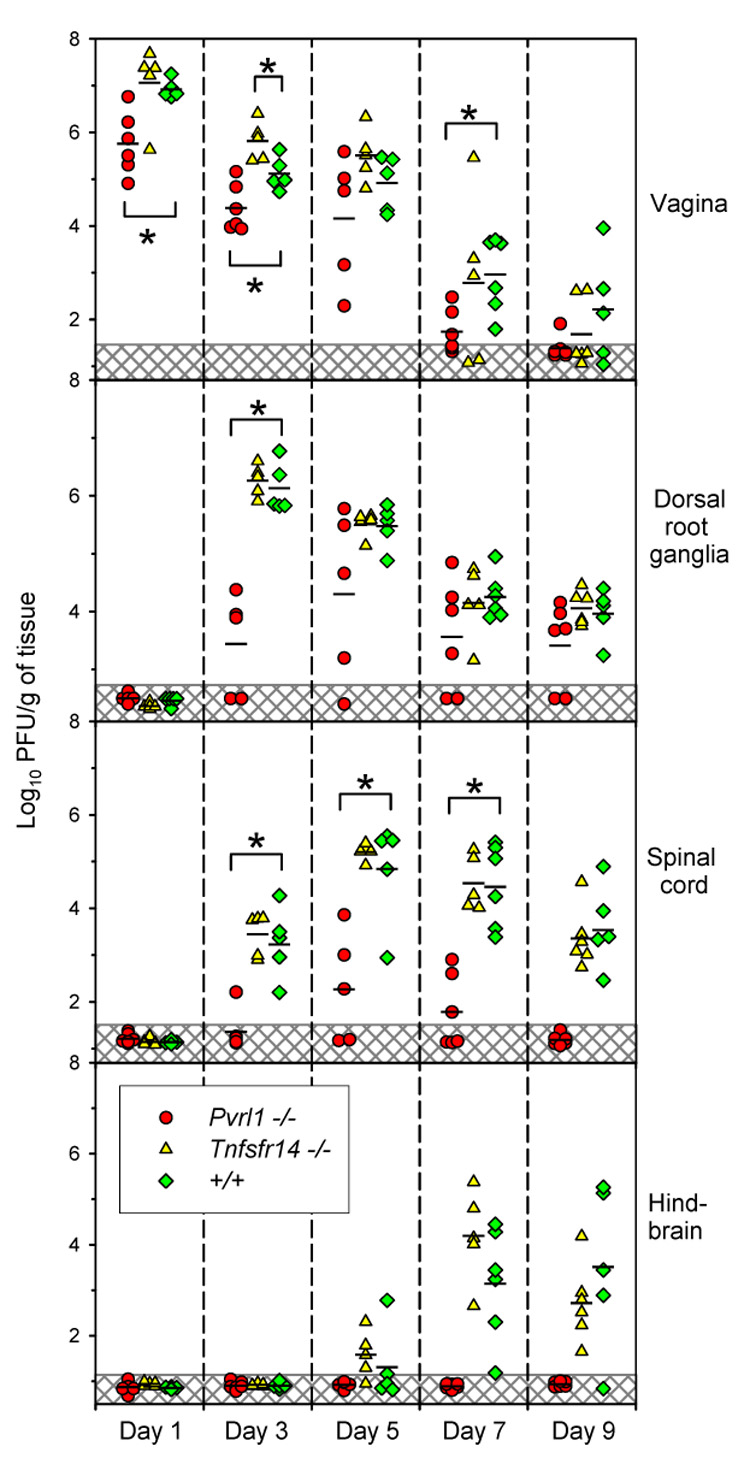
The mice were treated with Depo-Provera, inoculated with HSV-2 at 6 × 105 PFU per mouse, sacrificed on the day indicated after inoculation and various tissues removed for preparation of extracts and virus titrations by plaque assay. Each symbol represents PFU per g of tissue from an individual mouse. Geometric means for each experimental group are indicated by horizontal black lines (n = 5–6 for all three genotypes). Symbols in the hatched regions of each panel represent the lower limit of virus detection; values for these samples were recorded as the actual low number of PFU detected or assigned as 0.5 PFU per g of tissue when no PFU were detected. Brackets marked by an asterisk indicate that means of the viral loads detected in the +/+ mice were statistically different from those in the mutant mice (P <0.05, t test).
To determine whether replication of HSV-2 could be detected in double-mutant mice, groups of these mice and control nectin-1-expressing Tnfrsf14−/− Pvrl1+/− mice were inoculated with the higher dose of HSV-2 (6 × 106 PFU). Five days after virus inoculation, the mice were sacrificed for quantification of progeny virus in homogenates of vaginas, DRG and spinal cords. The 5-day time point was selected because, by this time, input virus that failed to initiate infection would have been inactivated and, at this time, mice of all other genotypes had significant levels of virus in the organs tested (Fig. 6). Table 1 shows that, while significant levels of virus were present in all three organs of the nectin-1-expressing mice, no virus could be detected in the double-mutant mice (except for a few PFU near the lower limit of detection in one vaginal sample). Thus, double-mutant mice may support very low levels of HSV-2 replication in the vagina (as was noted also with HSV-2/Gal) but infection did not spread to the nervous system and did not lead to disease (Fig. 2).
Table 1.
No infectious virus detected in dorsal root ganglia or cords of double-mutant mice at 5 days after HSV-2 inoculation
| Log10 PFU/g of tissue (lower limit of detection)* |
||||
|---|---|---|---|---|
| Mouse genotype | Mouse number | Vagina | Dorsal root ganglia | Spinal cord |
| Tnfrsf14−/− Pvr11+/− | 2438 | 3.57 | 4.86 | 3.40 |
| 2439 | 3.22 | 4.62 | 3.67 | |
| 2441 | 3.60 | 4.59 | 2.62 | |
| Tnfrsf14−/− Pvrl1−/− | 2440 | 1.89 (1.42) | none (2.49) | none (1.17) |
| 2546 | none (1.40) | none (2.40) | none (1.17) | |
| 2547 | none (1.23) | none (2.40) | none (1.11) | |
Mice of the genotypes indicated were treated with DepoProvera and 6 days later were inoculated with 6 × 106 PFU of HSV-2. Five days after virus inoculation the mice were sacrificed and the organs indicated taken to prepare homogenates for titrations of infectious virus. Virus was undetectable in nervous system tissues of the double-mutant mice and in vaginal tissues of 2/3 of these mice. For all three organs the mean virus titers detected in mice expressing nectin-1 were statistically different from the actual titer or limits of detection in the double-mutant mice (P < 0.005, t test).
DISCUSSION
A major conclusion emerging from this study is that HSV-2 infection of the vaginal epithelium of mice requires expression of either nectin-1 or HVEM, based on findings that double-mutant Tnfrsf14−/− Pvrl−/− mice were largely resistant to infection via the vaginal route of inoculation whereas both Tnfrsf14−/− and Pvrl−/− mice were susceptible. Absence of nectin-1 expression reduced the efficiency of infection of the vaginal epithelium whereas absence of HVEM expression appeared to be without effect. Possibly, nectin-1 is normally more accessible or efficient as an entry receptor than HVEM, at least in the stratified mouse vaginal epithelium. It was previously reported that nectin-1 mediated infection of the vaginal epithelium in mice, based on finding that a soluble form of nectin-1 could reduce or abolish infection (Linehan et al., 2004). However, this soluble nectin-1 would also be expected to abolish infection mediated by HVEM. Although nectin-1 and HVEM bind to different interfaces on gD (Spear et al., 2006), binding of either soluble receptor to HSV can inhibit infection of cells bearing either receptor (Geraghty et al., 1998).
A second major conclusion is that neither HVEM nor nectin-1 is essential for the spread of HSV-2 infection from the vaginal epithelium to the nervous system in mice, based on the detection of infectious HSV-2 and/or β-galactosidase expressed by HSV-2/Gal in DRG and spinal cords of wild-type, Tnfrsf14−/− and Pvrl−/− mice. Virus produced by non-neuronal cells in the vaginal epithelium could directly enter the endings of neurons that extend to the epithelium, leading to viral replication in DRG, whereas replication of virus in the spinal cord would probably require neuron-to-neuron transmission.
Since replication of HSV-2 in the vaginal epithelium of double-mutant mice was extremely limited, and therefore HSV-2 access to neurons would be limited, it was not possible to assess whether HSV-2 could infect neurons via the vaginal route in these mice. Therefore, a possible role for another as-yet-unrecognized entry receptor that is expressed in the nervous system, but not on the vaginal epithelium, cannot be ruled out. Use of mutant mice in which receptor expression is knocked out only in neurons could address this issue.
Clearly, absence of nectin-1, but not HVEM, delayed and reduced the efficiency of HSV-2 spread to DRG and the spinal cord and also delayed and reduced the incidence of mortality. These comparisons were made at an input dose of virus (6 × 105 PFU) sufficient to infect virtually 100% of wild-type, Tnfrsf14−/− and Pvrl−/− mice, but a dose that resulted in reduced involvement of the vaginal epithelium in Pvrl−/− mice. A 10-fold higher dose of HSV-2/Gal caused greater involvement of the vaginal epithelium in Pvrl−/− mice (Fig. 3) but inoculation of HSV-2 at this higher dose did not enhance disease (Fig. 2) or increase viral loads in the nervous system (Fig. 6 and Table 1).
A third conclusion is that the formation of external lesions, as depicted in Fig. 5A, depended on expression of nectin-1, as these typical lesions were not observed in Pvrl−/− mice. In wild-type or Tnfrsf14−/− mice, external hair loss, inflammation and lesions were invariably noted prior to any signs of lethal disease although not all mice exhibiting external lesions experienced lethal disease. In the Pvrl−/− mice, on the other hand, the typical external signs of disease were never observed, at either dose of virus, even in mice who succumbed to disease and despite levels of virus in various tissues of individual mice that were comparable to those observed in wild-type mice (Fig. 6). Since the secondary external lesions in nectin-1-expressing mice are associated with the spread of virus to perineal skin and surrounding areas, it seems likely that this spread is somehow blocked in nectin-1-deficient mice.
It has been established in a flank (zosteriform) model of HSV disease in mice that secondary lesions occur, beginning about 4 days after inoculation of virus at the primary site, and that these secondary lesions result from viral spread via neural pathways and are prevented by neurotomy (Simmons and Nash, 1984). In the vaginal model of disease, the secondary lesions occurring on the external skin after primary lesion development in the vaginal epithelium could also depend in part on neural routes of spread. The isolated microscopic and gross lesions containing HSV-2/Gal at sites non-contiguous to the perivaginal opening (Fig. 5) and occurring 5–9 days after virus inoculation are consistent with a neural route of spread but do not rule out horizontal cell-cell spread in epithelial cell layers contiguous from the vaginal mucosa to external skin (Parr and Parr, 2003). Expression of nectin-1 has been shown to promote cell-to-cell spread of HSV in non-neuronal cultured cells (Sakisaka et al., 2001). Consistent with this finding, our results show that (i) lesions on the vaginal epithelium of nectin-1-deficient mice are smaller than normal at 24 h and (ii) absence of nectin-1 expression prevents external lesions that are likely to depend on long-range cell-to-cell spread of infection whether this spread is via neural routes or between contiguous mucosal and skin epithelia or both.
Whereas absence of nectin-1 expression prevented the typical external lesions caused by either HSV-2/Gal or HSV-2, absence of HVEM reduced the incidence of these lesions, but only for HSV-2/Gal, at least at the virus dose used in Fig. 1. Adoptive transfer of immune cells has been shown to block zosteriform spread in the mouse flank model (Simmons and Nash, 1984). Possibly, enhanced immune responses to an attenuated virus expressing a bacterial antigen, i.e. HSV-2/Gal, could explain the reduced incidence of external lesions in the Tnfrsf14−/− mice, compared with wild-type control mice, inasmuch as absence of HVEM expression has been associated with enhanced immune responses (Wang et al., 2005).
To what extent are these results relevant to human genital disease? The three known human entry receptors for HSV-2 are nectin-1, nectin-2 and HVEM. Human mutations affecting expression of functional HSV entry receptors have been described only for nectin-1. Chain-terminating mutations in the Pvrl1 gene have been linked to an autosomal recessive cleft lip/cleft palate-ectodermal dysplasia syndrome in a few families from the Margarita Islands, Brazil and Israel (Suzuki et al., 2000). Unfortunately, access to persons with disruptions in the nectin-1 gene has been limited and it has not been determined whether these people are resistant to HSV infection and disease. The results presented here suggest that they would not be totally resistant, especially since nectin-2 as well as HVEM could be functional in situ as entry receptors. Nectin-1 is expressed in the human vaginal epithelium throughout the menstrual cycle (Linehan et al., 2004) and is also expressed in human neurons (Simpson et al., 2005; Takai et al., 2003). The patterns of expression of HVEM and nectin-2 have not been well-defined in the tissues of relevance to this study. It seems safe to predict that, as in the mouse, multiple entry receptors could permit infection via the vaginal epithelium and spread to the nervous system. If, as in the mouse, nectin-1 is required for secondary mucocutaneous lesions, it may also be required for recurrent lesions, one of the most troublesome aspects of HSV disease. Possibly, new approaches to blocking the interaction of HSV with nectin-1 could be effective for preventing recurrent disease.
EXPERIMENTAL PROCEDURES
Cells and Virus strain
Vero cells cultured in Dulbecco’s modification of Eagle’s (DME) medium plus 10% fetal bovine serum (FBS) were used for the propagation of virus. Plaque titrations were performed on Vero cells by standard methods. HSV-2 strain 333 was isolated from a genital lesion and underwent limited passage in human cells (Westmoreland and Rapp, 1976). The virus was plaque-purified and passaged no more than 3 times in Vero cells.
Construction of HSV-2/Gal
HSV-2/Gal has a lacZ expression cassette inserted between the convergently transcribed genes, UL3 and UL4. This virus was generated by co-transfecting Vero cells with genomic DNA from wild-type HSV-2(333) and plasmid pUL3UL4-CMVlacZ, followed by isolation and plaque-purification of a recombinant virus that expresses β-galactosidase. The insert in the plasmid used for recombination contains the following elements: nucleotides 11002-11911 from the HSV-2(333) genome, including the entire UL3 ORF and all but 20 nucleotides of the downstream UL3-UL4 intergenic region; the immediate-early promoter from human cytomegalovirus (CMV); the lacZ gene; nucleotides 11663-11823 from the HSV-1 (KOS) genome, including the entire UL3-UL4 intergenic region from HSV-1; nucleotides 11932-12531 from the HSV-2(333) genome, including all but the first few codons of the UL4 ORF. The intergenic regions, one from HSV-2 and the other from HSV-1 (different in sequence from the HSV-2 version), were placed upstream and downstream of the inserted CMV-lacZ cassette to ensure that polyadenylation signals would function properly for the UL3, lacZ and UL4 transcripts and to avoid homologous recombination between these repeats. The nucleotide sequences given are from the published sequences for HSV-1(17) (McGeoch et al., 1988) and HSV-2(HG52) (Dolan et al., 1998). HSV-2/Gal and HSV-2 were indistinguishable in levels of UL3 and UL4 expression, as determined by western blot analysis using antibodies previously described (Yamada et al., 1998; Yamada et al., 1999). Also, HSV-2/Gal was indistinguishable from parental HSV-2 in its ability to replicate in Vero cells, with respect to the kinetics of progeny production and yields of progeny virus during a single replicative cycle. In wild-type mice, the spread of HSV-2/Gal to various target organs was similar to that of HSV-2 but HSV-2/Gal was less virulent than HSV-2 (Fig. 1). The survival plots for days without lesions and days remaining alive differed significantly (Gehan-Breslow test: P = 0.004 and <0.001, respectively, for the C57BL/6 mice used as controls for the back-crossed Tnfrsf14−/− mice and P = 0.018 and 0.003, respectively, for the wild-type littermate mice used as controls for the Pvrl−/− mice).
Mouse lines
Animal care and use were in accordance with institutional and NIH guidelines and all studies were approved by the Animal Care and Use Committee of Northwestern University. Mice were maintained in specific-pathogen-free conditions until infection and then were transferred to a containment facility.
Generation of the Tnfrsf14−/− mice and back-cross to the C57BL/6 background have been described (Wang et al., 2005). The mice used here were obtained from homozygous mutant breeding pairs and their genotypes checked by PCR analysis of genomic DNA using forward primer HVEM1 (5’ACTCACAGACACCTACTATGG) and reverse primers HVEM2 (5’GGAGATGAGTGCTGGAGGAGA) and HVEM KO (5’CTGAAGAGGAGTTTACGTCCAG). Female C57BL/6 mice, purchased from Jackson Labs, served as wild-type controls for the Tnfrsf14−/− mice.
The generation and properties of Pvrl−/− mice were also previously described (Inagaki et al., 2005). Frozen heterozygous embryos (genetic background 50% 129/SV, 25% C57BL/6 and 25% DBA) at the two-cell stage were implanted into pseudopregnant C57BL/6 mothers. The mice obtained were genotyped by PCR analysis of genomic DNA as previously described (Inagaki et al., 2005). The colony was maintained by heterozygous mating. Wild-type and Pvrl+/− mice were used as littermate controls for the Pvrl−/− mice. Tnfrsf14−/− and Pvrl−/− mice and their progeny were crossed to obtain double-mutant mice.
Infection of mice and post-infection procedures
Female mice ranging in age from 11–18 weeks were injected with 0.1 ml of Depo-Provera diluted to 25 mg/ml with phosphate-buffered saline (PBS) (Parr et al., 1994). Histology performed on the vaginas of mice sacrificed 6 days after injection showed that, in both wild type and mutant mice, Depo-Provera altered the vaginal epithelium, causing remodeling to non-cornified thinned epithelia as described (Parr et al., 1994). At 6 days after DepoProvera injection, experimental mice were anesthetized with tribromoethanol (Papaioannou and Fox, 1993) and then inoculated intravaginally, using a positive-displacement micropipettor, with 20 µl of HSV-2 or HSV-2/Gal diluted in PBS containing 1% inactivated calf serum and 0.1% glucose. The virus solution remaining after inoculation was re-titered to ensure proper dosage (6 × 105 or 6 × 106 PFU/mouse). The lower dose was chosen because, in pilot experiments, 6 × 105 PFU/mouse was shown to cause 100% incidence of vaginal infection in +/+ mice, as assessed by staining the vaginal epithelium 24 hrs after HSV-2/Gal inoculation, and >90% incidence of external lesions in +/+ mice after inoculation with either HSV-2 or HSV-2/Gal. The 10-fold higher dose was the highest practical dose that could be administered in attempts to enhance or detect disease in some of the mutant mice.
Some mice were maintained after inoculation with HSV-2 or HSV-2/Gal solely to observe and document clinical signs of disease for 21 days or until death or sacrifice. These mice were examined daily for external lesions (hair loss, inflammation, and skin lesions around the vaginal opening, the rectum, tail base and down the legs) and neurological signs of disease (abdominal distention indicative of fecal or urine retention or, rarely, hind-limb paralysis) or other signs of morbidity (hunched posture, lethargy, dehydration, chills). Those exhibiting severe morbidity were sacrificed. The recorded day of death was when the mouse was found dead or had to be sacrificed.
Other mice were sacrificed at specific times after virus inoculation to assess the spread of virus infection to various tissues. For mice inoculated with HSV-2/Gal, the vagina and the entire spinal column, as shown in Fig. 4, were removed after sacrifice. The vagina was split open longitudinally and the vertebrae of the spinal column were cut to expose the spinal cord. These organs were fixed in 2% paraformaldehyde/0.02% Nonidet-P40 in PBS for 4–6 hrs and then stained overnight with X-gal (solution: 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside, 3 mM potassium ferrous cyanide, 3 mM ferric cyanide, 2 mM magnesium chloride, 0.1 mM EGTA, 0.01% sodium deoxycholate and 0.02% Nonidet P40). With the aid of a dissecting microscope, staining of the vaginal luminal surfaces was scored as follows: 0 = no blue staining; 1 = <5 blue foci; 2 = >5 foci; <30% of surface covered in blue lesions; 3 = >30%, <50%; 4 = >50%, <80%; 5 = >80% covered in lesions, cervical area infected. Staining of the ganglia, attached nerves and spinal cords was scored as follows: 0 = no staining; 1 = 2–3 positive ganglia in sacral region only; 2 = 3–5 positive ganglia in sacral region, nerves positive or negative; 3 = >30% of ganglia positive in lumbar/sacral region, nerves positive, spinal cord positive or negative; 4 = >50% of ganglia positive in lumbar/sacral region, nerves positive, spinal cord positive; 5 = >80% of ganglia positive in lumbar/sacral region, thoracic ganglia positive or negative, nerves positive, spinal cord positive in multiple areas.
For mice inoculated with HSV-2, after sacrifice several organs were removed, weighed, snap-frozen and stored at -80°C for virus titration by plaque assay. The organs were homogenized in 2.5 ml of cold PBS-1% calf serum-0.1% glucose, using disposable homogenizers, sonicated briefly, centrifuged at low speed to remove tissue debris and the supernatant serially diluted for PFU quantification on Vero cells. Results are presented as PFU per g of tissue.
Histology
C57BL/6, Tnfrsf14−/− and Pvrl−/− mice (n=3) were injected with Depo-Provera as described in the preceding section and control mice of each genotype (n=3) were left untreated. Six days after the injection, the mice were sacrificed and vaginas removed for fixation, embedding in paraffin, and sectioning. The sections were stained with H&E. Tissues from mice infected with HSV-2/Gal, including external skin stained with X-gal (Fig. 5), were also taken for histology to localize the X-gal stained cells in H&E-stained sections.
Statistical tests
Kaplan-Meier survival analysis was performed using the Gehan-Breslow test. Means of scores recorded for X-gal-stained tissues and geometric means of values for viral infectious units in tissues were compared using the unpaired t test or Mann Whitney rank-sum test.
ACKNOWLEDGMENTS
We thank M. Parr and E. Parr for advice on adapting the mouse vaginal model of HSV disease to this study, S. Manoj for performing the western blot analyses of UL3 and UL4 expression, Y. Nishiyama (Nagoya University, Japan) for the antibodies to these proteins and K. Mink for establishing the HVEM gene-targeted mouse line. This work was supported by grants from the National Institute for Allergy and Infectious Diseases including the Midwest Sexually Transmitted Infections and Topical Microbicides Cooperative Research Center U19 AI031494 (PGS) and AI048073 (CFW). E. Lin was supported by Medical Scientist Program T32 GM008152 and by National Research Service Award F30 NS049726. Histology services were provided by the Pathology Core supported by the Robert H. Lurie Comprehensive Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Carfi A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, Wiley DC. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol Cell. 2001;8:169–179. doi: 10.1016/s1097-2765(01)00298-2. [DOI] [PubMed] [Google Scholar]
- Cocchi F, Menotti L, Mirandola P, Lopez M, Campadelli-Fiume G. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J Virol. 1998;72:9992–10002. doi: 10.1128/jvi.72.12.9992-10002.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft M. The evolving crosstalk between co-stimulatory and co-inhibitory receptors: HVEM-BTLA. Trends in Immunology. 2005;26:292–294. doi: 10.1016/j.it.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Dolan A, Jamieson FE, Cunningham C, Barnett BC, McGeoch DJ. The genome sequence of herpes simplex virus type 2. JVirol. 1998;72:2010–2021. doi: 10.1128/jvi.72.3.2010-2021.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco D, Forghieri C, Campadelli-Fiume G. The pro-fusion domain of herpes simplex virus glycoprotein D (gD) interacts with the gD N terminus and is displaced by soluble forms of viral receptors. Proc Natl Acad Sci USA. 2005;102:9323–9328. doi: 10.1073/pnas.0503907102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science. 1998;280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- Haarr L, Shukla D, Rødahl E, Dal Canto MC, Spear PG. Transcription from the gene encoding the herpesvirus entry receptor nectin-1 (HveC) in nervous tissue of adult mouse. Virology. 2001;287:301–309. doi: 10.1006/viro.2001.1041. [DOI] [PubMed] [Google Scholar]
- Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. Crystal structure of glycoprotein B from herpes simplex virus 1. Science. 2006;313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- Honda T, Sakisaka T, Yamada T, Kumazawa N, Hoshino T, Kajita M, Kayahara T, Ishizaki H, Tanaka-Okamoto M, Mizoguchi A, et al. Involvement of nectins in the formation of puncta adherentia junctions and the mossy fiber trajectory in the mouse hippocampus. Mol Cell Neurosci. 2006;31:315–325. doi: 10.1016/j.mcn.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Hsu H, Solovyev I, Colombero A, Elliott R, Kelley M, Boyle WJ. ATAR, a novel tumor necrosis factor receptor family member, signals through TRAF2 and TRAF5*. J Biol Chem. 1997;272:13471–13474. doi: 10.1074/jbc.272.21.13471. [DOI] [PubMed] [Google Scholar]
- Inagaki M, Irie K, Ishizaki H, Tanaka-Okamoto M, Morimoto K, Inoue E, Ohtsuka T, Miyoshi J, Takai Y. Roles of cell-adhesion molecules nectin 1 and nectin 3 in ciliary body development. Development. 2005;132:1525–1537. doi: 10.1242/dev.01697. [DOI] [PubMed] [Google Scholar]
- Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005;24:4144–4153. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan MM, Richman S, Krummenacher C, Eisenberg RJ, Cohen GH, Iwasaki A. In Vivo Role of Nectin-1 in Entry of Herpes Simplex Virus Type 1 (HSV-1) and HSV-2 through the Vaginal Mucosa. J Virol. 2004;78:2530–2536. doi: 10.1128/JVI.78.5.2530-2536.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez M, Cocchi F, Menotti L, Avitabile E, Dubreuil P, Campadelli-Fiume G. Nectin2a (PRR2a or HveB) and nectin2d are low-efficiency mediators for entry of herpes simplex virus mutants carrying the Leu25Pro substitution in glycoprotein D. J Virol. 2000;74:1267–1274. doi: 10.1128/jvi.74.3.1267-1274.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez WM, Spear PG. Structural features of nectin-2 (HveB) required for herpes simplex virus entry. J Virol. 2001;75:11185–11195. doi: 10.1128/JVI.75.22.11185-11195.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch DJ, Dalrymple MA, Davison AJ, Dolan A, Frame MC, McNab D, Perry LJ, Scott JE, Taylor P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol. 1988;69:1531–1574. doi: 10.1099/0022-1317-69-7-1531. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A, Nakanishi H, Kimura K, Matsubara K, Osaki-Kuroda K, Katata T, Honda T, Kiyohara Y, Heo K, Higashi M, et al. Nectin: an adhesion molecule involved in formation of synapses. J Cell Biol. 2002;156:555–565. doi: 10.1083/jcb.200103113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–436. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- Murphy KM, Nelson CA, Sedy JR. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat Rev Immunol. 2006;6:671–681. doi: 10.1038/nri1917. [DOI] [PubMed] [Google Scholar]
- Ogita H, Takai Y. Nectins and nectin-like molecules: Roles in cell adhesion, polarization, movement, and proliferation. IUBMB Life. 2006;58:334–343. doi: 10.1080/15216540600719622. [DOI] [PubMed] [Google Scholar]
- Papaioannou VE, Fox JG. Efficacy of tribromoethanol anesthesia in mice. Lab Animal Sciences. 1993;43:189–192. [PubMed] [Google Scholar]
- Parr MB, Kepple L, McDermott MR, Drew MD, Bozzola JJ, Parr EL. A mouse model for studies of mucosal immunity to vaginal infection by herpes simplex virus type 2. Lab Invest. 1994;70:369–380. [PubMed] [Google Scholar]
- Parr MB, Parr EL. Intravaginal administration of herpes simplex virus type 2 to mice leads to infection of several neural and extraneural sites. J Neurovirol. 2003;9:594–602. doi: 10.1080/13550280390246499. [DOI] [PubMed] [Google Scholar]
- Rey FA. Molecular gymnastics at the herpesvirus surface. EMBO Reports. 2006;7:1000–1005. doi: 10.1038/sj.embor.7400807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richart SM, Simpson SA, Krummenacher C, Whitbeck JC, Pizer LI, Cohen GH, Eisenberg RJ, Wilcox CL. Entry of herpes simplex virus type 1 into primary sensory neurons in vitro Is mediated by nectin-1/HveC. J Virol. 2003;77:3307–3311. doi: 10.1128/JVI.77.5.3307-3311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakisaka T, Taniguchi T, Nakanishi H, Takahashi K, Miyahara M, Ikeda W, Yokoyama S, Peng YF, Yamanishi K, Takai Y. Requirement of interaction of nectin-1 alpha / HveC with afadin for efficient cell-cell spread of herpes simplex virus type 1. JVirol. 2001;75:4734–4743. doi: 10.1128/JVI.75.10.4734-4743.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99:13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- Shukla D, Spear PG. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J Clin Invest. 2001;108:503–510. doi: 10.1172/JCI13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons A, Nash A. Zosteriform spread of herpes simplex virus as a model of recrudescence and its use to investigate the role of immune cells in prevention of recurrent disease. J Virol. 1984;52:816–821. doi: 10.1128/jvi.52.3.816-821.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson SA, Manchak MD, Hager EJ, Krummenacher C, Whitbeck JC, Levin MJ, Freed CR, Wilcox CL, Cohen GH, Eisenberg RJ, Pizer LI. Nectin-1/HveC mediates herpes simplex virus type-1 entry into primary human sensory neurons and fibroblasts. J Neurovirol. 2005;11:208–218. doi: 10.1080/13550280590924214. [DOI] [PubMed] [Google Scholar]
- Spear PG. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol. 2004;6:401–410. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- Spear PG, Eisenberg RJ, Cohen GH. Three classes of cell surface receptors for alphaherpesvirus entry. Virology. 2000;275:1–8. doi: 10.1006/viro.2000.0529. [DOI] [PubMed] [Google Scholar]
- Spear PG, Manoj S, Yoon M, Jogger CR, Zago A, Myscofski D. Different receptors binding to distinct interfaces on herpes simplex virus gD can trigger events leading to cell fusion and viral entry. Virology. 2006;344:17–24. doi: 10.1016/j.virol.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Hu D, Bustos T, Zlotogora J, Richieri-Costa A, Helms JA, Spritz RA. Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia. Nature Genetics. 2000;25:427–430. doi: 10.1038/78119. [DOI] [PubMed] [Google Scholar]
- Takai Y, Shimizu K, Ohtsuka T. The roles of cadherins and nectins in interneuronal synapse formation. Curr Opin Neurobiol. 2003;13:520–526. doi: 10.1016/j.conb.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Wang Y, Subudhi SK, Anders RA, Lo J, Sun Y, Blink S, Wang J, Liu X, Mink K, Degrandi D, et al. The role of herpesvirus entry mediator as a negative regulator of T cell-mediated responses. J Clin Invest. 2005;115:711–717. doi: 10.1172/JCI200522982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2 and pseudorabies virus. Virology. 1998;246:179–189. doi: 10.1006/viro.1998.9218. [DOI] [PubMed] [Google Scholar]
- Westmoreland D, Rapp F. Host range temperature-sensitive mutants of herpes simplex virus type 2. J Virol. 1976;18:92–102. doi: 10.1128/jvi.18.1.92-102.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Jiang YM, Oshima S, Wada K, Goshima F, Daikoku T, Nishiyama Y. Characterization of the UL4 gene product of herpes simplex virus type 2. Arch Virol. 1998;143:1199–1207. doi: 10.1007/s007050050367. [DOI] [PubMed] [Google Scholar]
- Yamada H, Jiang YM, Zhu HY, Inagaki-Ohara K, Nishiyama Y. Nucleolar localization of the UL3 protein of herpes simplex virus type 2. J Gen Virol. 1999;80:2157–2164. doi: 10.1099/0022-1317-80-8-2157. [DOI] [PubMed] [Google Scholar]
- Yoon M, Zago A, Shukla D, Spear PG. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2 and 3-O-sulfated heparan sulfate but not with nectin-1. J Virol. 2003;77:9221–9231. doi: 10.1128/JVI.77.17.9221-9231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]