

Abstract
The chicken anemia virus protein apoptin induces a p53-independent, Bcl-2-insensitive type of apoptosis in various human tumor cells. Here, we show that, in vitro, apoptin fails to induce programmed cell death in normal lymphoid, dermal, epidermal, endothelial, and smooth-muscle cells. However, when normal cells are transformed they become susceptible to apoptosis by apoptin. Long-term expression of apoptin in normal human fibroblasts revealed that apoptin has no toxic or transforming activity in these cells. In normal cells, apoptin was found predominantly in the cytoplasm, whereas in transformed and malignant cells it was located in the nucleus, suggesting that the localization of apoptin is related to its activity. These properties make apoptin a potential agent for the treatment of a large number of tumors, also those lacking p53 and/or overexpressing Bcl-2.
Keywords: programmed cell death, tumor cells, antitumor agent
Apoptosis (or programmed cell death) is an active and programmed physiological process for eliminating superfluous, altered, or malignant cells (1). Apoptosis is characterized by shrinkage of cells, segmentation of the nucleus, condensation, and cleavage of DNA into domain-sized fragments in most cells followed by internucleosomal degradation. Finally, the apoptotic cells fragment into membrane-enclosed bodies, which are rapidly phagocytosed by neighboring cells (2–4).
The apoptotic process can be initiated by a variety of regulatory stimuli (5, 6). Changes in cell-survival rate play an important role in human pathogenesis, e.g. in cancer development, which is caused by enhanced cell proliferation but also by decreased cell death (7–10). A variety of chemotherapeutic agents and radiation have been demonstrated to induce apoptosis in tumor cells, in many instances via the action of wild-type p53 (11–15). Most tumors, however, acquire a mutation in p53 during their development, often correlated with poor response to cancer therapy (16–18). In several leukemias a high expression level of the proto-oncogene Bcl-2 is associated with a strong resistance to various apoptosis-inducing chemotherapeutic agents (19–22). Apoptin, a 14-kDa basic and proline-rich protein (23) derived from the chicken anemia virus, can induce apoptosis in chicken and human malignant cell lines (24, 25). We have established that apoptosis induced by apoptin is p53-independent (26) and cannot be blocked by Bcl-2 (27, 28). Therefore, apoptin is a potential antitumor agent. To explore this possibility further, we have examined the in vitro apoptotic activity of apoptin in normal human cells versus transformed human cells.
MATERIALS AND METHODS
Cell Culture.
Human primary T cells were isolated from six normal blood donors by Ficol centrifugation and grown in RPMI medium 1640 containing 6% human serum and 0.8 μg/ml phytohemagglutinin. After 3 days, the medium was changed, and 300 units/ml of interleukin-2 were added (29). Human umbilical-cord vascular endothelial cells (HUVEC) and smooth-muscle cells (HSMC) were isolated from umbilical cords as described (30). HUVEC were grown in M199 medium supplemented with 10% human serum, 10% newborn calf serum, 150 μg/ml epidermal growth factor, and 5 units/ml heparin. HSMC were grown in DMEM containing 10% fetal calf serum. Human keratinocytes were isolated from foreskin and grown in the presence of a feeder layer of lethally irradiated 3T3 fibroblasts. Primary cultures of human epidermal keratinocytes (FSK-1) were initiated in complete medium as described (31) with minor modifications by M. Ponec et al. (32) and cultured in keratinocyte serum-free medium afterward. For the experiments described here, passage no. 3 was used. Human diploid foreskin fibroblasts (VH10) (33) were grown in DMEM supplemented with 10% fetal calf serum.
Simian virus 40 (SV40)-transformed tumorigenic fibroblasts (NW-18) (34) were grown in MEM supplemented with 8% fetal calf serum. Tumorigenic keratinocytes (SCC-15) (35), derived from squamous-cell carcinoma, were cultured in DMEM/F12 (3:1) medium containing 5% fetal calf serum and 0.4 μg/ml hydrocortisone.
VH10 fibroblasts were transformed with SV40 early-region DNA, to generate nonimmortalized, precrisis (pre) cells, from which immortalized, postcrisis (post) cells were isolated, as described by B. Klein et al. (33). The cells were grown in MEM with 8% fetal calf serum. The spontaneously transformed keratinocyte strain HaCaT (36) was a gift from N. Fusenig (Deutsches Krebsforschungszentrum, Heidelberg, Germany). HaCaT cells were grown in DMEM supplemented with 10% fetal calf serum. The SV40-transformed keratinocyte strain SVK14 (37) was kindly provided by M. Ponec (Academic Hospital, Leiden, The Netherlands) and was cultured in the same medium as SCC-15 cells, but supplemented with 1 μM isoproterenol.
All culture media were obtained from GIBCO/BRL and contained the antibiotics penicillin and streptomycin.
Plasmids.
The expression plasmid pCMV-fs, formerly called pCMV-VP3, contains chicken anemia virus DNA sequences encoding apoptin exclusively (nt 427–868), plasmid pCMV-tr encodes a truncated apoptin that lacks the C-terminal 11 amino acids (24), and pCMV-des encodes desmin, a structural protein of muscle cells (T. van Laar, personal communication). All three genes are under the regulation of the cytomegalovirus (CMV) promoter. Plasmid DNA was purified by centrifugation in a CsCl gradient and column chromatography in Sephacryl S500 (Pharmacia).
Transient Transfection.
Human CD3-positive T cells, which were supported by interleukin-2 after phytohemagglutinin stimulation, were transfected in the presence of DEAE-dextran, as described (24). VH10, pre, post, and NW-18 cells, HUVEC and HSMC were transfected with plasmid DNA by calcium-phosphate precipitation as described (38). FSK-1, HaCaT, SVK14, and SCC-15 cells were transfected with transfection agent N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate (DOTAP) (Boehringer Mannheim), essentially as described by Fischer et al. (39).
Stable Transfection.
Human diploid foreskin fibroblasts (VH10) were stably transfected with pCMV-fs or pCMV-neo-Bam; the latter is the “empty” control plasmid expressing the neomycin-resistance gene only. Stable clones were selected with medium containing G418, grown on glass microscope slides, and fixed with 80% acetone. Expression of apoptin was analyzed by indirect immunofluorescence.
Indirect Immunofluorescence.
T cells were grown in suspension and fixed on glass microscope slides. All other cells were grown on glass microscope slides. The slides were either uncoated (VH10, pre, post, NW-18) or coated with 3-amino-propyltriethoxysilane (FSK-1, HaCaT, SVK14, and SCC-15) or with gelatin (HUVEC and HSMC). The cells were fixed with 80% acetone for 10 min at room temperature and used for indirect immunofluorescence as described (40). To demonstrate the presence and/or cellular localization of (truncated) apoptin in transfected cells mouse mAbs CVI-CAV-85.1 (85.1) (23) and CVI-CAV-111.3 (111.3) (G. Koch, personal communication) were used, and for human desmin the mouse mAb 33 (Monosan, Uden, The Netherlands) was used. Fluorescein isothiocyanate-labeled goat anti-mouse antibodies (Jackson Immunoresearch) were used as second antibodies. Nuclear DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) or propidium iodide (PI).
RESULTS AND DISCUSSION
In a first set of experiments, three types of normal diploid human cells, i.e. HUVEC, HSMC, and CD3-positive T cells, which were supported by interleukin-2 after phytohemagglutinin stimulation, were transiently transfected with a plasmid encoding full-sized apoptin (pCMV-fs). Cells expressing apoptin were screened via indirect immunofluorescence with mAb 85.1 (23) or 111.3 (G. Koch, personal communication). Induction of apoptosis in apoptin-positive cells was analyzed with the help of DAPI or PI, which cause a regular staining in intact nuclei, but an irregular and/or weak staining in apoptotic cells (41). Five days after transfection, approximately 20% of the apoptin-expressing normal cells stained abnormally with DAPI or PI (data not shown). This is significantly lower than the mean of 75% previously found in malignant cells (24, 26–28). The localization of apoptin in these normal cells also differed from the localization in tumor cells. In all of the normal cells, apoptin was found in the cytoplasm (Fig. 1), and not in the nucleus as has been observed for various tumor cell lines (24, 26–28). These data suggest that apoptin fails to induce apoptosis in cultured human nontransformed, nontumorigenic cells, and that the cellular localization of apoptin is important for its apoptotic activity.
Figure 1.
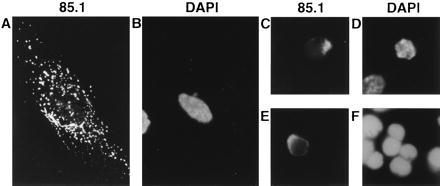
Localization of apoptin and staining of the chromatin in three normal human cell types, transiently transfected with pCMV-fs. Apoptin was stained with anti-apoptin mAb 85.1 (A, C, and E) and the DNA was stained with DAPI (B and D) or PI (F), in representations of identical cells: (A and B) HSMC, (C and D) HUVEC, and (E and F) T cells. Cells were fixed 5 days after transfection and analyzed by indirect immunofluorescence. (Original magnification: A and B, ×630; C–F, ×1,000.)
To verify these findings we have directly compared the effect of apoptin expression in tumor cell lines versus that in the normal, untransformed cells. To that end, diploid skin fibroblasts (VH10) and epidermal keratinocytes (FSK-1) from healthy individuals and their tumorigenic counterparts (NW-18 and SCC-15) were transfected transiently with pCMV-fs or pCMV-tr. The latter plasmid encodes a truncated apoptin protein, which lacks one of its putative nuclear-localization signals and is partially disturbed in its nuclear transport as is also evident from its reduced apoptotic activity (24, 26).
As a negative control, cells were transfected with a plasmid-encoding desmin (pCMV-des) (T. van Laar, personal communication), which does not have apoptotic activity (42). Five days after transfection, the percentage of apoptin-positive VH10 fibroblasts (Fig. 2A) and FSK-1 keratinocytes (Fig. 2B) that had become apoptotic was approximately 15%. This level of aberrantly DAPI-stained cells was similar to that of cells containing truncated apoptin or desmin. Consequently, the low level of apoptosis in these apoptin-positive cells is probably due to the transfection procedure but not to the action of apoptin. To examine whether tumorigenic fibroblasts and keratinocytes are susceptible to apoptin, NW-18 and SCC-15 cells were transfected with plasmids encoding apoptin (full-sized or truncated) or desmin. Apoptin and, to a lesser extent, truncated apoptin could induce apoptosis in the NW-18 (Fig. 2A) and SCC-15 (Fig. 2B) tumor cells. Up to 65–75% of the apoptin-positive cells were apoptotic at 5 days after transfection, which is in a similar range as reported for apoptin-induced cell death in osteosarcoma cell lines (26). The proportion of abnormally DAPI-stained desmin-positive NW-18 and SCC-15 cells was not significantly higher than that among normal fibroblasts and keratinocytes (Fig. 2). The differential activity of apoptin in normal and tumor cells cannot be explained by differences in proliferation rates, as these were similar for VH10 fibroblasts and Saos-2 osteosarcoma cells (data not shown) and for normal keratinocytes and SCC-15 cells (35).
Figure 2.
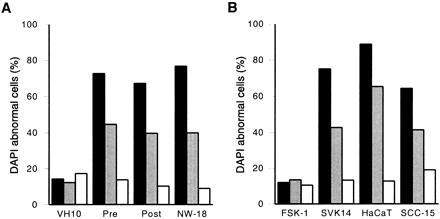
Apoptin activity in normal versus transformed/malignant cells. The percentage of cells that stained abnormally with DAPI is given as a relative measure for apoptosis in (A) normal (VH10) versus transformed (pre, post) and tumor (NW-18) fibroblasts and (B) normal (FSK-1) versus transformed (SVK14, HaCaT) and tumor (SCC-15) keratinocytes transiently transfected with pCMV-fs (solid bars), pCMV-tr (shaded bars), or pCMV-des (open bars). Cells were fixed 5 days after transfection and analyzed by indirect immunofluorescence. Results are the means of at least three independent experiments. In each experiment at least 200 cells were examined that were positive for full-sized or truncated apoptin, or for desmin.
Our analysis shows that apoptin does not induce apoptosis in normal fibroblasts and keratinocytes, but does so in tumorigenic derivatives of these cell types. To answer the question whether induction of cell death by apoptin requires a tumorigenic phenotype or whether a nonmalignant transformed phenotype is sufficient, we have examined the effect of apoptin in transformed, nontumorigenic fibroblasts and keratinocytes. Apoptin, truncated apoptin, and desmin were transiently expressed in SV40-transformed VH10 fibroblasts (derived from the untransformed VH10 cells), before (pre) or after (post) immortalization, in SV40-transformed and immortalized SVK14 keratinocytes, and in spontaneously transformed HaCaT keratinocytes. All of these cells are nontumorigenic and have retained a normal proliferation and differentiation pattern. Apoptin was able to induce apoptosis in all of these transformed cell types, to a similar extent as in tumor cells (Fig. 2). The percentage of apoptosis in cells containing (truncated) apoptin was now also significantly higher than in desmin-positive cells.
It is important to note that at 5 days after transfection the majority of tumor and/or transformed cells underwent apoptosis due to expression of apoptin. This percentage increased even further when cells were analyzed at 6–7 days, when almost 100% of the cells were affected, indicating that all cells expressing apoptin eventually will be killed.
To exclude that in normal cells apoptin has a delayed apoptotic activity that might not be visible in a transient transfection assay, normal VH10 cells were stably transfected with pCMV-fs or with pCMV-neo-Bam as a control. A similar number of colonies for pCMV-fs and the control was found in two independent experiments. Several pCMV-fs-transfected and control colonies were isolated and expanded according to the standard cell culture procedure (for diploid fibroblasts). The cultures obtained from these colonies then were passaged for 6 weeks; during this period the two types of cells behaved similarly and underwent about 10 population doublings. Analysis by immunofluorescence showed that the pCMV-fs-transfected cells, as opposed to the control cells, exhibited the characteristic cytoplasmic fluorescence with antiapoptin serum (data not shown). Subsequently, several passages later, both apoptin-expressing and control colonies became senescent. Therefore, it can be concluded that normal VH10 cells are resistant to apoptin-induced apoptosis and that their limited lifespan is not affected by apoptin. This also indicates that apoptin has no transforming activity in VH10 cells.
Our data imply that apoptin can induce apoptosis in transformed and malignant cells in vitro, but fails to do so in normal cells. In contrast, cytotoxic drugs and radiation preferentially kill proliferating cells in culture, irrespective of whether they are normal or transformed (43). Thus apoptin appears to be the first protein known to specifically induce apoptosis in tumor cells. The parvovirus structural protein NS1 also induces cell lysis preferentially in neoplastic cells, however, it is unclear as yet whether NS1 kills cells via apoptosis (44).
Apart from the different apoptotic activities of apoptin in both malignant and transformed cells versus normal cells, we also have observed differences in the cellular localization of apoptin in these cell types. In transformed and malignant cells, before the apoptotic morphological changes are noticeable, apoptin is distributed as fine granules, mainly in the nucleus (Fig. 3E). After the cells have undergone apoptosis, apoptin is aggregated in the nucleus (Fig. 3 G, I, and K). In contrast, the localization of apoptin in normal cells is mainly cytoplasmic, concentrated around the nucleus, both as small granules and larger aggregates (Figs. 1 and 3 A and C). Thus far, we have not encountered a single exception to this rule; in all cases apoptin has a nuclear localization in human transformed or tumorigenic cells and a cytoplasmic localization in normal cells. It has been proposed that transformed cells have undergone alterations in their transport system, as a result of which a normally functioning inhibitor has been switched off. Due to these changes, nuclear transport of proteins may be promoted or hindered in cancer cells (45). It can be hypothesized that in normal cells apoptin is associated to and/or modified by one or more cellular factor(s), resulting in its localization within perinuclear structures. The loss of such (functional) factor(s) in transformed/malignant cells might allow apoptin to enter the nucleus. Apoptin harbors not only two putative nuclear-import sequences (23–28), but also an amino acid region resembling nuclear export signals (position 33–46: IRIGIAGITITLSL), similar to that of the protein kinase inhibitor and the HIV-Rev protein (46–48). It might well be that this potential nuclear export signal cannot be recognized in the various malignant and transformed cell lines analyzed.
Figure 3.
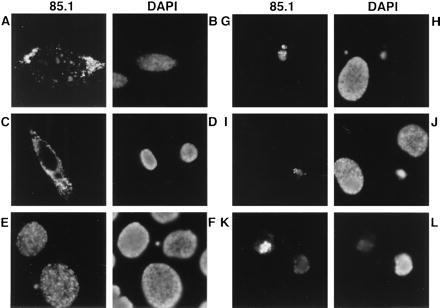
Localization of apoptin and staining of the chromatin in normal and transformed/malignant cells transiently transfected with pCMV-fs. Apoptin was stained with anti-apoptin mAb 85.1 and DNA with DAPI in representations of identical cells: (A and B) VH10, (C and D) FSK-1, (E and F) HaCaT, (G and H) post, (I and J) NW-18, and (K and L) SCC-15. Cells were fixed 2 (E and F) or 5 (A–D and G–L) days after transfection and analyzed by indirect immunofluorescence. (Original magnification: A–D, ×630; I–L, ×1,000; and E–H, ×1,250.)
The results described here suggest that the nuclear localization of apoptin is important for its ability to induce apoptosis. This is in agreement with the observation that truncated apoptin has reduced activity because it lacks one of the putative nuclear-localization signals and, therefore, is transported less efficiently into the nucleus as can be concluded from the fact that more than half of the staining is cytoplasmic (data not shown; ref. 26) (Fig. 2). Truncation does not completely block the apoptotic activity, but seems to slow down the rate of apoptosis (26). Electron microscopic studies with chicken mononuclear cells revealed that apoptin colocalizes with cellular chromatin (24). The interaction of chromatin with apoptin could result in unwinding of its superstructure. The latter phenomenon has been described for rat ventral prostate cells, which became apoptotic after castration of the rats (49).
Our present and earlier results indicate that apoptin is a potential antitumor agent for the following reasons. First, apoptin is specifically active in malignant and transformed cells, but not in normal cells, at least in vitro. Therefore, the toxic effects of apoptin treatment in vivo might be very low. Second, apoptin induces apoptosis in a p53-independent manner (26). The fact that several chemotherapeutic agents lack the capacity to induce apoptosis in a large number of tumors appears to be related to a disruption of p53 function (11–18). Therefore, induction of a p53-independent apoptotic pathway might be a useful alternative in tumor therapy. Furthermore, the activity of apoptin is not blocked by Bcl-2 (27, 28), which is known to be involved in the development of, for instance, leukemic tumors and which can inhibit apoptosis induced by chemotherapeutic agents (19–22). Recently, we have observed that BAG-1, a Bcl-2-related protein, cannot block apoptin-induced apoptosis, either (A.A.A.M.D.-V.O., unpublished data). At the moment, we are developing gene-therapy strategies to deliver apoptin into tumor cells in vivo.
Acknowledgments
We thank Prof. Dr. N. Fusenig (Deutsches Krebforschungszentrum, Heidelberg, Germany) for kindly providing HaCaT cells and K. van Bergen for isolating primary T cells. Dr. G. Koch (Institute for Animal Science and Health, Lelystad, The Netherlands) is acknowledged for the kind gift of mAbs 85.1 and 111.3, and Dr. H. van Ormondt for critically reading this manuscript. This research was partially made possible by grants from The Netherlands Ministry of Economic Affairs, Aesculaap Beheer BV, Boxtel, and the Dutch Cancer Foundation, Amsterdam, The Netherlands.
ABBREVIATIONS
- HUVEC
human umbilical-cord vascular endothelial cells
- HSMC
human smooth-muscle cells
- SV40
simian virus 40
- DAPI
4′,6-diamidino-2-phenylindole
- CMV
cytomegalovirus
- post
postcrisis cells
- pre
precrisis cells
- PI
propidium iodide
References
- 1.Earnshaw W C. Curr Opin Cell Biol. 1995;7:337–343. doi: 10.1016/0955-0674(95)80088-3. [DOI] [PubMed] [Google Scholar]
- 2.Wyllie A H, Kerr J F R, Currie A R. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 3.Arends M J, Wyllie A H. Int Rev Exp Pathol. 1991;32:223–254. doi: 10.1016/b978-0-12-364932-4.50010-1. [DOI] [PubMed] [Google Scholar]
- 4.Roy C. Exp Cell Res. 1992;200:416–424. doi: 10.1016/0014-4827(92)90190-j. [DOI] [PubMed] [Google Scholar]
- 5.Wyllie A H. Curr Opin Genet Dev. 1995;5:97–104. doi: 10.1016/s0959-437x(95)90060-8. [DOI] [PubMed] [Google Scholar]
- 6.White E. Genes Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- 7.Kerr J F R, Winterford C M, Harmon B V. Cancer. 1994;73:2013–2026. doi: 10.1002/1097-0142(19940415)73:8<2013::aid-cncr2820730802>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 8.Thompson C B. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 9.Bellamy C O C, Malcomson R D G, Harrison D J, Wyllie A H. Semin Cancer Biol. 1995;6:3–16. doi: 10.1006/scbi.1995.0002. [DOI] [PubMed] [Google Scholar]
- 10.Steller H. Science. 1995;267:1445–1449. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 11.Kaufman S H. Cancer Res. 1989;49:5870–5879. [Google Scholar]
- 12.Warters R L. Cancer Res. 1992;52:883–890. [PubMed] [Google Scholar]
- 13.McDonell T J, Meyn R E, Robertson L E. Semin Cancer Biol. 1995;6:53–60. doi: 10.1006/scbi.1995.0007. [DOI] [PubMed] [Google Scholar]
- 14.Lowe S W, Ruley H E, Jacks T, Housman D E. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 15.Fisher D E. Cell. 1994;78:539–542. doi: 10.1016/0092-8674(94)90518-5. [DOI] [PubMed] [Google Scholar]
- 16.Hooper M L. J Cell Sci Suppl. 1994;18:13–17. doi: 10.1242/jcs.1994.supplement_18.3. [DOI] [PubMed] [Google Scholar]
- 17.Lowe S W, Bodis S, McClatchey A, Remington L, Ruley H E, Fisher D E, Housman D E, Jacks T. Science. 1994;266:807–810. doi: 10.1126/science.7973635. [DOI] [PubMed] [Google Scholar]
- 18.Smith M L, Fornace A J., Jr Curr Opin Onc. 1995;7:69–75. [PubMed] [Google Scholar]
- 19.Hockenberry D M. J Cell Sci Suppl. 1994;18:51–55. doi: 10.1242/jcs.1994.supplement_18.7. [DOI] [PubMed] [Google Scholar]
- 20.Wyllie A H. Cancer Metastasis Rev. 1992;11:95–103. doi: 10.1007/BF00048057. [DOI] [PubMed] [Google Scholar]
- 21.Sachs L, Lotem J. Blood. 1993;82:15–21. [PubMed] [Google Scholar]
- 22.Dive C, Wyllie A H. In: Frontiers in Pharmacology: Cancer Chemotherapy. Hickman J A, Tritton T T, editors. Oxford: Blackwell; 1993. pp. 21–56. [Google Scholar]
- 23.Noteborn M H M, De Boer G F, Van Roozelaar D J, Karreman C, Kranenburg O, Vos J G, Jeurissen S H M, Hoeben R C, Zantema A, Koch G, Van Ormondt H, Van der Eb A J. J Virol. 1991;65:3131–3139. doi: 10.1128/jvi.65.6.3131-3139.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noteborn M H M, Todd D, Verschueren C A J, De Gauw H W F M, Curran W L, Veldkamp S, Douglas A J, McNulty M S, Van der Eb A J, Koch G. J Virol. 1994;68:346–351. doi: 10.1128/jvi.68.1.346-351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noteborn M H M, Koch G. Avian Pathol. 1995;24:11–31. doi: 10.1080/03079459508419046. [DOI] [PubMed] [Google Scholar]
- 26.Zhuang S-M, Shvarts A, Van Ormondt H, Jochemsen A G, Van der Eb A J, Noteborn M H M. Cancer Res. 1995;55:486–489. [PubMed] [Google Scholar]
- 27.Zhuang S-M, Landegent J E, Verschueren C A J, Falkenburg J H F, Van Ormondt H, Van der Eb A J, Noteborn M H M. Leukemia. 1995;9:S118–S120. [PubMed] [Google Scholar]
- 28.Zhuang S-M, Shvarts A, Jochemsen A G, Van Oorschot A A A M, Van der Eb A J, Noteborn M H M. Carcinogenesis. 1995;16:2939–2944. doi: 10.1093/carcin/16.12.2939. [DOI] [PubMed] [Google Scholar]
- 29.Marijt W A, Veenhof W F, Brand A, Goulmy E, Fibbe W E, Willemze R, Van Rood J J, Falkenburg J H. J Exp Med. 1991;173:101–109. doi: 10.1084/jem.173.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaffe E A, Nachman R L, Becker C G, Minick C R. J Clin Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rheinwald J G, Green H. Cell. 1975;6:331–343. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- 32.Ponec M, Kempenaar J A, De Kloet E R. J Invest Dermatol. 1981;76:211–214. doi: 10.1111/1523-1747.ep12525761. [DOI] [PubMed] [Google Scholar]
- 33.Klein B, Pastink A, Odijk H, Westerveld A, Van der Eb A J. Exp Cell Res. 1990;191:256–262. doi: 10.1016/0014-4827(90)90012-y. [DOI] [PubMed] [Google Scholar]
- 34.Weissman B E, Stanbridge E J. Cytogenet Cell Genet. 1983;35:263–268. doi: 10.1159/000131878. [DOI] [PubMed] [Google Scholar]
- 35.Rheinwald J G, Beckett M A. Cancer Res. 1981;41:1657–1663. [PubMed] [Google Scholar]
- 36.Boukamp P, Petrussevska R T, Breitkreutz D, Hornung J, Markham A, Fusenig N E. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor-Papadimitriou J, Purkis P, Lane E B, McKay I A, Chang S E. Cell Differ. 1982;11:169–180. doi: 10.1016/0045-6039(82)90008-2. [DOI] [PubMed] [Google Scholar]
- 38.Van der Eb A J, Graham F L. Methods Enzymol. 1980;65:826–839. doi: 10.1016/s0076-6879(80)65077-0. [DOI] [PubMed] [Google Scholar]
- 39.Fischer D F, Gibbs S, Van De Putte P, Backendorf C. Mol Cell Biol. 1996;16:5365–5374. doi: 10.1128/mcb.16.10.5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van den Heuvel S J L, Van Laar T, Kast W M, Melief C J, Zantema A, Van der Eb A J. EMBO J. 1990;9:2621–2629. doi: 10.1002/j.1460-2075.1990.tb07444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Telford W G, King L E, Fraker P J. Cytometry. 1992;13:137–143. doi: 10.1002/cyto.990130205. [DOI] [PubMed] [Google Scholar]
- 42.Menke, A. L., Shvarts, A., Riteco, N., Van Ham, R. C. A., Van der Eb, A. J. & Jochemsen, A. G. (1997) Cancer Res. 57, in press. [PubMed]
- 43.Hill R P, Tannock I F. In: The Basic Science of Oncology. Tannock I F, Hill R P, editors. New York: McGraw-Hill; 1992. pp. 256–291. [Google Scholar]
- 44.Vanacker J-M, Rommelaere J. Semin Virol. 1995;6:291–297. [Google Scholar]
- 45.Csermely P, Schnaider T, Szanto I. Biochim Biophys Acta. 1995;1241:425–451. doi: 10.1016/0304-4157(95)00015-1. [DOI] [PubMed] [Google Scholar]
- 46.Wen W, Meinkoth J L, Tsien R Y, Taylor S S. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- 47.Fischer U, Huber J, Boelens W C, Mattaj I W, Lührmann R. Cell. 1995;82:475–483. doi: 10.1016/0092-8674(95)90436-0. [DOI] [PubMed] [Google Scholar]
- 48.Gerace L. Cell. 1995;82:341–344. doi: 10.1016/0092-8674(95)90420-4. [DOI] [PubMed] [Google Scholar]
- 49.Kyprianou N, Isaacs J T. Endocrinology. 1989;122:522–532. [Google Scholar]