
Fig. 7. Proposed mechanism for heterologous desensitization of β2-AR by KC following RSV infection.
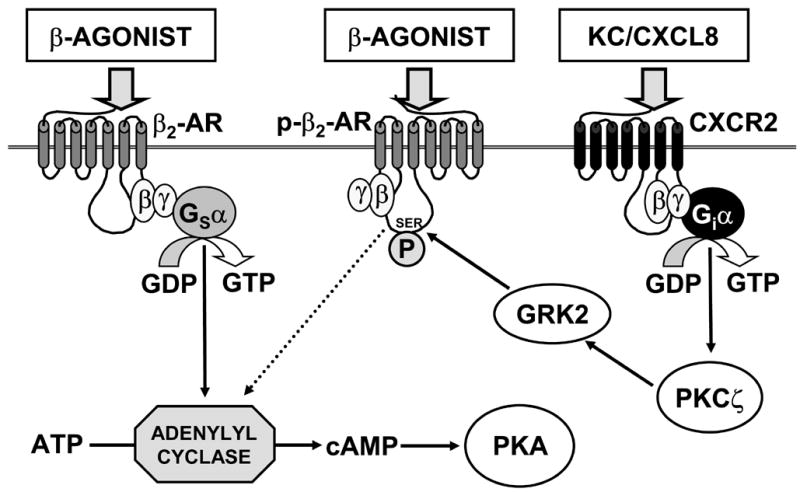
Normally, binding of β-agonist to β2-AR results in hydrolysis of GTP by the stimulatory α subunit of the receptor-associated heterotrimeric G protein (Gsα), which then dissociates and activates adenylyl cyclase. Adenylyl cyclase in turn catalyzes formation of cAMP from ATP. cAMP then activates cAMP-dependent protein kinase (protein kinase A, PKA), which stimulates AFC via effects on ENaC and the Na+/K+ ATPase (and possibly CFTR). However, following RSV infection, KC/CXCL-8 is produced. Upon binding to its receptors (CXCR2, and in humans also CXCR1), KC promotes hydrolysis of GTP by the inhibitory α subunit of the receptor-associated heterotrimeric G protein (Giα), which then activates PKCζ. In turn, PKCζ phosphorylates and activates G protein-coupled receptor kinase 2 (GRK2), which either serine phosphorylates β2-AR (p-β2-AR), uncoupling them from Gsα, or which can directly sequester Gsα (not shown). Upon binding of β-agonist, these phosphorylated receptors are unable to activate adenylyl cyclase and no AFC response is detected.