

Abstract
The lymphocyte activation gene-3 (LAG-3), selectively transcribed in human activated T and NK cells, encodes a ligand for major histocompatibility complex (MHC) class II molecules. Like CD4, LAG-3 ectodomain is composed of four Ig-like domains (D1–D4). Nothing is known about the LAG-3 regions or residues required to form a stable MHC class II binding site. In contrast to CD4, soluble LAG-3 molecules stably interact with MHC class II molecules expressed on the cell surface. In addition, the first two N-terminal domains of soluble LAG-3 (D1 and D2) molecules, alone, are capable of binding MHC class II. From a LAG-3 model structure, we designed mutants and tested their ability to bind MHC class II molecules in an intercellular adhesion assay. We found residues on the membrane-distal, CDR1–2-containing top face of D1 that are essential for either binding or repulsing MHC class II proteins. Most of these residues are clustered at the base of a large extra-loop structure that is a hallmark of the LAG-3 D1 Ig-like domain. In addition, as for CD4, oligomerization of LAG-3 on the cell surface may be required to form a stable MHC binding site because mutation of three residues in the ABED β-strands containing side of D1 results in a dominant negative effect (i.e., binding inhibition of coexpressed wild-type LAG-3).
The lymphocyte activation gene-3 (LAG-3), expressed in human activated T and NK cells, encodes a type I membrane protein with four extracellular immunoglobulin superfamily (IgSF) domains (1). Analysis of this sequence has revealed notable patches of identity with stretches of amino acid sequences found at corresponding positions in CD4, although the overall amino acid sequence homology with human CD4 is barely above background level. There are also some internal sequence homologies in the LAG-3 molecule between domains 1 and 3 (D1 and D3) as well as between D2 and D4, suggesting that LAG-3 has evolved similarly to CD4 by gene duplication from a preexisting two-IgSF structure (1). LAG-3 and CD4 can therefore be regarded as evolutionary “first cousins” within IgSF (2).
Using a quantitative cellular adhesion assay, we have shown that rosette formation between LAG-3-transfected COS-7 cells and major histocompatibility complex (MHC) class II+ B lymphocytes is specifically dependent on LAG-3/MHC class II interaction (2). Direct, specific binding of the soluble four-domain LAG-3 extracellular segment to various human class II molecules (including different alleles and isotypes), as well as to murine and monkey class II molecules has also been observed with a LAG-3D1D4Ig fusion protein (3). This dimeric LAG-3D1D4Ig recombinant globulin binds MHC class II monomorphic residues with a much higher avidity (Kd = 60 nM at 37°C) than CD4Ig (4) and, accordingly, is able to block CD4/MHC class II interaction in an intercellular adhesion assay (4).
LAG-3 is triggered into action only after lymphocyte activation and therefore is not instrumental in the induction phase of the immune response, unlike other MHC ligands such as CD4 and CD8. Indeed, LAG-3 is not expressed in peripheral blood mononuclear cells (PBMCs), except in patients given high-dose IL-2 (5). The interaction of LAG-3 in vitro with its ligands leads to down-regulation of CD4+ T cell clone activation (3, 6). This interaction is mediated through T–T cell contacts, presumably via negative MHC class II signaling into T cells (3). In addition, because LAG-3 expression is strongly associated with production of IFN-γ (7), a potent MHC class II inducer, a need for the coordinated expression of both LAG-3 and MHC class II molecules on T cells to down-regulate the ongoing immune response in vivo cannot be ruled out.
Interactions of CD8 with class I molecules and CD4 with class II molecules are thought to increase the strength of interaction between T cell antigen receptor (TCR)-associated molecules on the T cell and antigen-associated ligands on the presenting cell. The results of mutagenesis experiments with CD8 as well as the dimeric structure of this molecule support the view that the stochiometry of CD8/class I interactions could be 1:2 (8). For CD4, mutagenesis studies have delineated different faces of the D1 and D2 Ig-like domains in the binding interaction (9, 10). Some of the residues involved may in fact interfere with CD4 lattice formation on the cell surface, leading to the stabilization of the CD4/MHC class II complex (11). Overall, these results suggest that interaction of coreceptors with their MHC ligands results in ordered oligomerization, which is required for full TCR activation.
In this paper, we further characterize a third MHC ligand, the LAG-3-encoded protein, and present a three-dimensional model of the LAG-3 two N-terminal Ig-like domains, which stably bind MHC class II molecules when solubilized. In addition, we report the effects of a panel of 20 mutants on their binding with class II MHC molecules.
MATERIALS AND METHODS
mAb and Fusion Proteins.
Anti-LAG-3 mAb, 17B4, 4F4 (LAG-3.1 epitope), 11E3 (LAG-3.2), and 15A9 (LAG-3.3) (2, 6) have previously been described. Dimeric LAG-3IgD1D4 and LAG-3IgD1D2 were prepared as previously described (3). COS-7 cell conditioned media were used as a source for Ig fusion proteins.
Cloning of Murine LAG-3.
mLAG-3 cDNA was isolated from activated thymocytes by “fishing” with biotinylated hLAG-3 cDNA. This novel homologous gene isolation strategy is being published elsewhere (12).
LAG-3 Site-Directed Mutant Expression Plasmid and Adhesion Assay.
LAG-3 site-directed mutants were prepared by encoding the desired mutation in overlapping oligonucleotide primers and generating the mutants by PCR (13), or by inserting mutated synthetic double-strand oligonucleotides. Sequencing of PCR products was performed with a Sequenase kit according to the manufacturer’s recommendations (United States Biochemical).
COS-7 cells were grown in RPMI medium supplemented with 10% fetal calf serum. COS-7 cells were transfected with the pCDM8-LAG-3C plasmid (2) by electroporation using a Cellject apparatus (Eurogentec, Brussels). Electroporation was carried out in 500 μl at 200 V, 1,500 μF, and infinite shunt resistance with 30 μg/ml of plasmid in the culture medium at a concentration of 5 × 106 cells per ml. B cells (2 × 108) were incubated for 45 min at 37° with 2 mCi (1 Ci = 37 GBq) of 51Cr (final volume = 3 ml). Cells were washed three times and subsequently used for binding. Two days after transfection, COS-7 cells were treated with PBS 1×, EDTA 1 mM and replated at 0.05 × 106 cells per well in 12-well flat-bottom tissue culture plates. Twenty four hours later, 5.5 × 106 51Cr-labeled Daudi B cells were incubated in duplicate wells on this COS-7 cell monolayer (final volume = 550 μl) for 1 hr. The Daudi cells were then aspirated and the wells washed five to seven times by slowly dropping 1 ml of medium. The well edges were washed by aspiration using a Pasteur pipette. Remaining cells were lysed in 1 ml PBS, 1% Triton for 45 min at 37°C. Lysates were then centrifuged at 10,000 rpm for 10 min and 500 μl of the resulting supernatant was counted. The number of rosettes (more than five Daudi cells) obtained with LAG-3 mutants was determined relative to the number of rosettes obtained with wild-type LAG-3 (wtLAG-3) transfected cells. COS-7 cells, transfected with pCDM8 containing the LAG-3 insert in the reverse orientation (termed GAL), served as negative controls for rosette formation. Evaluation of cell binding employing radiolabeled Daudi cells correlated well with enumeration of rosetting (data not shown).
Immunofluorescence Staining and Flow Cytometry of Transfected Cells.
The level of mutant LAG-3 expression was studied with 15A9 reactivity because this epitope (LAG-3.3) was never affected by mutations. The level was calculated with arbitrary fluorescence units (FU) defined by the following formula: FU = % of positive cells × mean fluorescence of positive cells. To compare the LAG-3 expression level of mutant LAG-3 and wtLAG-3, relative levels of expression were calculated as follows using 15A9 mAb and a control mAb (termed neg):
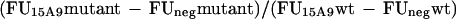 |
mAb reactivity to mutated LAG-3 molecules was calculated as follows:
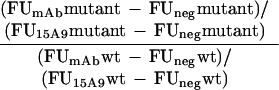 |
Molecular Modeling of LAG-3 Domains 1 and 2.
The LAG-3 amino acid sequence was aligned with that of human CD4 and then manually refined to maximize sequence similarity matches in the presumed β-strand segments. Coordinates for these segments were obtained from the published CD4 structure (14). An approximate three-dimensional model of the first two amino-terminal domains of LAG-3 (LAG-3 D1-D2) was generated, based on conservation of the key residues in IgSF variable (V) and constant (C) LAG-3 domains. Using such consensus residues as “anchor points” for sequence alignments, D1 LAG-3 residues were assigned to the A, B, C, C′, C", D, E, F, and G strands of an Ig V fold and the connecting loop regions. Side-chain replacement and loop conformations were approximated using conformation researching. Molecular mechanics and molecular dynamics calculations were carried out using the CVFF force field, with the Discover module of the insight package (Biosym Technologies, San Diego). The overall version of the model, with modified assignments of residues to β-strands or loops, was tested to improve the alignment of the LAG-3 D1-D2 region sequence with both IgSF V and C folds using the Bence–Jones protein, REI, and CD4 as template structures (15, 16). The geometry of this model was energetically minimized while constraining the backbone of the β-strands and then submitted to molecular dynamics calculations at 300 K. The molecular motions observed did not lead to disruption of the protein Ig-like tertiary arrangement. The 30-residue extra loop (G48-R77) deserved special attention because no template structure was available. We applied the simulated annealing technique from 1,000 K to 300 K on the isolated 30-residue peptide. Low-energy conformations of the isolated fragment were studied and energetically minimized. In the end, a structure with an intermediate stability range was retained.
RESULTS
Comparison of Human and Murine LAG-3 and CD4 Sequences.
Microsequencing of the native LAG-3 protein expressed in phytohemagglutinin blasts (following immunoprecipitation and SDS/PAGE purification, not shown) revealed six additional amino acids (LQPGAE) in the N-terminal sequence of the mature hLAG-3 protein compared with the predicted hLAG-3 amino acid sequence (1). Accordingly, the mature hLAG-3 N-terminal sequence (+1 in Fig. 1) starts at amino acid Leu23 of the LAG-3 precursor (1). In addition, we found, by sequencing both genomic and cDNA hLAG-3 clones derived from different individuals, an additional C nucleotide at position 1709 (a GC-rich region) of the original LAG-3 FDC sequence (1). This frameshift in the 3′ coding region leads to an extended cytoplasmic region in hLAG-3 that features numerous “EP” amino acid repeats (see Fig. 1). The mature hLAG-3 protein sequence includes 502 aa, taking into account changes at both N and C termini. The mLAG-3 cDNA, recently identified (accession no. X98113X98113), was isolated from activated thymocytes (12), and the amino acid sequence identity between mLAG-3 and hLAG-3 was found to be 70%.
Figure 1.

Sequence alignment of human and murine LAG-3 and CD4. Identical amino acid residues (at least 3 out of 4) through the LAG-3 and CD4 sequences are shown in bold. Numbers refer to the mature human LAG-3 sequence.
Fig. 1 shows an alignment of human and murine LAG-3 and CD4 amino acid sequences. Comparison of the new mLAG-3 amino acid sequence to hLAG-3, hCD4, and mCD4 further strengthens the structural homology between the “first cousins” LAG-3 and CD4 with critical positions in V or C2 Ig-like domains being conserved in at least three of the four molecules analyzed (see Fig. 1). These molecules belong, definitively, to a distinct subgroup of structurally related IgSF proteins with four extracellular Ig-like domains and a W × C signature motif in β-strand F of D2 and D4 (1). Homology is with reference to all domains except the extra loop in D1 and the intracytoplasmic proline-rich motif, which are both characteristic of LAG-3 molecules.
Comparison of the LAG-3 Model with the CD4 Structure.
Residues referred to are numbered according to the N-terminal sequencing of the mature protein. Conformational stability of the LAG-3 patterns during a long molecular dynamics simulation trajectory (100 ps at 300 K) supports the reliability of this model. Due to the low sequence identity with any known three-dimensional structure, the resolution of the model gives only an approximate conformation of the complementarity determining region (CDR)-like regions. The LAG-3 D1 sequence is globally consistent with an IgV fold and the D2 sequence with an IgC fold (2). In the CD4 D1 domain, BED strands make up one β-sheet, and AGFCC′C" strands are in the other sheet of the β-sandwich. In LAG-3, the corresponding β-strands are ABED and GFCC′C", respectively (1). The assignment of the CD4 A strand to an unusual β-sheet may be derived from the analysis of a truncated CD4 structure (14) (i.e., lacking 6 aa at its N terminus; by analogy with the mature LAG-3 protein). Strand A in LAG-3 begins in the first sheet alongside strand B and passes across to the second alongside strand G. Note that the second cysteine involved in the D1 disulfide bond (C138) is located in a different β-strand (strand G) than that found in the equivalent residue (C84) in CD4 (strand F) (1). This led to a different positioning of the LAG-3 FG loop (Ig CDR3) which, in contrast to the shortened CD4 FG loop, has a similar size to the κ light chain (e.g., REI) and is therefore prominent. A peculiarity shared between the LAG-3 and CD4 D1 domains is a lengthening of the C′C" loop (Ig CDR2), a region that seems to be important in CD4 for interactions with class II MHC molecules and HIV gp120. This lengthening in LAG-3 is particularly dramatic because it includes the 30-aa-long extra loop. Overall, the D1 domains of LAG-3 and CD4 appear to be quite different, although both are consistent with an Ig V fold.
The extra loop in D1 is fully encoded by exon 3 of LAG-3, in addition to the rest of the V domain. This genomic organization explains why the extra loop is constitutively expressed on LAG-3+ lymphocytes. We searched for an evolutionary remnant of this structure in the CD4 gene, given that both genes are thought to derive from a common ancestor. In fact, cells from African Green monkey-3 have been found to express an infrequent CD4 transcript including an additional exon inserted by alternative splicing (17). This exon encodes a 25-aa peptide that is similarly positioned (CDR2 region) to the LAG-3 extra loop on the first Ig-like domain and contains numerous Pro, a His, a Tyr, and a Trp residue, as does the LAG-3 extra loop. This alternative splicing is a rare event in monkey cells, and the fact that a similar event might occur in human cells cannot be ruled out. Accordingly, the RNA from 10 individual donors’ PBMCs were amplified through reverse transcription–PCR (40 cycles) using primers adjacent to the CD4 CDR2 sequence. A second PCR using nested primers (40 cycles) was performed, and no fragments or an additional CD4 extra-loop segment was detected (data not shown). It is therefore unlikely that any human CD4 molecules express a long extra loop that would most likely be present in the two-domain IgSF ancestor from which CD4 and LAG-3 are derived (2, 5).
Although D2 of CD4 has a tertiary folding that can be readily recognized as resembling Ig constant domains, details of the folding are considerably divergent: (i) shorter strand lengths partly due to a small Ig domain (≈80 residues), (ii) absence of a D strand, which is replaced by a C′ strand, and (iii) an unusual disulfide bond between C and F strands, which is in the same sheet (15). In contrast, D2 of LAG-3 has similar size, β-strand composition (ABED and GFC), and disulfide bond connection to the usual Ig constant domains.
In contrast to CD4, a major flexibility point in the LAG-3 structure is predicted to be formed by the long β-strand joining the first and second domain giving bulk flexibility to these two quite rigid substructures. In CD4, the hydrophobicity of the hinge side-chains is believed to be responsible for its rigidity, as bulky hydrophobic side-chains lead to extended conformations minimizing interresidue steric hindrance. In the less hydrophobic LAG-3 hinge region, two polar side-chains (R141 and R143) can be found that may fold the segment when attempting to form hydrogen bonds.
A Truncated Soluble Fusion Protein Reveals a Critical Role of LAG-3 Domains 1 and 2 in Binding MHC Class II Molecules.
To test whether the first two domains of LAG-3 contain a class II binding site, we generated a fusion protein consisting of extracellular domains 1 and 2 of LAG-3 (termed LAG-3D1D2Ig; for details see ref. 3) joined to a human IgG1 Fc portion. Binding of this fusion protein to class II+ Daudi cells was as intense as LAG-3D1D4Ig (including domains 1, 2, 3, and 4 of LAG-3) (3) or 9.49 anti-class II mAb (data not shown). Thus, the structure formed by Ig-like domains 1 and 2 is sufficient for binding to class II molecules. In addition, deletion of the Ig-like domain 2 leads to inhibition of rosette formation between class II+ Daudi cells and COS-7 transfectants (see Table 1) in spite of adequate surface expression on COS-7 cells of epitopes expressed on domain 1. These data suggest that the D2 domain is either involved in LAG-3/class II binding or in positioning the D1 domain for class II binding.
Table 1.
Effect of LAG-3 mutations on LAG-3/class II MHC interaction
| Mutation | Localization of residue | Anti-LAG-3 mAb reactivity
|
Level of LAG-3 expression | Relative adhesion | ||
|---|---|---|---|---|---|---|
| 17B4 | 11E3 | 15A9 | ||||
| Wt | — | + | + | + | 1.0 | 1 |
| GAL | Neg. control | − | − | − | 0 | 0.09 ± 0.06 |
| Q13A | AB | + | + | + | 1.0 | 0.92 ± 0.06 |
| D30A | BC | + | + | + | 0.81 | 0.41 ± 0.17 |
| H56A | Extra loop | − | + | + | 0.7 | 0.45 ± 0.28 |
| H56F | Extra loop | − | + | + | 0.76 | 0.89 ± 0.36 |
| H63A | Extra loop | + | + | + | 0.89 | 0.83 ± 0.10 |
| H63F | Extra loop | + | + | + | 0.95 | 0.91 ± 0.18 |
| R73E | Extra loop | + | − | + | 1.1 | 3.25 ± 1.00 |
| R75A | Extra loop | + | + | + | 0.67 | 4.20 ± 0.66 |
| R75E | Extra loop | + | + | + | 1.4 | 4.10 ± 0.90 |
| R76E | Extra loop | + | + | + | 0.85 | 2.95 ± 1.00 |
| Y77F | C" | + | + | + | 0.75 | 0.05 ± 0.06 |
| R88A | C"D | + | + | + | 0.82 | 0.18 ± 0.07 |
| R103A | DE | + | + | + | 0.93 | 0.49 ± 0.03 |
| R107K | E | + | + | + | 1.1 | 1.02 ± 0.19 |
| D109E | E | + | + | + | 0.81 | 0.18 ± 0.19 |
| R115A | EF | + | + | + | 0.74 | 0.23 ± 0.05 |
| D133A/R134A | FG | + | + | + | 0.99 | 0.78 ± 0.24 |
| D225L | FG (D2) | + | + | + | 0.85 | 0.89 ± 0.13 |
| (D2)− | D2 deletion | + | + | + | 0.6 | 0.11 ± 0.09 |
| (54/66)− | Extra-loop deletion | − | ± | + | 0.6 | 0.08 ± 0.12 |
The table shows the binding of 51Cr-labeled Daudi B cells to COS-7 cells expressing LAG-3 mutants. All mutations were in Ig-like domain 1 except for D225L. Nonspecific binding to control-transfected cells was not subtracted from experimental mean cpm. The results expressed relative to wt value should be compared with the background level as defined by the GAL (the LAG-3 insert cloned in reverse orientation in pCDM8)-transfected control cells. Relative expression level of mutant LAG-3 versus wtLAG-3 is also shown. The level of mutant LAG-3 expression was studied with 15A9 reactivity, and relative mAb reactivity was measured as follows: +, 1–0.6; +/−, 0.2–0.6; −, 0–0.2.
LAG-3 Mutants Analyzed in a Cell–Cell Adhesion Assay.
Amino acid substitutions were performed to map regions thought to be exposed (e.g., the extra loop, CDR-1, -2, and -3), so as to assess their contribution to class II MHC binding. Focusing on residues that, by analogy with CD4, might interact with class II molecules, single amino acid substitutions were introduced into the LAG-3 molecule by site-directed mutagenesis (except for the D133A/R134A double mutant and the 54/66 extra-loop deletion mutant). For instance, the R88A mutation was chosen according to the CD4 K46A effect on class II binding (10). Correspondences with CD4 residues are: Q13/K7, R88/K46, R103/R58, R59, R115/K72, D133, R134/Q89, and D225/Q165 (9, 10). In general, single amino acids were replaced by alanine. Other substitutions were made to preserve or modify hydrophobicity, hydrogen bonding function, and charge.
Wt and mutant forms of LAG-3 were expressed in simian virus 40-transformed COS-7 monkey cells and then tested for their ability to bind to the class II+ Daudi cell line (2). Because the level of cell surface expression varied from mutant to mutant, expression of each mutant was determined and compared with wtLAG-3 expression in each experiment. In most cases, the surface expression and structural integrity of each mutant protein were confirmed by fluorescence-activated cell sorter analysis using 17B4, 11E3, and 15A9 mAb (Table 1). Indeed, most of the mutations showed no effect on mAb binding. Deletion of the tip of the extra loop (54/66) partially or completely disrupted the LAG-3.2 (11E3) and LAG-3.1 (17B4) epitopes, respectively. The latter epitopes were partly formed by residues R73 and H56, respectively, as assessed by the absence of reactivity of the LAG-3 point mutants R73E, H56F, and H56A with the corresponding mAb. Because reduction or loss of mAb binding was restricted to only one mAb, we interpreted this effect as a direct consequence of the mutation on an exposed residue and not as a result of major structural alteration of the molecule.
The ability of each LAG-3 mutant to bind to class II+ Daudi cells was determined in a quantitative cellular adhesion assay (Table 1). B cell binding after LAG-3 transfection was restricted to LAG-3+ COS-7 cells exclusively, as observed after fixation and immunoperoxidase staining using an anti-LAG-3 mAb (data not shown). In addition, the level of mutant LAG-3 expression was determined in each experiment by immunofluorescence with 15A9 mAb (the LAG-3.3 epitope was not altered by any mutation). Approximately 50% of cells transfected with wt or mutant LAG-3 cDNA expressed LAG-3 in each individual experiment (not shown). In most cases, mutant LAG-3 molecules were expressed at similar levels to wtLAG-3, and, therefore, the binding of Daudi cells to these mutants could be directly compared with binding to wtLAG-3. The binding of Daudi cells to LAG-3 mutants was analyzed quantitatively with 51Cr-labeled Daudi cells in three separate experiments.
Unexpectedly, some mutations were able to reduce class II binding while other mutations increased the affinity of LAG-3 for class II molecules (Table 1). Y77F (strand C′), R88A (CDR2), D109E (RGD motif in strand E), and R115A (EF turn) mutants strongly inhibited binding, while D30A (CDR1), H56A (extra loop), and R103A (DE turn) mutants only diminished binding 2-fold. In contrast, R73E, R75A, R75E, and R76E mutants increased binding 3-fold or more. All residues that affected the binding are found only on one side of domain 1 including the top, CDR-1, CDR-2 containing face, and the side of domain-1 containing β-strands ABED. Note that the positively charged residues (R73, R75, and R76) whose replacement either by alanine (R75A) or an amino acid varying in charge (R73E, R75E, or R76E) results in increased binding belong to the base of the extra loop and are clustered on the top face of domain 1.
Further support for the potential involvement of the LAG-3 extra loop in binding is provided by analysis of the mutant H56A, which decreased binding at least 2-fold, while substitution by a similar amino acid (H56F) did not result in altered binding. The H56 residue, which is part of the 17B4 epitope (see Table 1), is therefore likely to make direct contact with class II molecules. This assumption is also supported by the deletion mutant (54/66)−, which lacks the tip of the extra loop and also failed to bind to class II molecules.
The mutants D30A, H56A, Y77F, and R115A, and deletion mutants (D2)− and (54/66)− were expressed at lower levels than wtLAG-3 and therefore reexamined in experiments in which we ruled out the possibility that the effects on MHC class II binding might be due to the difference in expression level of LAG-3 molecules. In these experiments (see example in Fig. 2), wtLAG-3 expression was modulated by using varying amounts of DNA in the transfections to match mutant LAG-3 expression. All six mutants mentioned above showed <40% of bound cpm obtained with cells expressing wtLAG-3 at comparable levels. Therefore, diminished binding function is an effect of amino acid substitution or fragment deletion and not a consequence of reduced surface expression.
Figure 2.

MHC class II binding of selected LAG-3 mutants compared with that of wtLAG-3 expressed at different levels. COS-7 cells were transfected with GAL (30 μg/ml) and 3, 10, 30, and 100 μg/ml of pCDM8-wtLAG-3 DNA, respectively (○). Mutant LAG-3 DNA was used at 30 μg/ml. LAG-3 expression is shown as arbitrary fluorescence units (FU) using 15A9 mAb, and class II binding was determined by cpm counting in a cell–cell adhesion assay using 51Cr-labeled Daudi cells.
The residues R107 and D109 were replaced by an amino acid with a similar charge but differently sized side-chain to test solvent accessibility as well as the putative dipolar electrostatic interaction mediated by the RGD motif (β-strand E). The D109E mutant failed to bind to class II molecules whereas the R107K mutation did not affect class II binding. It is concluded that the RGD motif in LAG-3 domain 1 did not behave as a typical dipole to enhance the strength of this putative adhesiotope.
In addition, we found a set of mutations that did not affect LAG-3/MHC class II interactions. These residues were chosen in positions where contacts with class II molecules might occur, as shown for the corresponding positions in CD4 (e.g., Q13A, D133A/R134A, or D225L), or in unique LAG-3 motifs (e.g., H63A or H63F in the extra loop, or R107K in the RGD motif). In particular, we mutated a residue, D225, in a region thought to be exposed to solvent (FG loop of domain 2) and that, in CD4, is known to interact with class II molecules (9). This change had no measurable influence on class II MHC interactions. Although other candidate residues may be changed in this region, it is dubious that LAG-3 domain 2 interacts with class II molecules in the same way as CD4 domain 2.
Dominant Negative LAG-3 Mutations for MHC Class II Binding Affect Residues Embedded into the ABED β-Strands Containing Face.
Oligomerization of CD4 on the cell surface may be required to form a stable MHC class II binding site (11). We tested whether oligomerization of LAG-3 molecules on the cell surface might increase the avidity of interaction between LAG-3 and MHC class II molecules, leading to the stabilization of the complex. If LAG-3 oligomers are required for stable MHC class II binding, some mutations of LAG-3 that individually reduce MHC class II binding by themselves would also be expected to inhibit the ability of cotransfected wtLAG-3 molecules to bind class II molecules because both wt and mutant LAG-3 would be incorporated into the same oligomers.
We found three dominant negative LAG-3 mutations—R88A, D109E, and R115A—in the ABED β-strands containing face that were incapable of binding to MHC class II and also that prevent the binding of wtLAG-3 to MHC class II molecules (Fig. 3A). This inhibition was dependent on the amount of mutant LAG-3 DNA transfected (Fig. 3B). In contrast, other negative mutations, Y77F and R103A, affecting residues located on the top of the LAG-3 D1 structure or silent mutation (Q13A, not shown), did not prevent the binding of wtLAG-3 to MHC class II molecules (Fig. 3A). The number of cpm was even greater for Y77F + wt and R103A + wt than for wt alone, given the ability of these expressed mutant proteins (mutant/wt DNA ratio used in these transfection experiments was 4:1) to bind, even poorly, to MHC class II molecules.
Figure 3.

Coexpression of R88A, D109, or R115A mutant LAG-3 molecules with wtLAG-3 molecules prevents MHC class II Daudi cells from binding to COS-7 cell transfectants. DNA amounts used for transfection were 10 μg/ml for wtLAG-3 (wt) and the negative control (GAL), and 40 μg/ml for mutants (A). Class II binding was determined by cpm counting in a cell–cell adhesion assay using 51Cr-labeled Daudi cells. B shows the inhibition of Daudi cell binding as a function of the amount of R88A, D109E, or R115A DNA transfected. wtLAG-3 DNA (10 μg/ml) and various amounts of mutant DNA (DNA ratio mutant/wt shown in abscissa) were mixed, and pCDM8 DNA was added to maintain the total amount of transfected DNA at 50 μg/ml.
In these experiments, similar amounts of LAG-3 molecules were expressed (as assessed using the 15A9 LAG-3-specific mAb) when one of these three LAG-3 mutants—R88A, D109E, or R115A—was coexpressed with wtLAG-3 compared with other negative (Y77F and R103A) or silent (Q13A) mutations (data not shown). However, we could not check whether similar amounts of wtLAG-3 were expressed on COS-7 cells, regardless of the coexpression of R88A, D109E, or R115A, given the lack of specific mAb discriminating between wtLAG-3 and these point mutants. Nevertheless, it is likely that these mutant LAG-3 molecules located on the side of D1, and not the other negative mutants (Y77F and R103A) on the top of D1, used here as controls, oligomerize with wtLAG-3 and, hence, interfere with the self-association of wtLAG-3, which stabilizes the complex.
To test the LAG-3 oligomerization hypothesis, LAG-3-transfected COS-7 cells or MHC class II+ B cells were treated with paraformaldehyde prior rosetting (2). In this bioassay, treatment of COS-7 but not B cells with 0.5 or 1% paraformaldehyde abolished rosetting (data not shown), suggesting that structural rearrangement of LAG-3 molecules, possibly induced by MHC class II contacts, is a prerequisite for stable LAG-3/class II interaction.
DISCUSSION
We designed a LAG-3 model structure and selected mutants that were tested for their ability to bind to MHC class II molecules in an intercellular adhesion assay. The V-type Ig-like D1 domain appears to include the MHC class II binding site on LAG-3. In this domain, we found residues on the membrane-distal, CDR1-CDR2-containing top face that are essential for either binding (D30, H56, Y77, R103) or repulsing (R73, R75, R76) MHC class II proteins. In addition, we found three dominant negative LAG-3 mutants (R88A, D109E, and R115A) that themselves were incapable of binding to MHC class II molecules but, at the same time, prevented the binding of wtLAG-3 to MHC class II molecules. These residues (R88, D109, and R115) are located on the same ABED β-strands containing side of D1 and may therefore contribute to the formation of a second interaction site for LAG-3 homodimer formation on the surface of COS-7 cells.
The finding that three dominant negative mutants (R88A, D109E, and R115A) were clustered on the same face of LAG-3 domain 1, and that other negative mutants (Y77F and R103A) on the top face did not interfere with wtLAG-3 binding, support a model in which LAG-3 oligomerization occurs on COS-7 cells. To explain our results shown in Fig. 3A, we speculate that a single LAG-3 molecule exhibits only one binding site for class II molecules on the top face of domain 1 (site 1; see Fig. 4A) and a second binding site for another LAG-3 molecule (homotypic interaction) on the ABED β-strands containing face where the residues R88, D109, and R115 are embedded (site 2). This second binding site would lead to the formation of LAG-3 homodimers or even give the capacity of LAG-3 molecules to participate in lattice formation on the cell surface. Induction of conformational changes in the LAG-3 D1 structure by homodimers formation would explain why a site 1 mutant–wt combination is still active in binding MHC class II molecules (Fig. 4B), whereas a site 2 mutant–wt combination, for instance, is not (Fig. 4C). In the latter case, inappropriate conformational rearrangement due to the mutation in the homotypic binding site (e.g., R88, D109, and R115) leads to the inactivation of both mutant and wt MHC class II binding sites. In such a model, it is proposed that oligomerization of LAG-3 molecules induces structural changes in the binding site that enhance complementarity at the interface between LAG-3 and MHC, and these structural perturbations increase stability of the interaction on the cell surface. Oligomerization may take place constitutively or be initiated as in CD4 oligomerization (11) following a transient interaction with MHC class II molecules. Alternatively, preferential competition between some negative mutants and wt molecules would also explain our results, without a need for oligomerization. In fact, this possibility could not be ruled out given that rosette formation in this quantitative cellular adhesion assay depends, at the single-cell level, on the number of functional LAG-3 molecules expressed, with an undefined threshold effect.
Figure 4.

A hypothetical model of LAG-3-MHC class II interaction. Ovals, D1–D4 of LAG-3 ectodomain; □, active MHC class II binding site (site 1); ♦, inactive MHC class II binding site; •, homotypic interaction binding site (site 2); ★, a mutation with a negative effect on MHC class II binding. The MHC class II heterodimer with the LAG-3 binding site is shown at the top of the figure, facing each LAG-3 molecule. For ease of illustration, molecules are not drawn to scale. (A) A wt–wt LAG-3 homodimer binding two MHC class II heterodimers. (B) A site 1 mutant (Y77F, R103A)–wt LAG-3 homodimer still able to bind MHC. (C) A site 2 mutant (R88A, D109E, R115A)–wt LAG-3 homodimer, with an inappropriate conformational rearrangement in D1, unable to bind MHC.
The occurrence of LAG-3/MHC class II structure aggregation on T cells following T–T cell contacts would explain why the down-modulation of T cell clone proliferation and cytokine secretion by soluble LAG-3 is obtained only in cross-linking conditions (3). However, LAG-3 (gp70) is associated with gp45 protein(s) on the surface of both activated T and NK cells but not on COS-7 cells (2). Therefore, it is possible that oligomerization might be restricted to COS-7 cells with the formation first of LAG-3 homodimers and not LAG-3/gp45 heterodimers.
In addition to signals transduced in T cells by MHC class II aggregation, LAG-3 clustering may directly contribute to positive or negative triggering of effector cells. For instance, the LAG-3 protein has been proposed to transduce a positive signal in effector cells, because transgenic mice with a LAG-3 null mutation have a defect in the NK cell compartment (18). The short LAG-3 intracytoplasmic region does not contain, as do CD4 and CD8, a C-X-X-C motif known to interact with p56lck, an associated tyrosine kinase. On the other hand, the corrected hLAG-3 C terminus sequence reported here includes a specific proline-rich region (with many Glu-Pro repeats) at the C terminus of the cytoplasmic domain that may participate in signal transduction.
The surface of CD4, over which residues influencing class II binding have been found, is much larger than the LAG-3/class II interface (10). CD4 residues that affect class II binding can be found on all lateral surfaces of domain 1 and on the neighboring parts of the lateral surfaces of domain 2. In addition, the top surface of domain 1 does not contain such critical residues, in contrast to LAG-3. Finally, CD4/class II interactions (in the absence of CD4 oligomerization) seem to be much weaker than LAG-3/class II interactions (4). Indeed, we found that purified monomeric (following gel filtration) sLAG-3 D1–D4 molecules produced in fly cells (19) have the capacity to bind to class II+ cells, as assessed in an indirect immunofluorescence assay using a D3-D4-specific mAb (data not shown), in contrast to monomeric sCD4, which has a reported affinity of <10−4 M (11).
Our study suggests that LAG-3/class II interaction may be more analogous to CD8/class I than CD4/class II interaction. LAG-3 (gp70), like CD8 (8), may contact its ligand through the top of the D1 V-like domain and may be expressed as either a homo- (gp70/gp70) or a heterodimer (gp70/gp45). Further studies will have to establish whether oligomerization on the T cell surface in the presence of MHC is a common feature shared by these three MHC ligands.
Acknowledgments
We thank Arnaud Ythier for his valuable comments; Drs. G. Bismuth, C. Ronsin, N. Borie, and S. Fleury for carefully reviewing the manuscript; Drs. Hochstrasser and S. El-Ougouti for determining the N-terminal sequence of hLAG-3; and Muriel Tournier for her technical assistance. This work was funded by research grants from Association pour la Recherche contre le Cancer and Ares Services S.A.
ABBREVIATIONS
- LAG-3
lymphocyte activation gene-3
- CDR
complementarity determining region
- MHC
major histocompatibility complex
- wt
wild type
Footnotes
References
- 1.Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, Hercend T. J Exp Med. 1990;171:1393–1405. doi: 10.1084/jem.171.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, Auffray C, Triebel F, Piatier-Tonneau D. J Exp Med. 1992;176:327–337. doi: 10.1084/jem.176.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huard B, Prigent P, Pages F, Bruniquel D, Triebel F. Eur J Immunol. 1996;26:1180–1186. doi: 10.1002/eji.1830260533. [DOI] [PubMed] [Google Scholar]
- 4.Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. Eur J Immunol. 1995;25:2718–2721. doi: 10.1002/eji.1830250949. [DOI] [PubMed] [Google Scholar]
- 5.Huard B, Gaulard P, Faure F, Hercend T, Triebel F. Immunogenetics. 1994;39:213–217. doi: 10.1007/BF00241263. [DOI] [PubMed] [Google Scholar]
- 6.Huard B, Tournier M, Hercend T, Triebel F, Faure F. Eur J Immunol. 1994;24:3216–3221. doi: 10.1002/eji.1830241246. [DOI] [PubMed] [Google Scholar]
- 7.Annunziato F, Manetti R, Tomasevic L, Guidizi M-G, Biagiotti R, Gianno V, Germano P, Mavilia C, Maggi E, Romagnani S. FASEB J. 1996;10:769–776. doi: 10.1096/fasebj.10.7.8635694. [DOI] [PubMed] [Google Scholar]
- 8.Giblin P A, Leahy D J, Mennone J, Kavathas P B. Proc Natl Acad Sci USA. 1994;91:1716–1720. doi: 10.1073/pnas.91.5.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleury S, Lamarre D, Meloche S, Ryu S E, Cantin C, Hendrickson W A, Sekaly R P. Cell. 1991;66:1037–1049. doi: 10.1016/0092-8674(91)90447-7. [DOI] [PubMed] [Google Scholar]
- 10.Moebius U, Pallai P, Harrison S C, Reinherz E L. Proc Natl Acad Sci USA. 1993;90:8259–8263. doi: 10.1073/pnas.90.17.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakihama T, Smolyar A, Reinherz E L. Proc Natl Acad Sci USA. 1995;92:6444–6448. doi: 10.1073/pnas.92.14.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mastrangeli R, Micangeli E, Donini S. Anal Biochem. 1996;241:93–102. doi: 10.1006/abio.1996.0382. [DOI] [PubMed] [Google Scholar]
- 13.Mikaelian I, Sergeant A. Nucleic Acids Res. 1992;20:376–378. doi: 10.1093/nar/20.2.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryu S E, Truneh A, Sweet R W, Hendrickson W A. Curr Biol. 1994;2:59–74. doi: 10.1016/S0969-2126(00)00008-3. [DOI] [PubMed] [Google Scholar]
- 15.Ryu S E, Kwong P D, Truneh A, Porter T G, Arthos J, Rosenberg M, Dai X, Xuong N H, Axel R, Sweet R W, Hendrickson W A. Nature (London) 1990;348:419–426. doi: 10.1038/348419a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Yan Y, Garrett T P J, Liu J, Rodgers D W, Garlick R L, Tarr G E, Husain Y, Reinherz E L, Harrison S C. Nature (London) 1990;348:411–418. doi: 10.1038/348411a0. [DOI] [PubMed] [Google Scholar]
- 17.Fomsgaard A, Hirsch V M, Johnson P R. Eur J Immunol. 1992;22:2973–2981. doi: 10.1002/eji.1830221132. [DOI] [PubMed] [Google Scholar]
- 18.Miyazaki T, Dierich A, Benoist C, Mathis D. Science. 1996;272:405–408. doi: 10.1126/science.272.5260.405. [DOI] [PubMed] [Google Scholar]
- 19.Jackson M R, Song E S, Yang Y, Peterson P A. Proc Natl Acad Sci USA. 1992;89:12117–12121. doi: 10.1073/pnas.89.24.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]