

Abstract
Elucidating the structures of intermediates in the reduction of O2 to water by cytochrome c oxidase is crucial to understanding both oxygen activation and proton pumping by the enzyme. In the work here, the reaction of O2 with the mixed-valence enzyme, in which only heme a3 and CuB in the binuclear center are reduced, has been followed by time-resolved resonance Raman spectroscopy. The results show that O=O bond cleavage occurs within the first 200 μs after reaction initiation; the presence of a uniquely stable Fe—O—O(H) peroxy species is not detected. The product of this rapid reaction is a heme a3 oxoferryl (FeIV=O) species, which requires that an electron donor in addition to heme a3 and CuB must be involved. The available evidence suggests that the additional donor is an amino acid side chain. Recent crystallographic data [Yoshikawa, S., Shinzawa-Itoh, K., Nakashima, R., Yaono, R., Yamashita, E., Inoue, N., Yao, M., Fei, M. J., Libeu, C. P., Mizushima, T., et al. Science, in press; Ostermeier, C., Harrenga, A., Ermler, U. & Michel, H. (1997) Proc. Natl. Acad. Sci. USA 94, 10547–10553] show that one of the CuB ligands, His240, is cross-linked to Tyr244 and that this cross-linked tyrosyl is ideally positioned to participate in dioxygen activation. We propose a mechanism for O—O bond cleavage that proceeds by concerted hydrogen atom transfer from the cross-linked His—Tyr species to produce the product oxoferryl species, CuB2+—OH−, and the tyrosyl radical. This mechanism provides molecular structures for two key intermediates that drive the proton pump in oxidase; moreover, it has clear analogies to the proposed O—O bond forming chemistry that occurs during O2 evolution in photosynthesis.
The molecular mechanism of dioxygen activation and reduction by the terminal respiratory enzyme, cytochrome c oxidase (CcO), is accessible because of its unique kinetic properties. Elucidation of this mechanism is of fundamental importance in understanding O2 chemistry in biological systems and necessary for insight into the function of the protein as a redox-linked proton pump. CcO uses four redox-active metal centers, CuA, heme a, and the heme a3/CuB binuclear center, to sustain mitochondrial electron transport by reducing molecular oxygen to water. This reaction ensures a constant flow of electrons through the respiratory chain and the coupled generation of a proton gradient across the mitochondrial membrane, which is required for ATP synthesis. The oxygen chemistry catalyzed by CcO contributes directly to the build-up of the proton gradient because of its redox-linked proton pumping function (1). Thus, the overall reaction catalyzed by CcO may be written as
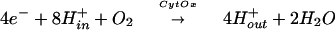 |
1 |
where Hin+ and Hout+ indicate protons on the matrix (in) and cytosolic (out) sides of the membrane. Because its reaction kinetics is controlled by its proton-pumping function, unique insights into oxygen activation mechanisms are possible (2, 3).
A number of reaction intermediates in dioxygen reduction have been identified recently (3, 4). Nonetheless, the timing and mechanism of the critical O=O bond cleavage process in the heme a3/CuB binuclear center and the structure of a key intermediate at the peroxy level are poorly understood. In one model, a heme-peroxy adduct [Fea3III—O—O(H)] is uniquely stable (2, 3, 5–8). This model contrasts with peroxidases (9) and catalases (10), in which the peroxy O—O bond is spontaneously cleaved to yield an oxoferryl (FeIV=O) product and a radical. Recent Raman work on the reaction of CcO with H2O2 provided an alternative view of the peroxy species that is more in line with the catalase/peroxidase mechanism because it showed the occurrence of an oxoferryl heme (νFe=O = 804 cm−1) at the peroxy oxidation level (11, 12). Although this observation is in agreement with optical absorption and MCD data (13–15), the assignment of an O—O bond-cleaved species in the formal peroxy intermediate is the subject of considerable debate (3, 4, 7, 8).
CcO catalyzes several different reactions with O2 and H2O2 that share some or all intermediate steps. Mixed-valence CcO (MV-CcO) is a two-electron reduced form of the enzyme that carries out O2 reduction only to the controversial peroxy oxidation level (3, 16, 17). The final product of this reaction exhibits a characteristic difference optical absorption spectrum (2–4).† This intermediate is clearly linked to the proton pump function because it has been shown to appear on reversal of the forward reaction in energized mitochondria (22). Determining the structure of the product of the MV-CcO/O2 reaction will resolve controversial aspects of the oxygen activation mechanism and provide a structural basis for formulating molecular mechanisms of the pump. In the experiments reported here, we have used time-resolved resonance Raman techniques to address these issues.
MATERIALS AND METHODS
Sample Preparation.
Bovine heart CcO was purified as described (23) with minor modifications. MV-CcO—CO was obtained by anaerobic incubation of 100 μM CcO in 100 mM sodium phosphate buffer, pH 7.4, containing 0.21% Brij 35, under a CO atmosphere for 3 hr at 37°C. Samples were recovered after the spectroscopic experiment, reconcentrated, and used again, up to a total of three times.
The Time-Resolved Pump–Probe Raman Spectroscopy.
The experimental apparatus that was used for resonance Raman measurements is illustrated in Fig. 1. Two gas-tight syringes containing equal amounts of MV-CcO—CO and O2-saturated sodium phosphate buffer were driven by the infusion pump at a rate of 110 μl/min and combined in an active mixer (unpublished). The mixing point and the interconnecting lines were enclosed in a jacket, through which a solution thermostated at 20 ± 1°C was circulated. The reaction mixture was delivered to the rectangular quartz flow cell (cross section 0.2 × 1.0 mm) directly attached to the mixer body. The estimated volume between the beginning of mixing and the sample point was 6 ± 2 μL.
Figure 1.

Experimental apparatus. ENZ, enzyme solution; BUF, O2-saturated buffer; MIX, active mixer; JCK, thermostated jacket; FC, flow cell; LB, laser beams; L1, L2, L3, lenses; F1, notch filter; F2, short-pass filter; F3, depolarizer; SL, entrance slit; POL, polychromator; CCD, CCD detector.
The excitation laser beams were focused with an L1 lens at the flow cell to a diameter of ≈100 μm, the scattering volume was 1.0-mm long, and the laser repetition was 25Hz. The reaction of MV-CcO with oxygen was initiated by CO photolysis with 500- μJ, 10-ns pulses of 595 nm light. Raman scattering at a desired delay time was excited with 30-μJ pulses at 416 nm; the delay was controlled by a digital delay generator. Scattered light was collected in a 90o geometry by using a 50-mm F1.1 camera lens (L2). The notch filter was used to reject the excitation wavelength and the short-pass filter to reject scattered light from the photolysis pulse. The light selected by the filter combination was focused at the entrance slit (spectral slit width of 9 cm−1) of the polychromator with the L3 lens. The polychromator (Instruments S.A., Edison, NJ, Spex model 500M) was equipped with a 1024 × 256 CCD detector (Spex, model Spectrum1); the depolarizer was used to nullify the polarization sensitivity of the detector.
Spectral Measurements.
The Raman spectrum of CcO at 10 ns after photolysis of CO in the presence of oxygen was collected for several minutes immediately before and after each accumulation at a desired delay time to ascertain that the reacting form of the enzyme was the mixed-valence species. The high-frequency region of the spectrum (Fig. 2a) exhibited characteristic modes at 1467 cm−1 and 1368 cm−1, indicative of ferric heme a. On the other hand, the spectrum of the fully reduced CcO immediately after photolysis of CO (Fig. 2b) exhibited characteristic bands at 1613 cm−1 and 1519 cm−1, which are absent in the spectrum of MV-CcO. Optical absorption spectra of the sample recorded before the Raman measurement showed absorption maxima at 432 nm and 592 nm that are indicative of MV-CcO—CO complex. The shape of the absorption spectrum at 444 nm and 605 nm showed no indication of the presence of the fully reduced CcO. Separate difference absorption measurements, vs. resting enzyme, showed compound C as the sole product of aerobic oxidation of our MV-CcO—CO samples. These observations exclude detectable contamination of the starting MV-CcO with fully reduced CcO.
Figure 2.

High-frequency Raman spectra of mixed-valence (a) and fully reduced (b) CcO at 10 ns after photolysis of CO. Trace a is an average of several 10-ns spectra obtained as described in Materials and Methods. Trace b was obtained in a separate experiment under the same conditions as used for trace a; the CO complex of the fully reduced CcO was prepared by reduction with 2 mM ascorbate/1 μM cytochrome c under a CO atmosphere.
Time-resolved spectra of 16O2 and 18O2 derivatives of MV-CcO were measured in 210-min periods in alternating order. Measurements were repeated by using independent sample preparations of the enzyme for each delay time. Data processing and analysis were carried out by using xdsoft (by D.P.) and origin 5.0 (Microcal Software, Northampton, MA) programs for the IBM personal computer.
RESULTS
16O2/18O2 Raman difference spectra at various times during the reaction of MV-CcO with dioxygen are shown in Fig. 3. At 25 μs, the Fe—O2 stretching vibration (νFe—O2) of the oxy intermediate at 568 cm−1 (545 cm−1 for 18O2) is the only isotope sensitive species present (3, 4). This mode decays after 50 μs, and by 400 μs, only a weak signal is seen. As the 568-cm−1 band decays with time, another oxygen-isotope-sensitive mode at 804 and 768 cm−1 for 16O- and 18O-derivatives, respectively, gains intensity. At 25 μs, the 804/768-cm−1 mode is below noise level; at 50 μs, a weak band is seen, and after 100 μs, it is clearly observed in the spectrum. The frequency, bandwidth and isotope shift of the 804/768-cm−1 band match closely those of the 804/769-cm−1 oxoferryl species observed in the reaction of the oxidized CcO with H2O2 (11, 12, 23).
Figure 3.

Reaction between MV-CcO and O2 at 20oC. Difference Raman spectra were calculated at the indicated times after initiation of the reaction between MV-CcO and O2 by subtracting the spectrum recorded with 18O2 as a substrate from that obtained with 16O2. All porphyrin vibrations cancel in the difference spectra, whereas vibrations of bound oxygen appear as derivative-shaped features. Full scale of the ordinate axis is 28% of the porphyrin ν7 mode in the absolute spectra. Each trace is an average of several isotope-difference spectra (1–1.5 × 106 shots per isotope total) obtained in independent experiments.
A third oxygen vibration is observed at 358/340 cm−1 for 16O/18O derivatives in the 400-μs spectrum. A weak signal at these frequencies can be recognized at 200 μs but not at earlier times. This mode has been observed previously in other reactions of CcO (12, 23–25), but its molecular origin is unclear. It was suggested to arise either from the Fe—O stretching mode of a peroxy intermediate (Fe—O—O—H) (24) or from the Fe=O-bending motion of a oxoferryl structure at the peroxy oxidation level (12, 25). The low intensity of the 358-cm−1 mode (Fig. 3) precludes a quantitative analysis of its temporal behavior in this work. However, from visual inspection of our data, the 358-cm−1 band does not develop before the 804-cm−1 band, which suggests that it does not arise from a Fe—OO(H) structure.
The temporal behavior of the oxy (568 cm−1) and oxoferryl (804 cm−1) species, normalized to the porphyrin ν7 mode, is shown in Fig. 4. Because no other intermediates were substantially populated between the decay of the oxy species and the rise of the oxoferryl, the reaction of MV-CcO with O2 was modeled as two sequential irreversible steps:
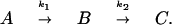 |
2 |
Experimental profiles for the 568-cm−1 and 804-cm−1 species were individually fitted to B and C, respectively; A represents ligand-free enzyme. A value for k1 of 9 × 104 s−1 was found to satisfy the rapid development we observe for the 568-cm−1 band, consistent with earlier data (2, 3). The decay of the 568-cm−1 band at k2 = 4.9 ± 0.1 × 103 s−1 was obtained from the fit, consistent with earlier work on the temporal behavior of the oxy intermediate (26). Analysis of the kinetics of the 804-cm−1 band gave k2 = 6.1 ± 1.2 × 103 s−1. The essentially identical values for k2 obtained for the disappearance of the 568-cm−1 mode and the appearance of the 804-cm−1 mode indicate that the decay of the oxy and the rise of the oxoferryl species occur in the same reaction step.
Figure 4.

Time course of the reaction between MV-CcO and O2 at 20oC. Relative intensities of the 568/545 cm−1 (○) and 804/768 cm−1 (•) difference bands were extracted from the data set in Fig. 3. Solid lines represent the results of the kinetic fitting procedure described in the text. Error bars denote uncertainty in the intensities from the signal/noise ratio in the corresponding difference spectra.
DISCUSSION
Mechanism of the O=O Bond Cleavage.
The point at which the O—O bond is cleaved during the CcO reaction cycle has been a matter of considerable debate. The rapid formation of the oxoferryl species during the MV-CcO/O2 reaction in Fig. 3 addresses this issue and demonstrates that bond cleavage takes place when only two reducing equivalents are added to the enzyme. For this to occur, one additional electron must be donated by the enzyme. In peroxidases (9) and catalases (10), the source of this electron is often the heme macrocycle. This does not occur in CcO, however, because neither optical (13–15) nor Raman (12) spectroscopy support the formation of a porphyrin π-cation radical. Oxidation of heme iron from Fea3IV to Fea3V has been proposed recently (4, 25), but this possibility is unprecedented among heme enzymes, and evidence to support it has not been reported so far. A third potential source of the additional reducing equivalent is CuB, which could be oxidized to the +3 valence state. The occurrence of trivalent copper has been demonstrated in model compounds (27, 28) but has not been observed in copper-containing proteins. Thus, oxidation of an amino acid residue to a radical, as in cytochrome c peroxidase (9) and prostaglandin H synthase (29), is the most likely possibility.
Tyr244 (in beef heart enzyme nomenclature; Tyr288 in Rhodobacter sphaeroides and Tyr280 in Paracoccus denitrificans) is a strong candidate for the redox-active residue. It is located in the immediate vicinity of the binuclear center (6, 30), within hydrogen-bonding distance of heme a3-ligated O2, and is conserved among several types of oxidases (31). Its likely function as a hydrogen atom donor during O—O bond cleavage has been strengthened considerably by the recent finding (59) of a cross-link between it and His240 in the mammalian enzyme and of the analogous cross-link in the bacterial enzyme (32). The covalent linkage will modulate the redox potential, the bond dissociation energy, and the pKa of the tyrosine O—H, as occurs for the redox-active, covalently cross-linked tyrosine residue in galactose oxidase (33, 34), to produce important energetic consequences (see below).
With the postulated hydrogen atom transfer function for the cross-linked His—Tyr structure, the molecular events at the binuclear center upon dioxygen binding and O=O bond cleavage are summarized in Fig. 5. After transient ligation to CuB1+ in the reduced binuclear center (Fig. 5a), molecular oxygen binds to the heme to produce the spectroscopically detectable Fe—O2 intermediate (Fig. 5b). Subsequent electron transfer from CuB1+ produces a transient peroxy species (Fig. 5c), which may bind to the positively charged CuB2+ to maintain electroneutrality and to preserve bond order. The peroxy species, however, does not accumulate to detectable levels; rather it rapidly abstracts a hydrogen atom from the nearby Tyr244 (Fig. 5d). At the same time, the O—O bond cleaves in a concerted manner, the distal oxygen forms the hydroxyl bound to CuB2+, while the proximal oxygen oxidizes Fea3III to form a ferryl heme (Fea3IV=O). Two bonds (O—O and H—OTyr) are broken in this step and two bonds (Fe=O and H—O−) are formed.
Figure 5.

A model for dioxygen bond cleavage by MV-CcO. Only the chemically essential sites (heme a3, CuB, His240, and Tyr244) are shown for clarity. Brackets denote transition states; details are given in the text.
Following the bond cleavage process, the binuclear center relaxes to the stable oxoferryl intermediate (Fig. 5e), with characteristic vibration at νFe=O = 804 cm−1, that has been detected here in the MV-CcO/O2 and previously in the oxidized CcO/H2O2 (11, 12) reactions. The CuB2+—OH− will probably remain hydrogen bonded to the phenol oxygen of the neutral tyrosyl radical. Another hydrogen bond is likely to be formed between the oxo atom and the hydroxyl, as hydrogen bonding of the oxoferryl oxygen has been demonstrated for the 804-cm−1 species (11). The unique CuB—His—Tyr structure in the binuclear center of CcO also may provide an explanation for the unusual and controversial oxygen vibration at 358 cm−1. The assignment of this mode to the CuB—OH structure, which is resonance enhanced via absorption by the CuB(OH)—His—Tyr system, is currently under consideration. We conclude that the final tyrosine radical/oxoferryl species in Fig. 5e is the chemical structure of the stable intermediate referred to as P.†
In Fig. 5, CuB is in its cupric state in the final product (Fig. 5e) of the MV-CcO/O2 reaction and the His—Tyr structure exists as a free radical, yet neither is detectable by EPR (16). The oxoferryl species will most likely occur in its low-spin S = 1 state in the stable product and also be undetectable by conventional, perpendicular mode EPR. The absence of EPR signals from the S = ½ CuB2+ and the S = ½ Tyr244 radical can be attributed to spin-coupling between the two species that is mediated by the ligated, cross-linked His240. The spin-coupling may also serve to delocalize the oxidizing equivalent, which is shown in Fig. 5 as Tyr244•, so that the CuB species assumes partial +3 character. In this regard, the structure in (Fig. 5e) can be considered as one resonance form of a more delocalized electronic system that involves CuB, His240, and Tyr244. Interestingly, formation of 7% of a radical species in the reaction of oxidized CcO with H2O2 has been reported for some conditions recently (15), which may represent partial uncoupling of the CuB/Tyr244 system.
The formation of the hydroxyl at the binuclear center in Fig. 5 requires the addition of a proton, which is provided by the tyrosine during the O—O bond cleavage chemistry. This residue is likely to remain oxidized as a deprotonated neutral tyrosyl radical until arrival of the third electron, which is not available when the reaction is started with MV-CcO. In fact, it has been shown that dioxygen chemistry in CcO up to the peroxy oxidation level does not involve charge uptake or release to bulk medium (35). Thus, neither is another proton taken up from bulk solution to form water, nor is the hydroxyl anion released from the binuclear center at the peroxy level.
Analogies Between Oxygen Evolution and Respiration.
The mechanism in Fig. 5 has striking analogies to a recent proposal for the O—O bond forming step in photosynthesis (36). In the photosynthetic system, the key elements are two terminal Mn ions at the open ends of a C-shaped (Mn)4 cluster, a tyrosyl radical, and Mn-bound substrate oxo and hydroxo groups. In the postulated bond-forming step, the tyrosyl radical abstracts the hydroxo hydrogen in a concerted process in which the O—O bond of a transient peroxo species forms:
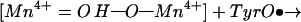 |
3 |
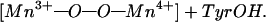 |
The proposed photosynthetic process is the exact reverse of that shown for oxidase in Fig. 5 because two bonds (Mn=O and O—H−) are broken and two bonds (O—O and H—Otyr) are formed. The spatial disposition of the product species in photosystem II—the two Mn ions, the transient peroxy intermediate, and the tyrosine phenol side chain—are essentially identical to that of the reacting CuB, heme a3 iron, transient peroxy species, and tyrosine side chain shown in Fig. 5. As in photosystem II, the concerted bond-cleavage process in the binuclear center in MV-CcO is electroneutral and proceeds without significant development of charge. Accordingly, we expect that it will proceed with low overall activation energy, consistent with the facile low temperature formation of compound C (18, 37).
The analogy to photosystem II provides a basis for understanding why oxidase uses a modified tyrosine to carry out the chemistry shown in Fig. 5. To abstract hydrogen from a Mn-bound hydroxyl requires that the redox potential of the tyrosine be sufficiently high to drive the process (Eq. 3). To reverse this, as in Fig. 5, so that the tyrosine functions as a hydrogen atom donor, requires that the potential of its phenol headgroup be lowered sufficiently to allow the reaction to proceed spontaneously. Electron transfer to the product His—Tyr• species will be used subsequently to drive proton translocation, however, which demands that the bond-cleaving reaction not be too strongly driven. The use of a cross-linked tyrosine, which is expected to reduce the potential of the radical only modestly from that of the unmodified residue, looks to be an efficient strategy by which to accomplish this.
Oxygen Intermediates and the H+-Pump.
The final product of the reaction between MV-CcO and dioxygen shown in Fig. 5e was first observed by Chance and coworkers (18) and called compound C. Its optical properties match closely those of the “607 nm” form of the enzyme that occurs in the oxidized CcO/H2O2 reaction (12, 13, 19, 20, 23, 38, 39) and the peroxy species, also called P, that is formed by reversed electron transfer through the enzyme (21, 22). In earlier work by Proshlyakov et al. (11, 12), an oxoferryl structure (νFe=O = 804 cm−1) was assigned to the “607 nm” species formed as the initial intermediate in the oxidized CcO/H2O2 reaction, although this assignment has been disputed, as noted above. The results in Fig. 3 for the MV-CcO/O2 reaction are consistent with those from the oxidized CcO/H2O2 work (11, 12) and strongly support the assignment of the bond-cleaved structure to P as shown in Fig. 5. In the oxidized CcO/H2O2 reaction, P is followed by a species,† F, that also has an oxoferryl structure (12, 23) (νFe=O = 785 cm−1). An intermediate with optical properties similar to those of F in the oxidized CcO/H2O2 reaction (12, 13, 19, 20, 23, 38, 39) has been detected by reversed electron transfer through oxidase, (21) and an analogous species can be observed during the reduction of O2 by fully reduced CcO (2, 3).
Wikström (40) has shown that the steps preceding the formation of P in cytochrome oxidase are not linked to the proton pump. Rather, the one electron redox transitions, P → F and F → oxidized enzyme, are each coupled to the translocation of two protons. Within the context of Fig. 5, the reaction chemistry can be understood as involving two basic reactions in series. In the first, O2 binds and the O—O bond is cleaved to produce two highly oxidizing species, the CuB2+—His240—Tyr244 radical center and the oxoferryl heme a3 species. With the bond cleavage chemistry complete, the two subsequent electron transfers are then used to drive the pumping process. Earlier mechanistic and thermodynamic considerations had shown that the committed step in oxidase catalysis involves the formation of the P intermediate. The steps before this reaction proceed with little driving force so that P formation essentially traps the O2 substrate (2, 7). A similar conclusion is reached in reversed electron flow experiments (40). Oxidized CcO can be converted to P under strongly oxidizing conditions, but the reaction cycle cannot be reversed further, indicating that the formation of P is irreversible. With the formulation of P as the bond-cleaved, radical-containing structure in Fig. 5, we can now associate irreversibility in the oxidase mechanism with the rupture of the O—O bond. Moreover, two fully reduced oxygen species, oxo and hydroxo, are formed as products, preventing dissociation of partially reduced oxygen species from the enzyme and avoiding potential cytotoxicity of respiration (2).
Differences Between P and F.
The mechanism proposed here for O—O bond cleavage and subsequent proton pumping stipulates that both P and F have FeIV=O structure at the a3 site. Despite this structural similarity, P and F have distinct optical absorption properties (12, 13, 19–21, 23, 38, 39). The absorption difference spectrum of P, relative to the resting CcO, shows a maximum at 607 nm, whereas that of F has a maximum in the 580-nm region and is considerably broader (12, 19, 20, 22, 38, 41); its extinction coefficient is less by ≈7 mM−1 cm−1 than that of P (21). The extinction coefficient difference may be overestimated somewhat, however, because the calculated spectrum of F (21) does not match that observed experimentally (12, 19, 20, 22, 38, 41), particularly as a characteristic band at 530 nm and a trough at 660 nm are missing. These optical differences have been used to argue that P and F must differ structurally at the heme a3 site (7).
Several observations, however, compromise this conclusion. Although there has been little model compound work done on oxoferryl heme species, we can gain some insight into the variability of the optical properties of oxoferryl hemes by considering the class of ferryl intermediates that occurs as compounds II in peroxidase catalysis. Table 1 summarizes data on several oxoferryl species that provide insight into the influence of the local protein environment on their optical properties. The spectral parameters of P and F in CcO also are included. This compilation shows that the optical properties of heme FeIV=O are sensitive functions of the structure of the macrocycle, of the basicity of the endogenous axial ligand, and of the hydrogen bonding status of the oxo oxygen. The data for myeloperoxidase show, for example, that a hydrogen bond, present at pH 7 and absent at pH 11, can shift λmax by 7 nm and alter ɛ by 7 mM−1 cm−1. Species spectrally similar to P and F of mammalian aa3-type oxidase also are developed by bacterial bo3-type quinol oxidase (52). Their difference absorption spectra are ≈25 nm blue shifted from those of bovine CcO, which brings them within range of the other histidine-ligated protoheme compounds in Table 1. Their absolute spectra, however, cannot yet be reliably estimated.
Table 1.
Visible absorption properties of several oxoferryl species and cytochrome c oxidase compounds P and F
| Protein | λmax (nm)/ ɛ(mM−1 cm−1) | λmax (nm)/ ɛ(mM−1 cm−1) | Ref. |
|---|---|---|---|
| Protoheme/histidine axial ligation | |||
| Horseradish peroxidase | 527/9.5 | 554/9.65 | 42 |
| Prostaglandin synthase | 527/10.1* | 557/9.3* | 43 |
| Cytochrome c peroxidase | 529/12.2* | 561/13.2* | 44 |
| Arthromyces ramosus peroxidase | 530/13.0* | 553/12.9* | 45 |
| Lactoperoxidase | 530*/9.8* | 564*/9.0* | 46 |
| Myoglobin | 549/9.8 | 47 | |
| Protoheme/tyrosinate axial ligation | |||
| Catalase | 531*/8.8* | 566*/9.6* | 48 |
| Protoheme/cysteinyl axial ligation | |||
| Cloroperoxodase | 542/16.3* | 571/13.8* | 49 |
| Modified protoheme/histidine axial ligation | |||
| Myeloperoxidase, pH 7 | 628/18 | 50 | |
| Myeloperoxidase, pH 11 | 635/25 | 50 | |
| Heme a/histidine axial ligation | |||
| Cytochrome c oxidase, P | ≈565†/≈7† | ≈605†/≈15† | 21, 51 |
| Cytochrome c oxidase, F | ≈585†/≈7† | 21, 51 | |
The extinction coefficient of P at its red absorption maximum, ≈15 mM−1 cm−1, is within the range of other oxoferryl heme chromophores, consistent with our structural interpretation of P in Fig. 5. For F, however, its peak extinction is less than that for other oxoferryl species, despite its unequivocal assignment as an FeIV=O species. The most likely rationale for the depressed extinction coefficient of F is that its reported wavelength maximum varies between 575 and 585 nm (19, 23, 38, 39, 53), suggesting that different forms of F exist. The origin of this variation may lie in heterogeneous hydrogen bonding and/or proximal base interactions, which broaden the observed spectrum and diminish the peak extinction coefficient.
Both electronic and structural factors are likely to contribute to the difference between P and F in absorption maxima. The CuB—His240—Tyr244• structure postulated in P is likely to absorb in the visible region (33, 34). Its reduction to form F will quench this absorption. Thus, the possibility of an electronic interaction between the metal/radical chromophore and heme a3 exists uniquely for P. Given the proximity of the CuB—His240—Tyr244• species to a3, we expect that a measurable perturbation to the heme a3 optical spectrum exists for P that is absent in F.
The electronic effects of the radical site are likely to be complemented by axial ligation effects because the observed νFe=O stretching frequency is considerably different in the two intermediates. For P, νFe=O = 804 cm−1, whereas for F, νFe=O = 785 cm−1. Both distal and proximal effects can contribute to these frequency shifts and each will modulate absorption properties. The presence of a weak hydrogen bond between the oxo oxygen and the distal site has been demonstrated for P (11), which is shown in Fig. 5 as originating from the CuB2+ hydroxo ligand. The decrease in νFe =O in F suggests formation of a second hydrogen bond to the bound oxo as the hydrogen bond network in the binuclear center rearranges during the P → F transition. Uptake of a proton from solution (35) and the protonation of Tyr244 upon this transition is most probably the source for this additional interaction. Consistent with this interpretation, νFe=O in F shows a more substantial H2O/D2O frequency shift (24, 54) and the oxo oxygen is more readily exchanged with solvent water (23). The hydrogen-bonding change at the Fe=O moiety in F relative to P will affect both the optical properties of the heme chromophore and the vibrational properties of the His—FeIV=O structure (50, 55–58) (Table 1). Variation in the basicity of the proximal histidine is also a likely contributor to the optical and vibrational differences between the two intermediates. Earlier model compound work has shown that the FeIV=O bond strength is a sensitive function of proximal basicity (56); the vibrational frequency decreases as the ligand becomes more electron donating to the FeIV=O center. For the peroxidase class of the enzymes, communication between the distal pocket and the proximal environment is well established in the phenomena that are characterized as heme-linked ionisation (57). A similar mechanism may well function in CcO to communicate events in the distal site to the proximal ligand. Both hydrogen-bonding differences and variation in proximal ligand basicity may contribute to the P/F disparity in νFe=O; each of these effects also will selectively modify the optical properties of the two intermediates.
The analysis above indicates that there are significant difference in the structures and local interactions of the P and F intermediates and that these are likely to modulate their optical properties substantially. Thus, the difference in the visible absorption spectra of P and F provides a weak basis for inferring that they cannot each have FeIV=O structures; rather we conclude that the weight of data is consistent with their assignment as oxoferryl species. Moreover, formulation of P as an FeIV=O-containing intermediate invokes O—O cleavage as the irreversible step in oxidase catalysis, which is attractive from both thermodynamic and kinetic points of view. With these structures and the additional proposal of the CuB2+—His240—Tyr244• species in P, we have a good basis on which to construct mechanisms for proton pumping. The model presented in Fig. 5, its extension to the reduction of O2 to water by the fully reduced CcO, and considerations of molecular intermediates in the pumping cycle provide specific structural species that are accessible to experimental tests.
Acknowledgments
This work was supported by Grants GM25480 and GM57323 from the U.S. National Institute of Health. We thank Dr. Curt Hoganson for helpful discussions.
ABBREVIATIONS
- CcO
cytochrome c oxidase
- MV-CcO
mixed-valence CcO
- P
spectral form of CcO at the 2e− reduced level
- F
spectral form of CcO at the 3e− reduced level
Footnotes
Several different approaches have been used to generate a form of CcO at the peroxy (2e−) oxidation level (11, 16, 18–21). In the literature, these species have been referred to variously as compound C, peroxy, “607 nm” form, or P. In this paper, these will be referred to collectively as P. Similarly, a form of CcO at the ferryl (3e−) oxidation level, which is referred to as ferryl, “580 nm” from, or F, will be designated as F to promote simplicity in comparison with earlier work. The P and F nomenclature is a formal construct and should not be taken as necessarily indicating the physical structure existing at the active site.
References
- 1.Wikström M. Nature (London) 1977;266:271–273. doi: 10.1038/266271a0. [DOI] [PubMed] [Google Scholar]
- 2.Babcock G T, Wikström M. Nature (London) 1992;356:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 3.Ferguson-Miller S, Babcock G T. Chem Rev (Washington, D C) 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 4.Kitagawa T, Ogura T. In: Progress in Inorganic Chemistry. Karlin K D, editor. Vol. 45. New York: Wiley; 1997. pp. 431–479. [Google Scholar]
- 5.Wikström M, Bogachev A, Finel M, Morgan J E, Puustinen A, Raitio M, Verkhovskaya M, Verkhovsky M I. Biochim Biophys Acta. 1994;1187:106–111. doi: 10.1016/0005-2728(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 6.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature (London) 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 7.Morgan J E, Verkhovsky M I, Wikström M. Biochemistry. 1996;35:12235–12240. doi: 10.1021/bi961634e. [DOI] [PubMed] [Google Scholar]
- 8.Sucheta A, Georgiadis K E, Einarsdóttir O. Biochemistry. 1997;36:554–565. doi: 10.1021/bi962422k. [DOI] [PubMed] [Google Scholar]
- 9.Poulos T L, Fenna R E. In: Metal Ions in Biological Systems. Sigel H, Sigel A, editors. Vol. 30. New York: Dekker; 1994. pp. 25–75. [Google Scholar]
- 10.Chuang W-J, Van Wart H E. J Biol Chem. 1992;267:13293–13301. [PubMed] [Google Scholar]
- 11.Proshlyakov D A, Ogura T, Shinzawa-Itoh K, Yoshikawa S, Appelman E H, Kitagawa T. J Biol Chem. 1994;269:29385–29388. [PubMed] [Google Scholar]
- 12.Proshlyakov D A, Ogura T, Shinzawa-Itoh K, Yoshikawa S, Kitagawa T. Biochemistry. 1996;35:76–82. doi: 10.1021/bi9511705. [DOI] [PubMed] [Google Scholar]
- 13.Weng L, Baker G M. Biochemistry. 1991;30:5727–5733. doi: 10.1021/bi00237a014. [DOI] [PubMed] [Google Scholar]
- 14.Watmough N J, Cheesman M R, Greenwood C, Thomson A J. Biochem J. 1994;300:469–475. doi: 10.1042/bj3000469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fabian M, Palmer G. Biochemistry. 1995;34:13802–13810. doi: 10.1021/bi00042a011. [DOI] [PubMed] [Google Scholar]
- 16.Clore G M, Andréasson L-E, Karlsson B, Aasa R, Malmström B G. Biochem J. 1980;185:155–167. doi: 10.1042/bj1850155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill B C, Greenwood C, Nicholls P. Biochim Biophys Acta. 1986;853:91–113. doi: 10.1016/0304-4173(86)90006-6. [DOI] [PubMed] [Google Scholar]
- 18.Chance B, Saronio C, Leigh J S., Jr J Biol Chem. 1975;250:9226–9237. [PubMed] [Google Scholar]
- 19.Wrigglesworth J M. Biochem J. 1984;217:715–719. doi: 10.1042/bj2170715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vygodina T V, Konstantinov A A. Ann N Y Acad Sci. 1988;550:124–138. doi: 10.1111/j.1749-6632.1988.tb35329.x. [DOI] [PubMed] [Google Scholar]
- 21.Wikström M, Morgan J E. J Biol Chem. 1992;267:10266–10273. [PubMed] [Google Scholar]
- 22.Verkhovsky M I, Morgan J E, Wikström M. Proc Natl Acad Sci USA. 1996;93:12235–12239. doi: 10.1073/pnas.93.22.12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Proshlyakov D A, Ogura T, Shinzawa-Itoh K, Yoshikawa S, Kitagawa T. Biochemistry. 1996;35:8580–8586. doi: 10.1021/bi952096t. [DOI] [PubMed] [Google Scholar]
- 24.Varotsis C, Zhang Y, Appelman E H, Babcock G T. Proc Natl Acad Sci USA. 1993;90:237–241. doi: 10.1073/pnas.90.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogura T, Hirota S, Proshlyakov D A, Shinzawa-Itoh K, Yoshikawa S, Kitagawa T. J Am Chem Soc. 1996;118:5443–5449. [Google Scholar]
- 26.Varotsis C, Woodruff W H, Babcock G T. J Biol Chem. 1990;265:11131–11136. [PubMed] [Google Scholar]
- 27.McDonald M R, Fredricks F C, Margerum D W. Inorg Chem. 1997;36:3119–3124. doi: 10.1021/ic9608713. [DOI] [PubMed] [Google Scholar]
- 28.McDonald M R, Scheper W M, Lee H D, Margerum D W. Inorg Chem. 1995;34:229–237. [Google Scholar]
- 29.Picot D, Loll P J, Garavito R M. Nature (London) 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 30.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 31.Hosler J P, Ferguson-Miller S, Calhoun M W, Thomas J W, Hill J, Lemieux L, Ma J, Georgiou C, Fetter J, Shapleigh J, et al. J Bioenerg Biomembr. 1993;25:121–136. doi: 10.1007/BF00762854. [DOI] [PubMed] [Google Scholar]
- 32.Ostermeier C, Harrenga A, Ermler U, Michel H. Proc Natl Acad Sci USA. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klinman J P. Chem Rev (Washington, D C) 1996;96:2541–2561. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]
- 34.Babcock G T, Espe M, Hoganson C, Lydakis-Simantiris N, McCracken J, Shi W J, Styring S, Tommos C, Warncke K. Acta Chem Scand. 1997;51:533–540. doi: 10.3891/acta.chem.scand.51-0533. [DOI] [PubMed] [Google Scholar]
- 35.Oliveberg M, Hallén S, Nilsson T. Biochemistry. 1991;30:436–440. doi: 10.1021/bi00216a019. [DOI] [PubMed] [Google Scholar]
- 36.Hoganson C W, Babcock G T. Science. 1997;277:1953–1956. doi: 10.1126/science.277.5334.1953. [DOI] [PubMed] [Google Scholar]
- 37.Blair D F, Witt S N, Chan S I. J Am Chem Soc. 1985;107:7389–7399. [Google Scholar]
- 38.Ksenzenko M Yu, Vygodina T V, Berka V, Ruuge E K, Konstantinov A A. FEBS Lett. 1992;297:63–66. doi: 10.1016/0014-5793(92)80328-e. [DOI] [PubMed] [Google Scholar]
- 39.Vygodina T V, Schmidmaier K, Konstantinov A A. Biol Mem. 1993;6:883–906. [Google Scholar]
- 40.Wikström M. Nature (London) 1989;338:776–778. doi: 10.1038/338776a0. [DOI] [PubMed] [Google Scholar]
- 41.Mitchell R, Mitchell P, Rich P R. Biochim Biophys Acta. 1992;1101:188–191. [PubMed] [Google Scholar]
- 42.Dunford H B, Stillman J S. Coord Chem Rev. 1976;19:187–251. [Google Scholar]
- 43.Lambeir A-M, Markey C M, Dunford H B, Marnett L J. J Biol Chem. 1985;260:14894–14896. [PubMed] [Google Scholar]
- 44.Yonetani T. J Biol Chem. 1965;240:4509–4514. [PubMed] [Google Scholar]
- 45.Proshlyakov D A, Paeng I R, Paeng K-J, Kitagawa T. Biospectroscopy. 1996;2:317–329. [Google Scholar]
- 46.Courtin F, Deme D, Virion A, Michot J L, Pommier J, Nunez J. Eur J Biochem. 1982;124:603–609. doi: 10.1111/j.1432-1033.1982.tb06637.x. [DOI] [PubMed] [Google Scholar]
- 47.George P, Irvine D H. Biochem J. 1952;52:511–517. doi: 10.1042/bj0520511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chuang W-J, Heldt J, Van Wart H E. J Biol Chem. 1989;264:14209–14215. [PubMed] [Google Scholar]
- 49.Nakajima R, Yamazaki I, Griffin B W. Biochem Biophys Res Commun. 1985;128:1–6. doi: 10.1016/0006-291x(85)91635-3. [DOI] [PubMed] [Google Scholar]
- 50.Oertling W A, Hoogland H, Babcock G T, Wever R. Biochemistry. 1988;27:5395–5400. doi: 10.1021/bi00415a002. [DOI] [PubMed] [Google Scholar]
- 51.Vanneste W H. Biochemistry. 1966;5:838–848. doi: 10.1021/bi00867a005. [DOI] [PubMed] [Google Scholar]
- 52.Morgan J E, Verkhovsky M I, Puustinen A, Wikström M. Biochemistry. 1995;34:15633–15637. doi: 10.1021/bi00048a005. [DOI] [PubMed] [Google Scholar]
- 53.Wrigglesworth J M, Ioannidis N, Nicholls P. Ann N Y Acad Sci. 1988;550:150–160. doi: 10.1111/j.1749-6632.1988.tb35331.x. [DOI] [PubMed] [Google Scholar]
- 54.Han S, Ching Y-c, Rousseau D L. Nature (London) 1990;348:89–90. doi: 10.1038/348089a0. [DOI] [PubMed] [Google Scholar]
- 55.Kitagawa T, Mizutani Y. Coord Chem Rev. 1994;135/136:685–735. [Google Scholar]
- 56.Oertling W A, Kean R T, Wever R, Babcock G T. Inorg Chem. 1990;29:2633–2645. [Google Scholar]
- 57.Hayashi Y, Yamazaki I. Arch Biochem Biophys. 1978;190:446–453. doi: 10.1016/0003-9861(78)90297-7. [DOI] [PubMed] [Google Scholar]
- 58.Teraoka J, Kitagawa T. J Biol Chem. 1981;256:3969–3977. [PubMed] [Google Scholar]
- 59.Yoshikawa, S., Shinzawa-Itoh, K., Nakashima, R., Yaono, R., Yamashita, E., Inoue, N., Yao, M., Fei, M. J., Libeu, C. P., Mizushima, T., et al., Science, in press. [DOI] [PubMed]