

Abstract
Positional cloning has already produced the sequences of more than 70 human genes associated with specific diseases. In addition to their medical importance, these genes are of interest as a set of human genes isolated solely on the basis of the phenotypic effect of the respective mutations. We analyzed the protein sequences encoded by the positionally cloned disease genes using an iterative strategy combining several sensitive computer methods. Comparisons to complete sequence databases and to separate databases of nematode, yeast, and bacterial proteins showed that for most of the disease gene products, statistically significant sequence similarities are detectable in each of the model organisms. Only the nematode genome encodes apparent orthologs with conserved domain architecture for the majority of the disease genes. In yeast and bacterial homologs, domain organization is typically not conserved, and sequence similarity is limited to individual domains. Generally, human genes complement mutations only in orthologous yeast genes. Most of the positionally cloned genes encode large proteins with several globular and nonglobular domains, the functions of some or all of which are not known. We detected conserved domains and motifs not described previously in a number of proteins encoded by disease genes and predicted functions for some of them. These predictions include an ATP-binding domain in the product of hereditary nonpolyposis colon cancer gene (a MutL homolog), which is conserved in the HS90 family of chaperone proteins, type II DNA topoisomerases, and histidine kinases, and a nuclease domain homologous to bacterial RNase D and the 3′-5′ exonuclease domain of DNA polymerase I in the Werner syndrome gene product.
Significant progress has been recently achieved in positional cloning and sequencing of human genes mutated in individuals with specific diseases‖; as of Aug. 1, 1996, the list of such genes consisted of 70 items (refs. 1–3; http://www.ncbi.nlm.nih.gov/XREFdb/). These genes are of major interest, and not only because of their importance for understanding the mechanisms of the respective diseases. Positionally cloned genes have been isolated solely on the basis of the phenotypic effect of mutations, rather than biochemical properties of the product, tissue specificity, or expression level. Therefore, in spite of the relatively small number of the sequenced positionally cloned disease genes, they may be a representative sampling of human genes for simple Mendelian traits (ref. 4; http://www.ncbi.nlm.nih.gov/Omim/). Thus analysis of structural features and evolutionary conservation of the disease gene products may yield information of general significance.
The complete genome sequences of several bacteria, an archaeon, and a unicellular eukaryote, the yeast Saccharomyces cerevisiae, have been determined recently (5); over 50% of the genome sequence of a multicellular eukaryote, the nematode Caenorhabditis elegans, is also available (ref. 6; http://www.sanger.ac.uk/~sjj/C.elegans_Home.html). Crossreferencing of human disease genes with their homologs in model organisms whose genomes have been completely or nearly completely sequenced is now a major source of information for understanding functions of these genes (7, 8). A critical issue in using model organisms, which can be addressed in a definitive way only by analysis of complete genome sequences, is the identification of orthologs of human genes. Orthologs are genes in different species related by vertical descent from a common ancestor and normally performing the same function, as opposed to paralogs, which are genes in the same species related by duplication (9). Without complete genome sequences, identification of orthologs could be only preliminary, as the closest relative of any particular human gene could reside in the unsequenced portion of the model genome.
Most of the positionally cloned genes encode large, multidomain proteins, some of which do not contain known enzymatic domains. Detection of functionally relevant sequence similarities in such proteins requires careful delineation of distinct domains and sensitive procedures to detect conserved motifs (10–12). Case studies indicate that increasing the repertoire and sensitivity of methods for motif detection and structural modeling indeed tends to reveal putative functional sites that escape detection with standard approaches (e.g., refs. 13–15). Thus systematic characterization of all disease gene products by detailed computer analysis appears timely.
In this study, we pursued three main goals: (i) determine the general features of the disease gene products such as the arrangement of predicted globular and nonglobular domains, subcellular localization, and evolutionary conservation; (ii) identify orthologs and paralogs in model organisms, namely nematode, yeast, and bacteria; and (iii) detect previously uncharacterized domains and motifs and predict their functions.
MATERIALS AND METHODS
Databases.
Information on positionally cloned disease genes and various aspects of the respective diseases is available in the MIM database (ref. 4; http://www.ncbi.nlm.nih.gov/Omim/). The nonredundant (NR) protein sequence database at the National Center for Biotechnology Information (NCBI, National Institutes of Health, Bethesda, MD) was used for general purpose sequence similarity searches. The dbEST database (16) was used to detect similarities to expressed sequence tags. The database of C. elegans gene products, wormpep11 (7,299 sequences), was obtained from ftp://ftp.sanger.ac.uk/pub/databases/wormpep. The NR database of yeast proteins (6,141 sequences) was constructed at the NCBI using the data from the Saccharomyces Genome Database (Stanford University; http://genome-www.stanford.edu/Saccharomyces); the yeast genome sequence translated in six reading frames was searched to detect possible similarities to unannotated proteins. A bacterial protein sequence database (44,617 sequences) was constructed by selecting all eubacterial sequences from the NR database using Entrez (17). The sequences of positionally cloned human disease genes were extracted from the NR database; a fasta library of the encoded protein sequences is available at http://www.ncbi.nlm.nih.gov/Disease_Genes/.
Sequence Analysis: Strategy, Methods, and Significance Criteria.
For database searches, we adopted an iterative strategy whereby sequences similar to the protein of interest are extracted from the database, and the search procedures are iterated until convergence (12). Additional database searches were performed after partitioning protein sequences into distinct domains. Globular and nonglobular domains were predicted based on sequence complexity, using the seg program with the parameters L(1) = 45, K2(1) = 3.4, K2(2) = 3.75 (18). Database search then was performed individually with the sequence of each of the predicted globular domains. For detection of subtle sequence similarities, an important parameter is the size of the database searched; databases of nematode, yeast, and bacterial proteins were used in an attempt to increase the search sensitivity (12).
Protein sequence databases were searched for pairwise similarity using the blastp and blast2 programs (19, 20). Nucleotide sequence databases translated in six reading frames were searched using the tblastn program (19). The blast programs estimate the statistical significance of local alignments produced by database searches using the extreme value distribution statistics for a single alignment segment and the sum statistics for two or more compatible segments. In blast2, the statistics have been modified to include gapped local alignments (20). The values of the probability (P) of obtaining an alignment with a given score by chance computed by blast are reliable as long as regions of biased composition (low complexity) do not contribute to the alignment scores (21). In our analysis, filtering for low-complexity regions was applied twice, first with parameters tuned for the detection of nonglobular domains (see above) and then with the standard parameters optimal for eliminating short, widespread low-complexity segments [L(1) = 12, K2(1) = 2.2, K2(2) = 2.5]. With these parameters, a P value of 0.001 or less was considered a strong indication of homology. Alignments in the “twilight zone,” namely those that were detected with the default blast2 parameters but had an associated P value greater than 0.001, were further evaluated using iterative blast2 searches and methods for motif and multiple alignment analysis (12).
The program psiblast (Position-Specific Iterative blast) was used to search the database iteratively, until convergence, with position-specific matrices derived from the original blast2 output and modified after each iteration (S. F. Altschul, A. A. Schaffer, T. Madden, and D. J. Lipman, personal communication). The P values computed by psiblast are comparable to those produced by blast and blast2. Alternatively, conserved sequence blocks were extracted from blast outputs using the cap program (22), or from multiple sequence alignments, using the macaw program (23). The blocks were used for iterative motif searches with the MoST program, with the cutoff set as r = 0.02 or r = 0.01 (22). Hidden Markov models were constructed from multiple alignments and used for iterative database screening with the local version of the Needleman–Wunsch algortihm (24).
Other Methods.
Protein secondary structure and transmembrane helices were predicted using the phd program (refs. 25 and 26; http://www.embl-heidelberg.de/predictprotein/predictprotein.html). Signal peptides were predicted using the SignalP program (ref. 27; http://www.cbs.dtu.dk/services/SignalP/). Coiled-coil regions were predicted using the coils program (ref. 28; http://ulrec3.unil.ch/software/Coils_form.html).
RESULTS AND DISCUSSION
Protein Size and Domain Architecture.
The set of positionally cloned disease genes products is enriched in large proteins with more than one predicted globular domain separated by nonglobular spacers, compared with the 3,475 human protein sequences contained in swiss-prot, Release 33.0 (Fig. 1 A and B; ref. 29). This is not unexpected as certain classes of proteins, e.g., immunoglobulins, and metabolic enzymes, are over-represented in swiss-prot. In a number of disease gene products, the multidomain organization is manifest at the level of sequence conservation—distinct domains are homologous to single-domain proteins or to domains in proteins with different domain architectures (Table 1; http://www.ncbi.nlm.nih.gov/Disease_Genes).
Figure 1.
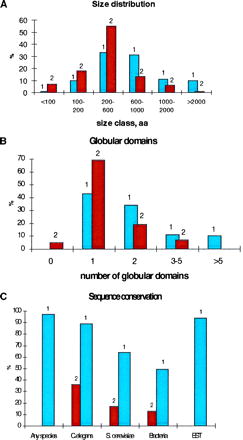
Features of human positionally cloned disease gene products. The vertical axis in each panel indicates the percentage of the analyzed protein set. (A) Protein size classes. 1, Positionally cloned disease gene products; 2, human proteins from swiss-prot. (B) Domain organization. 1, Positionally cloned disease gene products; 2, human proteins from swiss-prot. The horizontal axis indicates the predicted globular domains, with the lower size limit of 20 amino acid residues. (C) Sequence conservation. 1, All sequence similarities; 2, orthologs.
Table 1.
Previously undetected domains and motifs in human positionally cloned disease gene products
| Disease gene product/Segmentation/Signal peptide* GenBank/OMIM | Previously unknown domains or motifs | Method(s) of detection and significance; additional evidence† | Predicted function/activity | Other domains, motifs, and functions | Ortholog candidate‡ and paralogs in C. elegans (C), yeast (Y), bacteria (B); matches with ESTs (E)§ |
|---|---|---|---|---|---|
| Autosomal chronic granulomatosis disease protein/390:202-170-18 M55067M55067/306400 | Two conserved domains: i. in several phosphatidylinositol 3-kinases (U52192U52192; Z69660Z69660); ii. in yeast BEM1 and SCD2 proteins (not detected previously) | blast2 P = 3 × 10−6 (SCD2_SCHPO) | Intracellular signal transduction | SH3 domains; NADPH oxidase activator | C: ortholog not found; paralogs of SH3 domains only (e.g. CE01784) and of BEM1-SCD2 domain only (CE05832) Y: no ortholog; paralogs: BEM1 (Bem/Scd2- and SH3 domains are swapped as compared to human disease gene), several proteins with SH3 domain only, e.g. YHL002w B: not found E: Human +; mostly SH3 domains |
| Barth syndrome (tafazzin)/292: Signal peptide 32-33 X92762/302060 | A conserved motif in bacterial RadC | blast2 P = 0.31 (E. coli RadC, gil78795) HMM built from the alignment of tafazzin with nematode and yeast homologs—14 bits with RADC_BACSU compared to 11.5 bits for first false positive; conservation of putative active Glu and His | Hydrolytic enzyme; RadC is involved in DNA repair but such a role is unlikely for tafazzin given the presence of a signal peptide | Predicted secreted protein | C: CE03830 Y: P9659.5 B: ortholog not found; paralogs— RadC family E: Human + |
| Hereditary nonpolyposis colon cancer (mutL homolog)/756: 330-78-348 U07418/120436 | Putative ATP-binding domain shared with HSP90, signal transduction His kinases, and type II topoisomerases (Fig. 2A) | blast2 P = 0.055 (CHVG_AGRTU) psiblastP ≈ 10−4 (numerous histidine kinases, HSP90, and topoisomerases) MoST | ATPase, possibly with autophos- phorylating activity | Protein involved in mismatch repair | C: ortholog not found Y: MLH1_YEAST, PMS1_YEAST B: MutL/HexB family E: Human + Mouse + C. elegans + |
| Ocular albinism/424: 104-320 Z48804/300500 | 7TM receptor (previously thought to contain 6 transmembrane segments) | blast2 P = 0.006 (VIPS_HUMAN) P = 0.01 (CAR1_DICDI, cAMP receptor) PHDhtm (transmembrane helix prediction) | Putative G-protein-coupled receptor | None | C: ortholog not found; weak similarity to CE03862 Y: no ortholog or paralogs B: ortholog not found; limited similarity to YSCS_YERPE E: human + |
| Obesity factor (leptin)/167:96-71 Signal peptide 21-22 U18915/164160 | C-terminal motif conserved in inositol-phosphate synthases | blastP P = 0.91 (INO1_YEAST) MoST—motif from inositol-phosphate synthases detects leptins without false positives, r = 0.001 (P ≈ 0.003) | Possible involvement in inositol signaling | Secreted protein; helical cytokine | C: ortholog not found; conserved motif in inositol phosphate synthase (C47D12.9, gi e225658) Y: no ortholog; conserved motif in inositol- phosphate synthase (YJL153c) B: ortholog or paralogs not found E: Human + |
| Spinal muscular atrophy/294: 171-95-28 U18423/253300 | 2× repeat also found in Drosophila TUD (10× repeat) and HLS proteins, and in human p100 transcriptional coactivator | blast2 P = 0.012 (TUD_DROME) MoST—tudor repeat motif detects the spinal muscular atrophy protein without false positives, r = 0.01 | Repeat motif may be involved in regulatory interactions | None | C: ortholog not found; CE02626 (ortholog of p100) Y: no ortholog or paralogs B: ortholog or paralogs not found E: human + mouse + |
| Spinocerebral ataxia type-1 protein | Domain conserved in rat HBP1 transcription regulator | blast2 P = 10−5 (HBP1, gi|1488627) | Role in transcription regulation? | — | None |
| Thomsen disease¶ 988: 118-85-413-177-88-107 Z25884/160800 | A domain, in the cytoplasmic portion, also found in inositol-5-phosphate dehydrogenases (IMPDH) as a separate, noncatalytic subdomain in their known three-dimensional structure, in cystathionine β synthases, AMP-regulated protein kinases, and several other enzymes and uncharacterized proteins | psiblastP = 2 × 10−5 (hypothetical Methanococcus jannaschii protein, gi|1591551) | Unknown; effector binding? | Voltage-gated chloride channel | C: orthologous family of chloride channels, including CE01212; IMPDH-associated domain in several enzymes Y: YJR040w¶, the orthologous candidate, is a putative chloride channel with modified IMPDH-associated domain B: ortholog not found; YADQ_ECOLI is a putative chloride channel without IMDPH-associated domain E: Human + |
| Werner syndrome/1432: 274-47-57-148-528-106-272 L76937/277700 | N-terminal nuclease domain related to PM-SCL autoantigen, bacterial RNase D, and 3′-5′ proofreading exonuclease domain of bacterial DNA polymerase I (Fig. 2B) | blast2 P = 6 × 10−5 (RNAase D, Synechocystis sp., gi|1001530); psiblastP = 5 × 10−5 (DPO1_HAEIN, DNA polymerase I from H. influenzae) | Nucleasehelicase involved in repair | RecQ-like helicase domain | C: ortholog not found; both domains present, but in separate large proteins (highest similarity to YO63_CAEEL and YMR1_CAEEL) Y: no ortholog; RecQ-like helicases (e.g. SGS1_YEAST) B: ortholog not found; RecQ helicases and RNaseD E: Human + plants + (helicase domain only) |
The total length (number of amino acid residues) of the protein and the lengths of the predicted globular and nonglobular (underlined) domains are indicated; the position of the cleavage site for predicted signal peptides is shown; the GenBank accession number and the NCBI ID are indicated for each disease gene product.
The swiss-prot name (with underline) or the NCBI ID is indicated for homologs.
Orthologs are shown by bold type.
+ indicates 1–20 homologous expressed sequence tags (ESTs) from the given taxon; ++ indicates >20 ESTs.
Fanconi anemia gene codes for a protein with similar functions and domain structure (FACC_HUMAN); it is more likely to be the ortholog of YJR040w than the Thomsen disease gene product; the conserved domain, designated CBS (after cystathionine β synthase), has been recently described independently (30).
Sequence Conservation and Homologs in Model Organisms.
Nearly all of the disease gene products show significant sequence similarity to other proteins in current databases (Fig. 1C and Table 1), though for some, sequence conservation involves only short motifs. Most of the disease genes also are represented by highly similar homologs among the expressed sequence tags and in rodents (ref. 31; http://www.ncbi.nlm.nih.gov/Disease_Genes). Only 16 of the 70 proteins contain domains with significant similarity (P < 0.001) to proteins with known three-dimensional structure (ref. 32; http://www.pdb.bnl.gov). Thus in spite of the rapid growth in the number of known structures, homology modeling is not yet applicable to the majority of disease gene products, and sequence similarity search remains the principal route to functional inference (but see examples below, in which structural implications were made possible by iterative database screening).
We specifically assessed the similarity between the disease gene products and their homologs in extensively sequenced model organisms, namely the nematode C. elegans, yeast S. cerevisiae, and bacteria. In each case, we sought to show which homologs are direct counterparts of the given disease gene product with a high level of similarity and the same domain organization (orthologs), and which showed only a distant similarity and/or are similar only to distinct domains or motifs. The criteria for distinguishing orthologs from paralogs have been discussed (33). Briefly, in a comparison of proteins from two species, orthologs are expected to show: (i) significantly higher similarity to one another than to any other sequence from the second species; (ii) significantly higher similarity to one another than to any sequence from phylogenetically more distant organisms; (iii) alignment through the entire length of the proteins, i.e. conserved domain organization. Each of these criteria is critical for inferring the same function for orthologs of functionally characterized genes.
The majority of positionally cloned gene products showed sequence similarity to proteins from each of the model organisms (Fig. 1C); typically, this similarity was significant by the criteria used in blast2 searches (P < 0.001) but in several cases, only conserved motifs were detected (Table 1 and http://www.ncbi.nlm.nih.gov/Disease_Genes). In all cases when we inferred an orthologous relationship, the similarity between a disease gene product and its apparent ortholog was highly significant (P < 10−12 for C. elegans and yeast orthologs, and P < 10−4 for bacterial orthologs). There are pronounced differences in the representation of the disease gene set by apparent orthologs in the nematode, on one hand, and in yeast and bacteria on the other hand. Though the worm gene collection is only about 50% complete, 36% of the disease genes appear to have orthologs in C. elegans. It is likely that nearly all conserved domain families already are represented among the nematode sequences, but many individual genes are still missing. Thus, with the completion of the C. elegans genome, the fraction of human positionally cloned genes that have worm orthologs is expected to increase substantially.
By contrast, in yeast, and especially in bacteria, apparent orthologs were found only for a few of the disease genes (Fig. 1C, Table 1, and http://www.ncbi.nlm.nih.gov/Disease_Genes). More frequently, the yeast or bacterial homolog contains counterparts to only one domain or a subset of domains of a human protein, and conversely, the proteins from the model organisms may have additional domains of their own (Table 1 and http://www.ncbi.nlm.nih.gov/Disease_Genes).
Domain rearrangements are well documented for a number of protein families, primarily from vertebrates (34). In our analysis of the positionally cloned genes, which may be representative of a significant subset of human genes, we observed that, at large phylogenetic distances, such rearrangements appear to be a rule rather than an exception. This may have important implications for functional interpretation of the results obtained with homologs of disease genes in model organisms. Indeed, sequence comparisons for 29 human genes that have been isolated by functional complementation of yeast mutations indicated that 25 pairs of the human and yeast genes involved are likely orthologs with a conserved domain architecture; in a few cases, human paralogs of the respective yeast genes are also known but they do not complement the mutations (http://www.ncbi.nlm.nih.gov/Bassett/cerevisiae/). As most of the positionally cloned disease genes do not have yeast orthologs (Fig. 1C), they are unlikely to complement mutations in the yeast homologs. This is even more applicable to bacterial systems.
Protein Functional Categories.
More than one-half of the disease genes encode proteins involved in various forms of cell communication and signaling. These include intracellular regulators such as guanine nucleotide-releasing factors and GTPase activators, and membrane and secreted proteins, such as several receptors and transport system components. Genes involved in genome replication, repair, and transcription are represented by a few transcription regulators and DNA repair proteins, whereas proteins involved in translation are absent (http://www.ncbi.nlm.nih.gov/Disease_Genes).
For 11 proteins, no function has been reported or could be confidently predicted by sequence analysis. In most of the other proteins, even though some domains have known or strongly predicted functions, others remain uncharacterized. Only for a small subset of the disease genes, e.g., glycerol kinase or PAX-6 protein (the product of the gene mutated in aniridia), the functions are understood so thoroughly that further insights through computer analysis are unlikely.
Previously Unknown Motifs and Functional Predictions for Positionally Cloned Disease Gene Products.
In this work, previously undetected, conserved domains and motifs were detected in a number of disease gene products. Some of these findings suggest a new function and/or biochemical activity; other motifs await functional characterization. Table 1 contains the data on those of the disease gene products, for which biologically relevant inferences are possible based on the sequence conservation.
ATP-Binding Domain in a Colon Cancer Gene.
The hereditary nonpolyposis colon cancer gene product (MLH1), which is homologous to the bacterial mismatch repair protein MutL (35), is predicted to contain an ATP-binding domain conserved in the HSP90 family of chaperone proteins, type II DNA topoisomerases, and signal-transducing histidine kinases (Fig. 2A). A position-dependent weight matrix produced by psiblast from the part of the blast2 output that included only highly conserved sequences of MutL homologs retrieved numerous sequences of histidine kinases, type II (ATP-dependent) DNA topoisomerases, and HSP90 from the NR database selectively and with low P values of 10−4–10−5. Each of the three blocks with the highest sequence similarity between MutL, HSP90, topoisomerases, and histidine kinase families corresponds to one of the conserved motifs in the histidine kinases (44).
Figure 2.

Previously undetected conserved domains and motifs in positionally cloned disease gene products. Alignments were constructed using the macaw program. Unique identifiers for each sequence are shown. Distances to the ends of the proteins and distances between the aligned, conserved blocks are shown by numbers. Conserved bulky hydrophobic residues (I, L, M, V, F, Y, W) are indicated by yellow shading and by U in the consensus. Other conserved residues are shown in magenta. Other designations in the consensus: O, small residues (A, G, or S); +, basic residues (K and R); −, acidic residues (D and E). (A) A putative ATP-binding domain in hereditary nonpolyposis colon cancer gene product (HML1), MutL mismatch repair proteins, HSP90 chaperones, type II DNA topoisomerases, and bacterial histidine kinases. The sequences were from the swiss-prot database: MLH1__HUMAN, human MutL homolog, colon cancer susceptibility gene product; PMS1__YEAST and MLH1__YEAST, yeast mismatch repair gene products homologous to MutL; HEXB__STRPN, mismatch repair gene product from Streptococcus pneumoniae; MUTL__ECOLI, E. coli mismatch repair gene mutL product; HTPG__HAEIN, HTPG__BACSU, HS90__THEPA, HS90__CANAL, and HS9A__HUMAN, molecular chaperones of the HSP90 family from Haemophilus influenzae, Bacillus subtilis, Theileria parva, Candida albicans, and human; TOPB__HUMAN, TOP2__DROME, GYRB__HALSQ, PARE__ECOLI, and GYRB__ECOLI, type II topoisomerases from human, Drosophila melanogaster, Haloferax sp., and E. coli; SPHS__SYNP7 and PHOR__BACSU, histidine kinases involved in inducible alkaline phosphatase production from Synechococcus sp. and Bacillus subtilis; PILS__PSEAE, histidine kinase involved in fimbriae biogenesis from Pseudomonas aeruginosa; PHY1__TOBAC, tobacco phytochrome A1 (histidine kinase homolog); ENVZ__ECOLI, osmolarity sensor histidine kinase. Three motifs described in histidine kinases and phenotypes of E. coli envZ mutants (36) are shown below the alignment. Dominant-negative mutations in MutL protein (37) are indicated by gray shading. Asterisks indicate amino acid residues that in E. coli GyrB are in direct contact with ATP; two of such residues are in the spacer between motifs G1 and G2 (38). The secondary structure assignments are from the crystal structure of the N-terminal fragment of E. coli GyrB (38); h, α-helix, e, extended conformation (β-sheet), and l, loop. (B) A putative nuclease domain conserved in Werner syndrome gene product (WRNp), bacterial RNase D, and DNA polymerase I. The sequences were from swiss-prot (names with underlines) or from GenBank (National Center for Biotechnology Information accession numbers indicated below). PMSC__HUMAN, human polymyositis and scleroderma autoantigen; RND__HAEIN, RND__ECOLI, and RND/SYNSP (gi 1001530), RNase D from H. influenzae, E. coli, and Synechocystis sp.; ORF/SCHPO (gi 1256512), uncharacterized ORF product from Schizosaccharomyces pombe; DPO1__ECOLI, DPO1__HAEIN, DPO1/SYNSP, DPO1/TREPA, and DPO1/BPT5, DNA polymerase I from E. coli, H. influenzae, Synechocystis sp., Treponema pallidum, and bacteriophage T5. The structural assignments are from the Klenow fragment structure (39); the designations are as in A. The three aspartates that coordinate the two cations required for the 3′-5′ exonuclease reaction by the Klenow fragment are indicated by asterisks; the two residues directly involved in catalysis (40, 41) are indicated by exclamation marks. ExoI, ExoII, and ExoIII indicate the three motifs that are conserved throughout the nuclease superfamily (42, 43).
The ATP-binding site of E. coli DNA gyrase maps to the N-terminal fragment of the B subunit, and the crystal structure of this fragment complexed with an ATP analog has been determined (38). Each of the conserved motifs in our alignment contains residues that are in direct contact with ATP (Fig. 2A). In the gyrase, the loop in motif G2 interacts with the phosphates, the conserved Asp in G1 interacts with the amino group of adenine, and Asn in motif N is involved in Mg2+ coordination (ref. 45; Fig. 2A). The flexible loop in G1, which is the most conserved element in the four protein families, is not in direct contact with ATP, and its function will be of further interest.
These findings are compatible with the prediction of an ATP-binding domain in MLH1 and its homologs and suggest that the function of these proteins in DNA repair includes ATP hydrolysis and phosphoryl transfer; furthermore, MLH1 may be autophosphorylated as demonstrated for histidine kinases and HSP90 (46, 47). The recent analysis of random dominant-negative mutants of the E. coli MutL protein (37) showed that most of the mutations concentrated in the three conserved motifs in the predicted ATPase domain (Fig. 2A).
A Nuclease Domain in Werner Syndrome Protein.
A N-terminal nuclease domain homologous to bacterial RNAase D and 3′-5′ proofreading exonuclease domain of bacterial DNA polymerase I (PolA) was predicted in the Werner syndrome gene product (WRNp; ref. 48), which also contains a C-terminal helicase domain (Fig. 2B). The N-terminal, globular domain of WRNp showed significant similarity to the Synechocystis sp. RNase D (P value 6 × 10−5). The subsequent psiblast search detected the similarity to other RNases D and to the 3′-5′ proofreading exonuclease domain of PolA (Fig. 2B and Table 1); the latter belongs to a superfamily of exonuclease domains in a variety of DNA polymerases, which also includes bacterial RNAase T (42, 43). The crystal structure of the Klenow fragment of PolA including the 3′-5′ exonuclease domain has been determined (39), and the residues involved in the positioning of the two divalent cations required for catalysis and in the phosphodiester bond cleavage have been defined by structural analysis and site-directed mutagenesis (40, 41). All these residues are conserved in WRNp (Fig. 3B), suggesting that this protein possesses an active exonuclease domain, in addition to the helicase domain (Fig. 2B). The combination of predicted nuclease and helicase domains suggests that WRNp may be involved in DNA repair or RNA processing. Interestingly, a homologous nuclease domain was found in the human polymyositis/scleroderma autoantigen, a nucleolar protein (ref. 45; Fig. 2B); scleroderma symptoms are prominent in Werner syndrome patients (OMIM 277700).
CONCLUSION
Application of an iterative strategy, which combines protein sequence segmentation, enhanced versions of blast search, methods for motif analysis, and specialized databases, to the analysis of protein sequences encoded by positionally cloned human disease genes resulted in the detection of a number of previously undetected, to our knowledge, conserved motifs and several predictions of disease gene functions. Most of the disease gene products show significant similarity to proteins from the nematode C. elegans, yeast, and bacteria. Only in the nematode, apparent orthologs with conserved domain organization were detected for the majority of the disease gene products. In the yeast and bacterial homologs, changes in domain architecture are typical. The choice of an optimal system for studying human gene functions will depend on whether the human gene has an ortholog or only paralogs in a particular model organism.
Acknowledgments
We thank Roman Tatusov and Roland Walker for writing some of the programs used in this work; Stephen Altschul, David Lipman, Tom Madden, and Alejandro Schaffer for providing the unpublished program psiblast, and Victor McKusick for helpful discussions.
ABBREVIATION
- NR
nonredundant
Footnotes
These genes are frequently referred to as “disease genes,” and for the sake of brevity, we will use this expression in the rest of this paper.
References
- 1.Collins F S. Nat Genet. 1995;9:347–350. doi: 10.1038/ng0495-347. [DOI] [PubMed] [Google Scholar]
- 2.Bassett D E, Jr, Boguski M S, Spencer F, Reeves R, Goebl M, Hieter P. Trends Genet. 1995;11:372–373. doi: 10.1016/s0168-9525(00)89109-x. [DOI] [PubMed] [Google Scholar]
- 3.Bassett D E, Jr, Boguski M S, Hieter P. Nature (London) 1996;379:589–590. doi: 10.1038/379589a0. [DOI] [PubMed] [Google Scholar]
- 4.McKusick V A. Mendelian Inheritance in Man: Catalogs of Human Genes and Genetic Disorders. 11th Ed. Baltimore: Johns Hopkins Univ. Press; 1993. [Google Scholar]
- 5.Koonin E V, Mushegian A R. Curr Opin Genet Dev. 1996;6:757–762. doi: 10.1016/s0959-437x(96)80032-3. [DOI] [PubMed] [Google Scholar]
- 6.Waterston R, Sulston J. Proc Natl Acad Sci USA. 1995;92:10836–10840. doi: 10.1073/pnas.92.24.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tugendreich S, Bassett D E, Jr, McKusick V A, Boguski M S, Hieter P. Hum Mol Genet. 1994;3:1509–1517. doi: 10.1093/hmg/3.suppl_1.1509. [DOI] [PubMed] [Google Scholar]
- 8.Hieter P, Bassett D E, Jr, Valle D. Nat Genet. 1996;13:263–265. doi: 10.1038/ng0796-253. [DOI] [PubMed] [Google Scholar]
- 9.Fitch W M. Syst Zool. 1970;19:99–106. [PubMed] [Google Scholar]
- 10.Bork P, Gibson T J. Methods Enzymol. 1996;266:162–182. doi: 10.1016/s0076-6879(96)66013-3. [DOI] [PubMed] [Google Scholar]
- 11.Bork P, Koonin E V. Curr Opin Struct Biol. 1996;6:366–376. doi: 10.1016/s0959-440x(96)80057-1. [DOI] [PubMed] [Google Scholar]
- 12.Mushegian A R, Koonin E V. Genetics. 1996;144:817–828. doi: 10.1093/genetics/144.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meitinger T, Meindl A, Bork P, Rost B, Sander C, Haasemann M, Murken J. Nat Genet. 1993;5:376–380. doi: 10.1038/ng1293-376. [DOI] [PubMed] [Google Scholar]
- 14.Madej T, Boguski M S, Bryant S H. FEBS Lett. 1995;373:13–18. doi: 10.1016/0014-5793(95)00977-h. [DOI] [PubMed] [Google Scholar]
- 15.Koonin E V, Altschul S F, Bork P. Nat Genet. 1996;13:266–268. doi: 10.1038/ng0796-266. [DOI] [PubMed] [Google Scholar]
- 16.Boguski M S, Lowe T M, Tolstoshev C M. Nat Genet. 1993;4:232–233. doi: 10.1038/ng0893-332. [DOI] [PubMed] [Google Scholar]
- 17.Schuler G D, Epstein J A, Ohkawa H, Kans J A. Methods Enzymol. 1996;266:141–162. doi: 10.1016/s0076-6879(96)66012-1. [DOI] [PubMed] [Google Scholar]
- 18.Wootton J C, Federhen S. Methods Enzymol. 1996;266:554–573. doi: 10.1016/s0076-6879(96)66035-2. [DOI] [PubMed] [Google Scholar]
- 19.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 20.Altschul S F, Gish W. Methods Enzymol. 1996;266:460–480. doi: 10.1016/s0076-6879(96)66029-7. [DOI] [PubMed] [Google Scholar]
- 21.Altschul S F, Boguski M S, Gish W, Wootton J C. Nat Genet. 1994;6:119–129. doi: 10.1038/ng0294-119. [DOI] [PubMed] [Google Scholar]
- 22.Tatusov R L, Altschul S F, Koonin E V. Proc Natl Acad Sci USA. 1994;91:12091–12095. doi: 10.1073/pnas.91.25.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuler G D, Altschul S F, Lipman D J. Proteins Struct Funct Genet. 1991;9:180–190. doi: 10.1002/prot.340090304. [DOI] [PubMed] [Google Scholar]
- 24.Eddy S R, Mitchison G, Durbin R. J Comput Biol. 1995;2:9–23. doi: 10.1089/cmb.1995.2.9. [DOI] [PubMed] [Google Scholar]
- 25.Rost B, Sander C. Proteins Struct Funct Genet. 1994;19:55–72. doi: 10.1002/prot.340190108. [DOI] [PubMed] [Google Scholar]
- 26.Rost B, Casadio R, Fariselli P, Sander C. Protein Sci. 1995;4:521–533. doi: 10.1002/pro.5560040318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 28.Lupas A, Van Dyke M, Stock J. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 29.Bairoch A, Apweiler R. Nucleic Acids Res. 1996;24:21–25. doi: 10.1093/nar/24.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bateman A. Trends Biochem Sci. 1997;22:12–13. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- 31.Makalowski W, Zhang J, Boguski M S. Genome Res. 1996;6:846–856. doi: 10.1101/gr.6.9.846. [DOI] [PubMed] [Google Scholar]
- 32.Bernstein F C, Koetzle T F, Williams G J B, Meyer E F, Brice M D, Rodgers J R, Kennard O, Shimanouchi T, Tasumi M. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 33.Tatusov R L, Mushegian A R, Bork P, Brown N P, Hayes W, Borodovsky M, Rudd K E, Koonin E V. Curr Biol. 1996;6:279–291. doi: 10.1016/s0960-9822(02)00478-5. [DOI] [PubMed] [Google Scholar]
- 34.Doolittle R F. Annu Rev Biochem. 1995;64:287–314. doi: 10.1146/annurev.bi.64.070195.001443. [DOI] [PubMed] [Google Scholar]
- 35.Kolodner R. Genes Dev. 1996;10:1433–1442. doi: 10.1101/gad.10.12.1433. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y, Inouye M. J Mol Biol. 1993;231:335–342. doi: 10.1006/jmbi.1993.1286. [DOI] [PubMed] [Google Scholar]
- 37.Aronshtam A, Marinus M G. Nucleic Acids Res. 1996;24:2498–2504. doi: 10.1093/nar/24.13.2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wigley D B, Davies G J, Dodson E J, Maxwell A, Dodson G. Nature (London) 1991;351:624–629. doi: 10.1038/351624a0. [DOI] [PubMed] [Google Scholar]
- 39.Ollis D L, Brick P, Hamlin R, Xuong N G, Steitz T A. Nature (London) 1985;313:762–766. doi: 10.1038/313762a0. [DOI] [PubMed] [Google Scholar]
- 40.Derbyshire V, Grindley N D F, Joyce C M. EMBO J. 1991;10:17–24. doi: 10.1002/j.1460-2075.1991.tb07916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beese L S, Steitz T. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blanco L, Bernad A, Blasco M A, Salas M. Gene. 1991;100:27–38. doi: 10.1016/0378-1119(91)90346-d. [DOI] [PubMed] [Google Scholar]
- 43.Koonin E V, Deutscher M P. Nucleic Acids Res. 1993;21:2521–2522. doi: 10.1093/nar/21.10.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stock J B, Ninfa A J, Stock A M. Microbiol Rev. 1989;53:450–490. doi: 10.1128/mr.53.4.450-490.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bluthner M, Bautz F A. J Exp Med. 1992;176:973–980. doi: 10.1084/jem.176.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iuchi S, Lin E C C. J Bacteriol. 1992;174:5617–5623. doi: 10.1128/jb.174.17.5617-5623.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nadeau K, Das A, Walsh C T. J Biol Chem. 1993;268:1479–1487. [PubMed] [Google Scholar]
- 48.Lombard D B, Guarente L. Trends Genet. 1996;12:283–286. doi: 10.1016/0168-9525(96)30065-6. [DOI] [PubMed] [Google Scholar]