

Abstract
Two two-compound mixtures of candidate structures for the Phytophthora α1 mating hormone have been synthesized. The mixtures were designed to have differing configurations at C3, but proved to be identical. This suggests that epimerization occurred at C3, and comparison with the data for the natural samples suggest that this is also a mixture of isomers.
Phytophthora, derived from Greek “the plant destroyer”, is a parasitic genus of organism whose members live in the soil and attack the roots and basal stems of plants. These fungi-like parasites do considerable damage to assorted hosts.1 For example, Phytophthora infestans was responsible for the blight that precipitated the Irish potato famine in the mid-19th century. Other Phytophthora induce rot in plants as diverse as strawberries, tomatoes, tobacco, azaleas, coconut trees, and oak trees, among others. The control of these organisms remains difficult.
Phytophthora are classified as homothallic and heterothallic depending on whether they reproduce asexually or sexually. The sexual reproduction of heterothallic phytophthora is regulated by hormone secretion,2 and in 2005 Ojika and coworkers isolated about 1 mg of hormone α1 from the A1 mating type of Phytophthora nicotianae.3 Based on spectroscopic analysis of this sample and its bis-4-bromobenzoate derivative 2, Ojika proposed that the two-dimensional structure (constitution) of hormone α1 was 1,11,16-trihydroxy-3,7,11,15-tetramethylhexadecan-4-one 1 (Figure 1). With four stereocenters, 1 has sixteen stereoisomers, but no information on the three-dimensional structure (configuration) of 1 is yet available.
Figure 1.
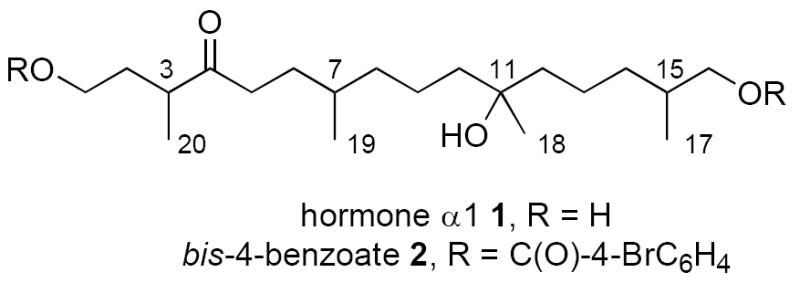
Two-dimensional structure of the Phythophera α1 mating hormone and its bis-4-bromobenzoate
Assigning the configuration of 1 is an especially important goal because Ojika has proposed that 1 is a universal hormone for all heterothallic species of Phytophthora. Yajima and coworkers recently made a mixture that presumably contains all sixteen stereoisomers of 1, and found that this mixture exhibited about 20% of the oospore-inducing activity of the natural sample.4 They also commented that it was impossible to discriminate between the diastereomers of 1 and 2 by NMR analysis.
We have recently used techniques of fluorous mixture synthesis to make stereoisomer libraries of a number of natural products.5,6 As a prelude to employing this technique to make a library of α1 hormones, we decided to execute a traditional synthesis of the hormone. The goals of this work were: 1) to put in place a route that could subsequently be used for fluorous mixture synthesis, 2) to prove the constitution of 1, and 3) to provide preliminary information about configuration of 1.
Herein we communicate the synthesis of four isomers of 1. Like Yajima,4 we confirm the constitution of 1. We also find the stereoisomers difficult to differentiate spectroscopically from each other and the natural product; however, we do find clear indications that there are small but meaningful differences in the spectra of some diastereomers. Based on our analysis, we suggest that the natural sample may not be a single isomer. We comment on the problems that need to be solved to provide a rigorous assignment of configuration of 1.
The latent symmetry of the C6–C16 fragment of 1 makes the retrosynthetic cleavage of the C5–C6 bond especially attractive. As summarized in Figure 2, we envisioned that the ene diyne 3 could be readily converted to 1 by hydrogenation and deprotection. Now cleavage of the C5–C6 bond of 3 provides a simple ketoalcohol 4 (C1–C5) and a symmetrical protected triol 5 (C6–C16). Triol 5 has four stereoisomers whose syntheses are facilitated by symmetry (two have a mirror plane while two have “pseudo-C2 symmetry”7). But using the symmetry in this way exacts a price because the symmetry of fragment 5 must be broken prior to the union of the two fragments.7 Purposefully refusing to pay that price, we decided to generate a two-compound mixture during the symmetry-breaking step of the synthesis to see if it were possible at any stage to differentiate or perhaps even separate the stereoisomers.
Figure 2.
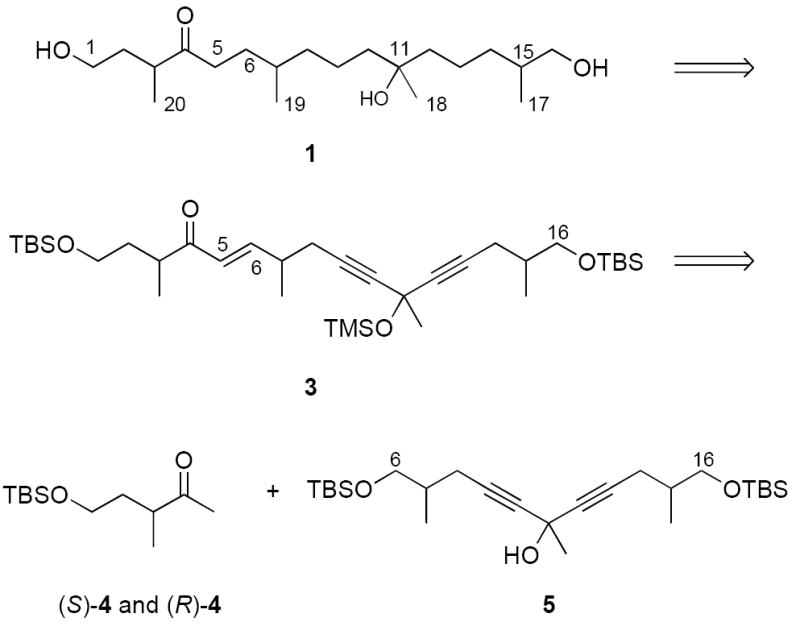
Retrosynthetic analysis of 1 based on the latent symmetry of the C6–C16 fragment
This unconventional decision was driven in part by our expectation that it might be difficult to decide whether an individual synthetic stereoisomer of 1 matched the natural sample or not. If we made two stereoisomers of 1 as a mixture, would we be able to differentiate them from each other? Assuming (without basis) that the natural product is a single isomer, then at least one of the two samples of the mixture (possibly both) would not be the natural product. Would it be possible to differentiate that isomer from the natural product?
Both enantiomers of protected ketoalcohol 4 were synthesized by standard Evans aldol methods,8 and the synthesis of the (S)-4 enantiomer is summarized in Scheme 1. Alkylation of readily available (S)-6 (see Supplementary Data) with NaHMDS and CH3I gave (S,S)-7 in 87% yield. Hydrolysis with lithium hydroperoxide, conversion to the Weinreb amide and addition of CH3MgBr provided (S)-4 in good overall yield.9 Likewise, (R)-4 was made by the same route starting from (R)-6 (not shown). Chiral GC analysis showed that the samples of 4 were enantiopure.
Scheme 1.
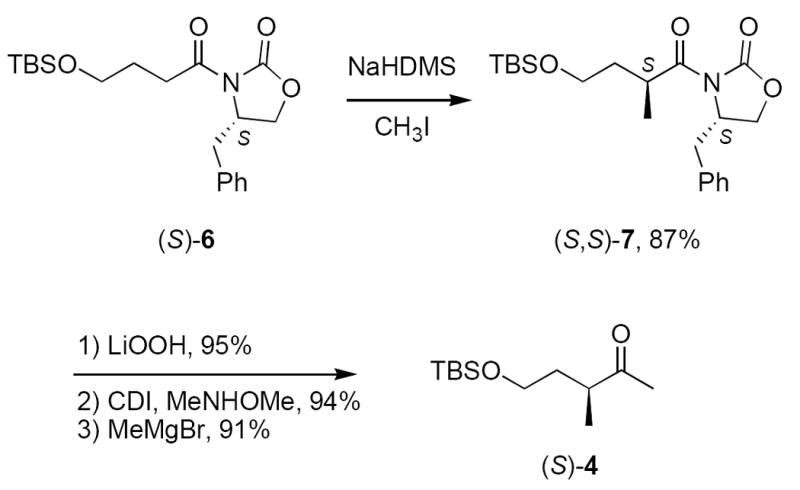
Synthesis of the C1–C5 fragment 4
Starting from readily available (R)-8 (see Supplementary Data), we prepared a two-compound mixture of C6–C16 fragment 10 as summarized in Scheme 2. Deprotonation of 2 equiv of (R)-8 and addition of 1 equiv acetyl chloride provided pseudo-C2 symmetric alcohol (R,R)-5. Removal of both TBS groups with TBAF gave a triol whose symmetry was broken in a non-selective fashion by statistical monosilylation. This provided an inseparable mixture of two compounds 9 whose configurations differed at C11 in 45% yield.10 Oxidation of the free primary alcohol of 9 with the Dess-Martin periodinane11 followed by silylation of the remaining tertiary alcohol with TMSCl provided 10, again as an inseparable mixture of epimers at C11.
Scheme 2.
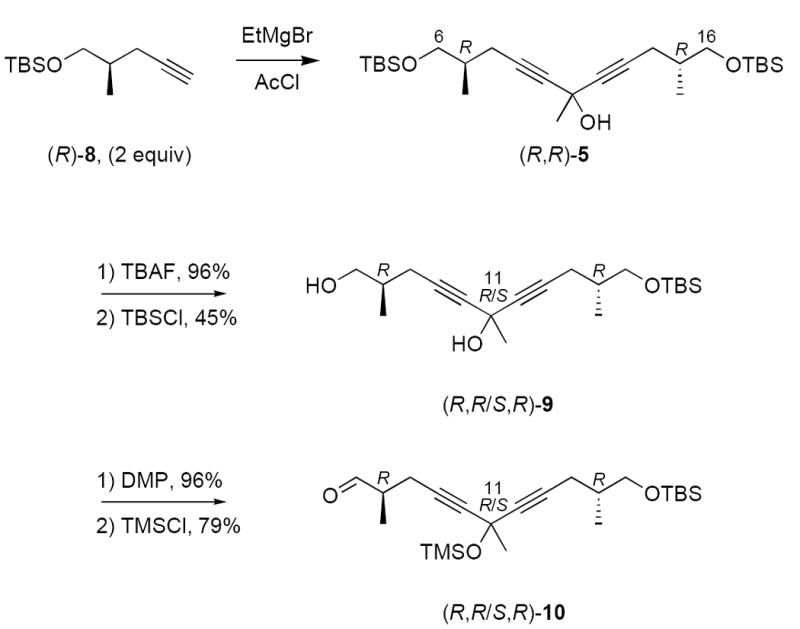
Synthesis of the C6–16 fragment 10
The coupling of the two fragments (Scheme 3) was accomplished by aldol reaction of (S)-4 and (R,R/S,R)-10 (LDA, THF, 80%), followed by mesylation and in situ dehydration (MsCl, Et3N, 85%). The resulting ene diyne (S,R,R/S,R)-3 must again be a mixture of two isomers. The alkene and alkynes were saturated by catalytic hydrogenation to provide a protected ketotriol, which was then deprotected with TBAF in THF at room temperature to give the first two-compound mixture (S,R,R/S,R)-1. The second two-compound mixture (R,R,R/S,R)-1 was prepared in the same way by coupling (R)-4 with (R,R/S,R)-10, then taking the coupled product through the same sequence of steps. Both samples of 1 were converted to the bis-4-bromobenzoate derivatives 2 by standard acylation (77%).
Scheme 3.
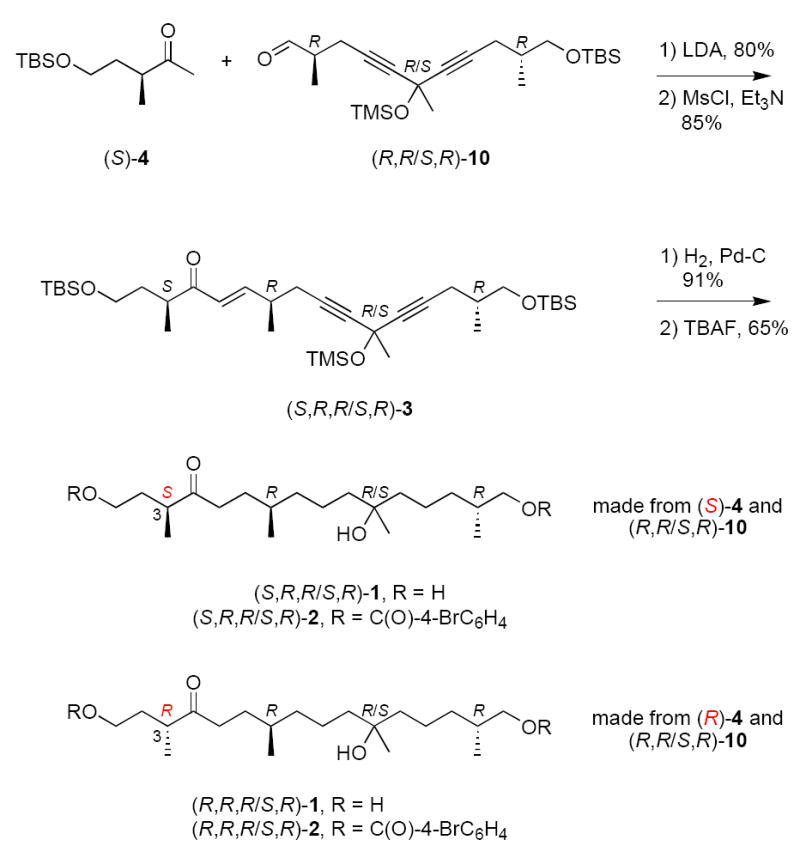
Fragment coupling and completion of the synthesis of 1
After detailed characterization, we then compared the spectroscopic data of the two two-compound mixtures of 1 with each other and with those reported for the natural product. Ojika and coworkers commented that the spectra of the natural product “suggested the presence of unknown trace components”,3 and we saw similar small resonances (<10%) in our spectra. Accordingly, we conclude that these do not come from an impurity per see, but instead arise from the hemiacetal 11 (addition of OH at C1 to ketone at C4) present at equilibrium (Figure 3).12
Figure 3.
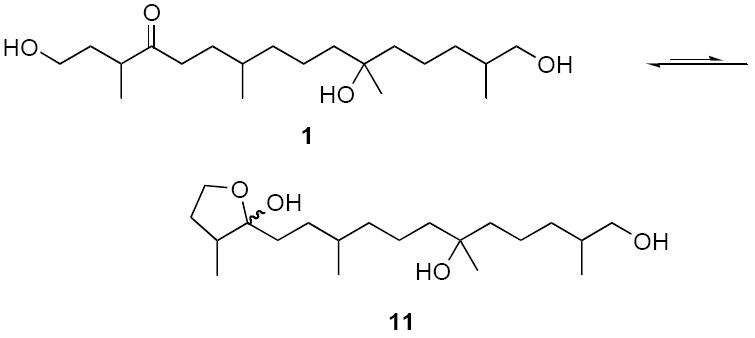
Proposed ketoalcohol – hemiacetal equilibrium
The 500 MHz 1H NMR spectra of the two synthetic samples of 1 were substantially identical, and there was no indication that these samples were isomer mixtures. Further, both of the spectra were substantially identical to that of the natural sample. The 150 MHz 13C NMR spectra of the two samples did show doubling of a few resonances (see Supplementary Data), but the spectra of the bis-4-bromobenzoate samples 2 were more informative.
Though the purified bis-4-bromobenzoate samples 2 exhibited a single spot on standard silica TLC and a single peak on reverse phase HPLC analysis, there were clear suggestions that they were not single components. The 1H NMR spectra of the two synthetic samples and the natural bis-4-bromobenzoate sample were substantially identical. All three spectra showed single resonances for all non-overlapping protons, with an important exception. The methyl group at C19 (bonded to C7) appeared as an apparent triplet, which we interpret as two overlapping doublets (δ 0.83 and 0.82), each arising from a different stereoisomer. The natural sample also showed this feature, and this suggests that it may not be isomerically pure.
The 13C spectra of the two synthetic samples 2 showed single resonances for many of the carbons. However, some of the resonances were clearly doubled, and one (C18) was even quadrupled. Figure 4 summarizes the doubled and quadrupled resonances (see Supplementary Data for a tabulation and overlaid, expanded spectra). In addition, HPLC analysis on a Chiralcel OD column showed four overlapping peaks for both samples of 2.
Figure 4.
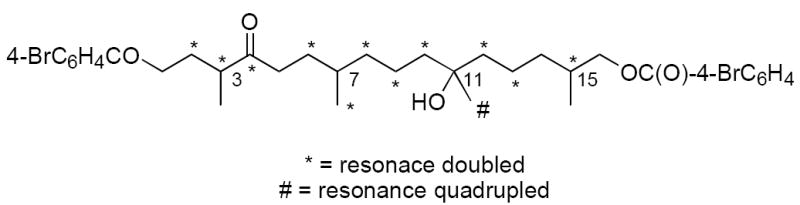
Doubled and quadrupled 13C NMR resonances of the two samples of 2
Taken together, these results suggests that epimerization occurred at some point during the synthesis and that the final products 1 and 2 are four-isomer mixtures rather than the expected two-isomer mixtures. We suggest that epimerization at C3 occurred during the aldol/elimination sequence (or thereafter), and this would mean that the two samples of 1 are identical. So far, all data that we have collected on the samples show them to be identical.
This work helps to advance the stereostructure assignment of the hormone α1. The 1H NMR spectra of the isomers of 1 that we have made are identical, while the 13C spectra are very similar but not identical. The spectra of the bis-4-bromobenzoate derivatives are better resolved and both 1H and 13C NMR spectra show small differences for stereoisomers. Because some of these differences also appear in the bis-4-bromobenzoate derived from the natural sample, it seems likely that that sample is also a mixture of stereoisomers, probably at C3. Finally, the purposeful synthesis of mixtures of isomers at C11 did prove helpful in looking for differences in spectra. But it is probably not a good idea for an expanded synthesis of a stereoisomer library by fluorous mixture synthesis or other methods because it did not prove practical to separate the isomers at any stage and because of the added complication posed by the observed epimerization at C3. To make a stereoisomer library will require a stereoselective synthesis of each isomer.
Supplementary Material
Contains full experimental and compound characterization details, along with copies of the NMR spectra of 1 and 2 and a tabular listing of resonances (78 pages). The Supplementary Data is available online with the paper in ScienceDirect.
Acknowledgments
We thank the National Institute of General Medical Science of the National Institutes of Health for funding of this work. We also thank Prof. M. Ojika and Dr. A. Yajima for sending copies of the NMR spectra of their samples.
Footnotes
Dedicated to the memory of Professor Yoshihiko Ito, 1937-2006
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Erwin DC, Bartnicki-Garcia S, Tsao PH. Phytophthora : its biology, taxonomy, ecology, and pathology. American Phytopathological Society; St Paul, Minn: 1983. [Google Scholar]; (b) Fry WE, Goodwin SB. Bioscience. 1997;47:363–371. [Google Scholar]
- 2.Chern LL, Tang CS, Ko WH. Bot Bull Acad Sin. 1999;40:79–85. [Google Scholar]
- 3.Qi J, Asano T, Jinno M, Matsui K, Atsumi K, Sakagami Y, Ojika M. Science. 2005;309:1828. doi: 10.1126/science.1114756. [DOI] [PubMed] [Google Scholar]
- 4.Yajima A, Kawanishi N, Qi J, Asano T, Sakagami Y, Nukada T, Yabuta G. Tetrahedron Lett. 2007;48:4601–4603. [Google Scholar]
- 5.Short overviews: Zhang W. Arkivoc. 2004:101–109.Curran DP. In: The Handbook of Fluorous Chemistry. Gladysz JA, Curran DP, Horvath IT, editors. Wiley-VCH; Weinheim: 2004. pp. 128–156.
- 6.(a) Luo ZY, Zhang QS, Oderaotoshi Y, Curran DP. Science. 2001;291:1766–1769. doi: 10.1126/science.1057567. [DOI] [PubMed] [Google Scholar]; (b) Dandapani S, Jeske M, Curran DP. Proc Nat Acad Sci. 2004;101:12,008–12,012. doi: 10.1073/pnas.0401677101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Curran DP, Zhang QS, Richard C, Lu HJ, Gudipati V, Wilcox CS. J Am Chem Soc. 2006;128:9561–9573. doi: 10.1021/ja061801q. [DOI] [PubMed] [Google Scholar]; (d) Curran DP, Zhang QS, Lu HJ, Gudipati V. J Am Chem Soc. 2006;128:9943–9956. doi: 10.1021/ja062469l. [DOI] [PubMed] [Google Scholar]; (e) Curran DP, Moura-Letts G, Pohlman M. Angew Chem Int Ed. 2006;45:2423–2426. doi: 10.1002/anie.200600041. [DOI] [PubMed] [Google Scholar]; (f) Yang F, Newsome JJ, Curran DP. J Am Chem Soc. 2006;128:14200–14205. doi: 10.1021/ja064812s. [DOI] [PubMed] [Google Scholar]
- 7.Poss CS, Schreiber SL. Acc Chem Res. 1994;27:9–17. [Google Scholar]
- 8.Evans DA, Takacs JM, McGee LR, Ennis MD, Mathre DJ, Bartroli J. Pure Appl Chem. 1981;53:1109–1127. [Google Scholar]
- 9.(a) Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]; (b) Garg NK, Caspi DD, Stoltz BM. J Am Chem Soc. 2004;126:9552–9553. doi: 10.1021/ja046695b. [DOI] [PubMed] [Google Scholar]
- 10.The spectra of the two mixed isomers in these compounds were substantially identical, and no doubling of resonances was observed. However, in related compounds with saturated alkynes, two resoances for C11 and C18 could sometimes be observed in the 13C NMR spectra.
- 11.(a) Dess DB, Martin JC. J Org Chem. 1983;48:4155–4156. [Google Scholar]; (b) Denmark SE, Fujimori S. Synlett. 2001:1024–1029. [Google Scholar]
- 12.Cavicchioli S, Savoia D, Trombini C, Umani-Ronchi A. J Org Chem. 1984;49:1246–1251. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Contains full experimental and compound characterization details, along with copies of the NMR spectra of 1 and 2 and a tabular listing of resonances (78 pages). The Supplementary Data is available online with the paper in ScienceDirect.