

Abstract
Prostate cancer is the most common cancer in men. The molecular mechanisms leading to its development are poorly understood. Maspin is a tumor-suppressing serpin expressed in normal breast and prostate epithelium. We have found that expression of maspin in normal and carcinoma-derived prostate epithelial cells is differentially regulated at the transcriptional level. We have identified two different kinds of cis elements, Ets and hormonal responsive element (HRE), in the maspin promoter. The Ets element is active in regulating maspin expression in normal prostate epithelial cells but inactive in tumor cells. The HRE site is a negative element that is active in both cell types. This negative DNA sequence can repress a heterologous promoter recognized by the androgen receptor. We conclude that expression of maspin is under the influence of both a positive Ets and a negative HRE element. Loss of maspin expression during tumor progression apparently results from both the absence of transactivation through the Ets element and the presence of transcription repression through the negative HRE element recognized by androgen receptor.
Prostate is an organ that continues to grow throughout life, setting the organ at the risk for events that lead to tumorigenesis. Most prostate tumors arise from the secretory epithelial cells that line the lumenal surface of the prostatic ducts and acini. Androgen plays a pivotal role in tumor development, and endocrine therapy was developed to treat those tumors responding favorably to androgen deprivation. Other oncogenic processes, such as mutation of Ras and activation of Bcl2 have been associated with prostatic carcinogenesis (1, 2). On the other hand, loss of function by tumor suppressor genes also contribute to tumor progression. Loss of heterozygosity studies in prostate have demonstrated that regions frequently deleted in prostate cancers, 8p, 10q, 16q, and 18q, contain candidate tumor suppressor genes (3–6).
Maspin is a tumor-suppressing serpin initially isolated from normal human mammary epithelial cells (7). Functional studies have demonstrated that maspin inhibits tumor invasion and motility of human mammary tumor cells in cell culture (8), as well as tumor growth and metastasis in the nude mice assay (7). The specific expression of maspin in normal mammary epithelial cells, but not in mammary carcinoma cell lines, was shown by Northern blot analysis. This regulation is controlled at the transcriptional level by the combination of elements including Ets and Ap1 elements in breast cells (9).
The prostate gland depends on androgenic hormones for its growth and development (10), analogous to the role of mammary hormones in development and morphology changes in the mammary gland (10). The molecular events leading to the development of prostate cancer may be similar to those in breast cancer. Because maspin functions as a tumor suppressor in the mammary gland (7, 8), we asked whether maspin plays a similar tumor-suppressing role in the prostate, and more importantly, what is the mechanism underlying gene regulation of maspin in prostate cells.
In this paper, we have shown that maspin expression is down-regulated in metastatic prostate cells. By promoter analysis, we have identified the Ets and hormonal responsive element (HRE) sites as cis elements involved in transcriptional activation and repression. Electrophoresis mobility shift assay (EMSA) experiments confirmed the binding of androgen receptor to the HRE site. Thus, our data demonstrate that in the prostate, expression of maspin is regulated by both positive and negative mechanisms at the transcriptional level.
MATERIALS AND METHODS
Cell Lines.
Normal human prostate epithelial cells (HPECs) (CF3, CF91, and MLC) are obtained from John Rhim (National Institutes of Health). Tumor cell lines LNCaP, PC3, and DU145 are from the American Type Culture Collection. Normal cells were cultured in keratinocyte medium supplemented with epidermal growth factor (5 ng/ml). Tumor cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum.
Northern Blot Analysis.
Total cellular RNA was prepared as described (11). Total RNA (20 μg) was fractionated on 1% agarose/1.7 M formaldehyde gels, transferred to Zeta-Probe (Bio-Rad) membranes in 20× standard saline citrate, and baked for 1 hr at 80°C. Blots were probed with a 2.5-kb EcoRI–XhoI fragment from the maspin cDNA plasmid. 36B4 was used as an internal loading and transfer control (12).
Oligonucleotides.
Oligonucleotides were synthesized by Amitof (Boston, MA). For annealing, pairs of sense and antisense oligonucleotides were mixed in equimolar amounts and annealed in 10 mM Tris⋅HCl, pH 8.0/200 mM NaCl/1 mM EDTA by heating to 95°C for 5 min and cooling to room temperature over a 3-hr period. The following oligonucleotides were used: for EMSA experiments: Maspin HRE, sense (OL1, AGTACTCTGATCTCCATTC) and antisense (OL2, GAATGGAGATCAGAGTACT); consensus GRE for competition, sense (OL3, CTAGGCTGTACAGGATGTTCTGCCTAG) and antisense (OL4, GATCCGACATGTCCTACAAGACGGATC); nonspecific oligonucleotide (NS) for competition, sense (OL5, CCTTGTCAGACAGGCAAGTCC)and antisense (OL6, GGAACAGTCTGTCCGTTCACGG); for pKT(297 mHRE) construction, sense (mHRE, AACTGCAGTTTACACAAAAAGAATGATATCCGGAGTAC) and antisense(OL7, GGTGGTATATCCAGTGATTTTTTTCTCC); for pBLAp1/HRE construction, sense (OL8, GATCCAGTACTCTGATCTCCATTCG) and antisense (OL9, GATCCGAATGGAGATCAGAGTACTG).
Constructs.
The pKT series vectors and pEtsCAT were constructed as described (9). For the pKT297 mHRE construct, a PCR fragment [using OL7/mHRE oligonucleotides and pKT(297) as the DNA template] was digested with HindIII and XbaI and subcloned into the XbaI and HindIII of pKTCAT promoterlesss vector.
For the construction of pBLAp1/HRE, pairs of OL8 and OL9 oligonucleotides were annealed as described above. The annealed product was phosphorylated by T4 polynucleotide kinase and ligated to the BamHI site of pBLAp1 (pBLCAT2 containing three copies of Ap1) to generate pBLAp1/HRE (see Fig. 6).
Figure 6.
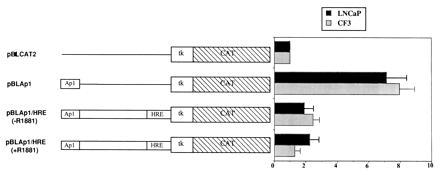
Effect of HRE on the promoter activity of pBLAP1. CAT constructs were transfected to CF3 cells and activity was normalized to pBLCAT2 control. Transfected cells were treated with R1881 (50 nM) or vehicle for 48 hr. Values are obtained from at least three repeated experiments. Error bars are standard errors.
Transfection and Chloramphenicol Acetyltransferase (CAT) Assay.
CAT constructs were made as illustrated in Fig. 4A. Cells were plated at 1 × 106 cells per p100 and grown to about 75% confluence. DNA was transfected by the method of modified DEAE-dextran (Promega). The amounts of DNAs used were 10 μg of reporter plasmid, except for pCMVCAT in which only 2 μg of DNA was used; 1 μg of pCMVβgal was used as an internal control for transfection efficiency. For the androgen treatment, 50 nM methyltrienolone (R1881, from Du Pont/New England Nuclear) or vehicle was added to the cultures after transfection. Forty-eight hours after transfection, cells were harvested in 0.25 M Tris⋅HCl, pH 8.5/15% glycerol. The extracts were made by using three cycles of freeze–thaw. The β-galactosidase activity in the extracts was calculated as described (11). Twenty units of extracts (calculated by β-galactosidase activity) was used for each CAT assay except for transfection with pCMVCAT-positive control in which only 10 units of extracts was used because of high activity. CAT assay was performed as described by Gorman et al. (13). Quantitation of acetylated CoA and nonacetylated chloramphenicol was performed by excising the appropriate regions of the silica gel TLC plate and measuring radioactivity in BioFluor (DuPont).
Figure 4.
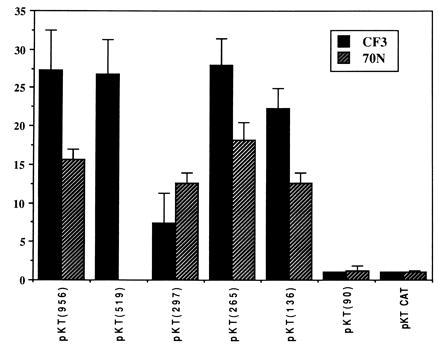
Comparison of CAT activity in both mammary epithelial 70N cells and prostate CF3 cells. Relative activity was represented by normalizing to pKTCAT. Error bars are from at least four experiments.
EMSA Experiments.
Nuclear extracts were made as described by Dignam et al. (14). Binding reactions were carried out at room temperature for 30 min in a mixture containing 4% glycerol, 1 mM MgCl2, 0.2 mM EDTA, 0.5 mM dithiothreitol, 50 mM NaCl, 10 mM Tris⋅HCl, 2 μg of poly(dI-dC), 50 nM R1881, 10 μg of nuclear extracts, and end-labeled oligonucleotide probe. Rat anti-AR monoclonal antibody (MAI-150) was purchased from Affinity Bioreagents, Golden, CO. Antibody against glucocorticoid receptor (GR) was from Santa Cruz Biotechnology. Rat IgG negative control was from Sigma. The complexes were subjected to electrophoresis in 5% polyacrylamide gels in 0.5× Tris/borate/EDTA buffer.
RESULTS
Differential Expression of Maspin in Human Prostate and Carcinomas.
Maspin is expressed in the mammary gland and the prostate, as well as stomach and upper gastrointestinal tract (9). The expression of maspin is limited to the epithelium component, as shown by the absence of maspin mRNA in fibroblasts, leukocytes, and neurons (9) and by immunostaining experiments using mammary gland tissue sections (7). To understand whether the expression pattern of maspin is altered during prostate tumorigenesis, we performed Northern blot analysis with RNAs from several human normal prostate and tumor cell lines. Maspin is highly expressed in CF3, CF91, and MLC normal prostate epithelial cells and down-regulated in LNCaP, PC3, and DU145 prostate tumors (Fig. 1). This expression pattern is similar to the findings in the normal mammary epithelial cells and carcinomas (7, 9), indicating that the down-regulation of maspin expression is a common phenotype of both breast and prostate tumors.
Figure 1.
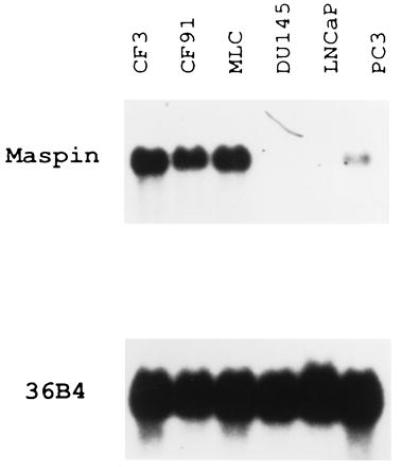
Northern blot analysis of maspin in human prostate epithelial cells. CF3, CF91, and MLC are human normal mammary epithelial cell strains. LNCaP, PC3, and DU145 are prostate tumor cells. Each lane contains 10 μg of total RNAs. The blots were hybridized with a 2.5-kb maspin cDNA probe. 36B4 was used as loading and transfer control.
Functional Analysis of Maspin Promoter in Prostate Cells.
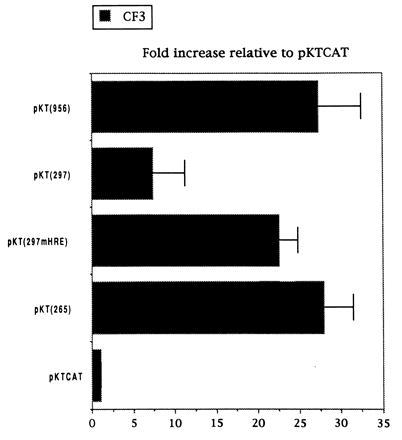
To examine the mechanism of maspin regulation in normal and tumor prostate cells, we made maspin promoter–CAT constructs to assay CAT activity in CF3 normal prostate cells and LNCaP prostate tumor cells (Fig. 2). CAT activity is expressed relative to that of pKTCAT in the same cells. Deletion from −956 bp to −475 bp did not change the activity. The deletion from −475 bp to −461 bp, which removed a distal Ets site, decreased the activity about 50%. This indicates the distal Ets site is involved in up-regulation of maspin in normal prostate CF3 cells. Further deletion up to −297 bp continued to decrease the activity to about 20% of the full-length promoter activity, indicating the presence of other unidentified positive cis element(s) in this region. The deletion from −297 bp to −265 bp, however, which removed the HRE element, completely restored the CAT activity in CF3 cells, indicating that HRE plays a negative role in transcription.
Figure 2.
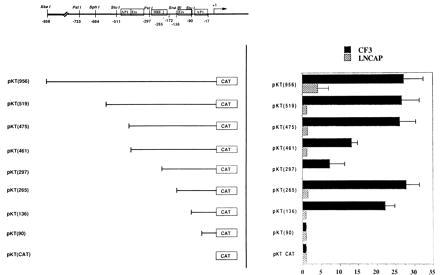
Maspin CAT constructs and CAT assays. (Left) CAT constructs and a schematic representation of maspin promoter with putative transcription factor binding sites are shown. (Right) CAT constructs were transfected into CF3 and LNCaP cells. Relative activity was represented by normalizing to pKTCAT. Error bars are from at least four experiments.
The deletion from −136 bp to −90 bp, which removed a proximal Ets site, completely abolished the CAT activity of CF3 extracts. The level of pKT(90) was comparable to that of the negative control vector, which does not contain a promoter. These data demonstrate that the proximal Ets site is the major positive cis element within 1 kb responsible for up-regulation of maspin in normal mammary epithelial cells.
The constructs were tested in prostate carcinoma LNCaP cell extracts (Fig. 2). The full-length promoter [pKT(956)] had very little activity (4-fold). Deletion from −956 bp to −519 bp decreased the activity to the level of negative control vector, indicating the presence of a weak positive activation site located within the region. Further deletions gave no CAT activity significantly higher than that of negative control vector, showing that the Ets site was not active in LNCaP tumor cells.
To further confirm the involvement of the Ets site in transcriptional activation of maspin, we investigated the ability of Ets to enhance transcription by cloning the Ets site (−120 bp to −90 bp) into the pBLCAT2 vector, which contains no enhancer but a minimal strength tk promoter. This construct was transfected into CF3 and LNCaP cells. As shown in Fig. 3, the presence of the proximal Ets site (single copy) increased the CAT activity of pBLCAT2 in CF3 cells. No enhancing function was observed in LNCaP cells.
Figure 3.
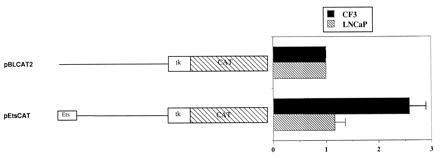
CAT assay of pEtsCAT construct in CF3 and LNCaP cells. The activity was normalized to pBLCAT2 control. Values are obtained from at least three repeated experiments. Error bars are standard errors.
Comparison of Maspin Promoter in Normal Prostate and Mammary Epithelial Cells.
Activities of the two cis elements in maspin promoter were compared in normal prostate and mammary epithelial cells. The proximal Ets site was identified in both cells as the dominant positive cis element, whereas the HRE element plays a negative role in transcription in prostate CF3 cells and is not active in 70N cells (Fig. 4). The distal Ets site is judged to play a positive role in transcription in prostate, because its effect is countered by the negative HRE. This balancing effect is shown by the fact that deletion of both distal Ets and the HRE sites [pKT(265)] resulted in regaining activity to the level of the full-length promoter [pKT(956)].
The HRE Site Is a Unique Negative Hormonal Response Element in the Maspin Promoter.
The HRE element has the consensus sequence 5′-GGTACANNNTGT(T/C)CT-3′ (15). This sequence can be recognized by multiple steroid receptors, such as the GR, androgen receptor (AR), and progesterone receptor. The HRE site (5′-GTACTCTGATCTCC-3′) in the maspin promoter is unique in that its sequence does not share very good homology with the consensus sequence. To further confirm the activity of this HRE in the maspin promoter, we mutated the HRE site in the pKT(−297) construct and transfected it into the CF3 cells (Fig. 5). Mutation at the HRE site alone specifically blocked the effect of transcription repression, confirming the observation from the deletion analysis that the HRE site is a negative hormonal response element.
Figure 5.
Effect of mutation at HRE site on the promoter activity. CAT constructs were transfected to CF3 cells and activity was normalized to pKTCAT control. Values are obtained from at least three repeated experiments. Error bars are standard errors.
To test whether the HRE element plays a general role as transcription repressor, we cloned the HRE element in front of a heterologous promoter pBLAp1 (pBLCAT2 vector containing the Ap1 enhancer; Fig. 6). The construct was transfected to CF3 cells. To test whether tumor cells retained the ability of transcription repression through the HRE, we transfected the pBLAp1/HRE into LNCaP cells. The transfected cells were treated with an androgen ligand (R1881) to test whether the repression is ligand-dependent. As shown in Fig. 6, pBLAp1 was active in both CF3 and LNCaP cells. The presence of HRE element effectively inhibited promoter activity. Little difference in inhibition was observed between R1881-treated or nontreated samples, indicating the repression mediated by HRE was ligand-independent. The extent of repression was similar in both CF3 and LNCaP, demonstrating the repression mechanism was intact in LNCaP tumor cells as in normal prostate CF3 cells. Hence, active repression through the HRE element contributed as importantly to turn off maspin gene in the tumors.
These data demonstrate that the HRE element in the maspin promoter is a general repression element, regardless of its presence in the native maspin promoter or other heterologous promoter.
AR Binds to the HRE Site of Maspin Promoter.
To confirm the presence of steroid receptor binding, oligonucleotides corresponding to the HRE region were end-labeled and used in EMSA experiments with nuclear extracts from CF3 normal epithelial cells, and LNCaP tumor cells. As shown in Fig. 7, a specific DNA–protein complex was identified with both CF3 and LNCaP nuclear extracts. The band can be competed by unlabeled HRE oligonucleotides but not by nonspecific oligonucleotides. Interestingly, it was not competed by a consensus GRE, indicating high affinity for the maspin HRE element.
Figure 7.
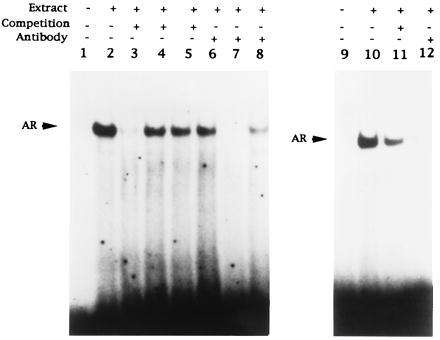
EMSA experiment. End-labeled oligonucleotides were incubated without extracts (lanes 1 and 8) or with extracts from CF3 cells (lanes 2–7) or LNCaP cells (lanes 9–12). Competition was carried out with 50× unlabeled wild-type HRE oligonucleotides (lanes 3 and 11), nonspecific oligonucleotides (lane 4), or GRE oligomers (lane 5). Antibodies against AR (lanes 7 and 12) or GR (lane 6) and rat IgG (lane 8) were added to the reaction mixture and incubated for 30 min at room temperature.
To test the hypothesis that AR binds to the HRE site, monoclonal antibody against AR was added in the reaction mixture. We have found this antibody completely blocked formation of the AR–DNA complex (Fig. 7.), whereas rat IgG did not. Another antibody against GR did not cause any supershift or block formation of the band.
These results indicate that AR, but not other receptors, binds to the HRE site of the maspin promoter.
DISCUSSION
To understand regulation of the maspin gene in prostate tissue, we have tested the promoter activity of maspin in both normal and tumor-derived prostate epithelial cells. We have demonstrated that the proximal Ets element in the promoter activates transcription of maspin in normal prostate epithelial cells. Deletion of this element abolished promoter activity. The enhancing function of the Ets element is not present in carcinoma cells as shown by CAT assays. Another element, HRE, represses transcription activity of maspin. AR binds to the HRE site in the EMSA.
The Ets binding proteins are transcription factors that bind to the consensus sequence GGAAGT. Several Ets proteins including PEA3 have been found to be involved in tumorigenesis and metastasis (16, 17). Although there are reports showing that numerous protease genes are regulated by Ets proteins (18, 19), to our knowledge, maspin provides a first example of a tumor suppressing protease inhibitor that is transcriptionally regulated through an Ets site.
Steroid receptors consist of AR, GR, progesterone receptor, and mineralocorticoid receptor as family members (12). They retain a highly homologous 80-amino acid region, the DNA binding domain, which recognizes a consensus DNA sequence the HRE. Studies of steroid action have focused on the GR and its DNA target sequence GRE. The effect of GR on transcription regulation varies from activation to repression. It has been reported that a specific base mutation at GRE and its promoter context (20, 21) could affect transcriptional regulation by GR. For GR-mediated transcriptional repression, several models have been proposed to account for the mechanisms. For example, the steric hindrance model proposed that the binding of GR to GRE repressed the OC gene and c-fos gene (22, 23). In another case, GR interacted with AP1 factor to block transcription of the collagenase gene (24).
While most studies of steroid function have used GR, the general conclusions probably hold true for other steroid receptors. Androgen is thought to promote tumor growth in androgen-responsive cells (25, 26), possibly by AR-mediated transcription activation of growth stimulating genes. Alternatively, we propose it could act by repressing the transcription of tumor-suppressing genes. Tumors arise from the net effect of both activation of oncogenes and inactivation of tumor suppressor genes. It is therefore critical to identify both positive and negative responsive genes of the AR.
We have identified a negative HRE site by using promoter analysis and gel shifting experiments. Transcription of maspin is repressed by HRE. Interestingly, this repression is androgen-independent in both CF3 normal prostate cells and LNCaP tumor cells. Steroid-independent mechanisms have been proposed previously (27–29). For example, Rosner and coworkers (28) have demonstrated that an alternative pathway stimulates the growth of prostate cells, through a protein called SHBG (sex-hormone binding globulin). Independently, Nazareth and Weigel (29) have shown that, in the absence of androgen, cAMP activates the transcription of androgen-responsive gen-probasin by activating existing ARs. This activation can be blocked by inhibitors of cAMP-dependent protein kinase (29). Repression of the maspin promoter by HRE may also adopt the same pathway independent of androgen.
Maspin may serve as a prognostic marker for prostate cancer. Loss of tumor-suppressing maspin in breast tumors is a progressive process. Maspin expression decreased with increasing malignancy of primary tumors and was absent from lymph node and distant metastases (7–9). Our data show that maspin is expressed in normal prostate cells and down-regulated in prostate tumor cells. Comparison of maspin promoter regulation in the prostate and mammary gland demonstrates that the regulation of maspin, at least at the transcriptional level, is similar in both organs. It is reasonable to speculate that maspin expression may decrease with increasing malignancy of primary prostate tumors. Recently, we and our collaborators have found that maspin is present in normal prostatic cells but not in tumor cells, by using in situ hybridization techniques (unpublished data). Thus, these data pose maspin as a potential marker and a promising target for therapeutic intervention in prostate cancer.
Prostate tumors are extremely heterogenic tumors with subpopulations exhibiting different levels of invasiveness in the same organ (30). From the therapeutic point of view, reexpression of maspin in the prostate tumors offers great hope for reversing the tumor phenotypes. Reexpression may be achieved by targeting both activation and repression modes. For primary tumors, it is likely that the activation is partially impaired, but the repression function is intact. Although it will probably be difficult to restore transcriptional activation of maspin through the Ets site, it may be more feasible to block the repression mediated by the AR binding HRE element. Treating tumors with ligands that block the binding of AR to the HRE or with reagents that compete strongly for binding to AR are possible methods of blocking HRE-mediated repression. Our discovery of HRE-mediated repression offers another opportunity to increase the expression of maspin in prostate tumors, which may in turn reduce the progressiveness of prostate cancer.
Acknowledgments
This work was supported in part by LXR Biotechnology, a grant from the National Institutes of Health (CA 61253 to R.S.), and a National Institutes of Health National Research Service Award fellowship (5-T32-CA09361) to M.Z.
ABBREVIATIONS
- HRE
hormonal responsive element
- EMSA
electrophoretic mobility shift assay
- CAT
chloramphenicol acetyltransferase
- GR
glucocorticoid receptor
- AR
androgen receptor
References
- 1.Carter B S, Epstein J I, Isaacs W B. Cancer Res. 1990;50:6830–6832. [PubMed] [Google Scholar]
- 2.Reed J C, Cuddy M, Slabiak T, Croce C M, Nowell P C. Nature (London) 1988;336:259–261. doi: 10.1038/336259a0. [DOI] [PubMed] [Google Scholar]
- 3.Bova G S, Carter B S, Bussemakers M J G, Emi M, Fujiwara Y. Cancer Res. 1993;53:3869–3873. [PubMed] [Google Scholar]
- 4.Isaacs W B, Isaacs J. In: Principles and Practice of Genitourinary Oncology. Raghaven D, Scher H, Leibel S, Lange P, editors. Philadelphia: Lippincott–Raven; 1997. pp. 403–408. [Google Scholar]
- 5.Bergerheim U S, Kunimi K, Collins V P, Ekmann P. Genes Chomosomes Cancer. 1991;3:215–220. doi: 10.1002/gcc.2870030308. [DOI] [PubMed] [Google Scholar]
- 6.Carter B S, Ewing C M, Ward W S, Treiger B F, Aalders T W, Schalken J A, Epstein J I, Isaacs W B. Proc Natl Acad Sci USA. 1990;87:8751–8755. doi: 10.1073/pnas.87.22.8751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zou Z, Anisowicz A, Hendrix M J C, Thor A, Neveu M, Sheng S, Rafidi K, Seftor E, Sager R. Science. 1994;263:526–529. doi: 10.1126/science.8290962. [DOI] [PubMed] [Google Scholar]
- 8.Sheng S, Carey J, Seftor E, Dias L, Hendrix M J C, Sager R. Proc Natl Acad Sci USA. 1996;93:11669–11674. doi: 10.1073/pnas.93.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang M, Maass N, Magit D, Sager R. Cell Growth Differ. 1997;8:179–186. [PubMed] [Google Scholar]
- 10.Schulze H, Isaacs J T, Coffey D S. Prog Clin Biol Res. 1987;243:1–19. [PubMed] [Google Scholar]
- 11.Swisshelm K, Ryan K, Lee X, Tsou H C, Peacocke M, Sager R. Cell Growth Differ. 1994;5:133–141. [PubMed] [Google Scholar]
- 12.Laborda J. Nucleic Acids Res. 1991;19:3998. doi: 10.1093/nar/19.14.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorman C M, Moffat L F, Howard B H. Mol Cell Biol. 1982;2:1044–1057. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dignam J, Lebovitz R, Roeder R. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beato M. Cell. 1989;56:335–344. doi: 10.1016/0092-8674(89)90237-7. [DOI] [PubMed] [Google Scholar]
- 16.Trimble M, Xin J, Guy C T, Muller W, Hassell J. Oncogene. 1993;8:3037–3042. [PubMed] [Google Scholar]
- 17.Kaya M, Yoshida K, Higashino F, Mitaka T, Ishii S, Fujinaga K. Oncogene. 1996;12:221–227. [PubMed] [Google Scholar]
- 18.Gutman A, Wasylyk B. EMBO J. 1990;9:2241–2246. doi: 10.1002/j.1460-2075.1990.tb07394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nerlov C, Rorth P, Blasi F, Johnsen M. Oncogene. 1991;6:1583–1592. [PubMed] [Google Scholar]
- 20.Starr D B, Matsui W, Thomas J R, Yamamoto K R. Genes Dev. 1996;10:1271–1283. doi: 10.1101/gad.10.10.1271. [DOI] [PubMed] [Google Scholar]
- 21.Drouin J, Sun Y L, Chamberland M, Gauthier Y, Lean A D, Nemer M, Schmidt T J. EMBO J. 1993;12:145–156. doi: 10.1002/j.1460-2075.1993.tb05640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stomstedt P-E, Poellinger L, Gustafsson J-A, Carlstedt-Duke J. Mol Cell Biol. 1991;11:3379–3383. doi: 10.1128/mcb.11.6.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakai D D, Helms S, Carlstedt-Duke J, Gustafsson J-A, Rottman F M, Yamamoto K R. Genes Dev. 1988;2:1144–1154. doi: 10.1101/gad.2.9.1144. [DOI] [PubMed] [Google Scholar]
- 24.Jonat C, Rahmsdorf H J, Park K K, Gebel S, Ponta H, Herrlich P. Cell. 1990;62:1189–1204. doi: 10.1016/0092-8674(90)90395-u. [DOI] [PubMed] [Google Scholar]
- 25.Goldenberg S L, Bruchovsky N. In: Principles and Practice of Genitourinary Oncology. Raghaven D, Scher H, Leibel S, Lange P, editors. Philadelphia: Lippincott–Raven; 1997. pp. 583–590. [Google Scholar]
- 26.Waxman J. Br Med Bull. 1991;47:197. doi: 10.1093/oxfordjournals.bmb.a072455. [DOI] [PubMed] [Google Scholar]
- 27.Denner L A, Weigel N L, Maxwell B L, Schrader W T, O’Malley B W. Science. 1990;250:1740–1743. doi: 10.1126/science.2176746. [DOI] [PubMed] [Google Scholar]
- 28.Nakhla A M, Khan M S, Romas N P, Rosner W. Proc Natl Acad Sci. 1994;91:5402–5405. doi: 10.1073/pnas.91.12.5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nazareth L V, Weigel N L. J Biol Chem. 1996;271:19900–19907. doi: 10.1074/jbc.271.33.19900. [DOI] [PubMed] [Google Scholar]
- 30.Hanks G E, Myers C E, Scardino P T. In: Cancer: Principles and Practice of Oncology. 4th Ed. Devita V, Hellman S, Rosenberg S, editors. Philadelphia: Lippincott; 1993. pp. 1073–1113. [Google Scholar]