

Abstract
Brain abscesses form in response to a parenchymal infection by pyogenic bacteria, with Staphylococcus aureus representing a common etiologic agent of human disease. Numerous receptors that participate in immune responses to bacteria, including the majority of TLRs, the IL-1R, and the IL-18R, use a common adaptor molecule, MyD88, for transducing activation signals leading to proinflammatory mediator expression and immune effector functions. To delineate the importance of MyD88-dependent signals in brain abscesses, we compared disease pathogenesis using MyD88 knockout (KO) and wild-type (WT) mice. Mortality rates were significantly higher in MyD88 KO mice, which correlated with a significant reduction in the expression of several proinflammatory mediators, including but not limited to IL-1β, TNF-α, and MIP-2/CXCL2. These changes were associated with a significant reduction in neutrophil and macrophage recruitment into brain abscesses of MyD88 KO animals. In addition, microglia, macrophages, and neutrophils isolated from the brain abscesses of MyD88 KO mice produced significantly less TNF-α, IL-6, MIP-1α/CCL3, and IFN-γ-induced protein 10/CXCL10 compared with WT cells. The lack of MyD88-dependent signals had a dramatic effect on the extent of tissue injury, with significantly larger brain abscesses typified by exaggerated edema and necrosis in MyD88 KO animals. Interestingly, despite these striking changes in MyD88 KO mice, bacterial burdens did not significantly differ between the two strains at the early time points examined. Collectively, these findings indicate that MyD88 plays an essential role in establishing a protective CNS host response during the early stages of brain abscess development, whereas MyD88-independent pathway(s) are responsible for pathogen containment.
Brain abscesses develop in response to a parenchymal infection with pyogenic bacteria, beginning as a localized area of cerebritis and evolving into a suppurative lesion surrounded by a well-vascularized fibrotic capsule (1). Brain abscesses are typified by extensive edema and tissue necrosis and tend to localize at white-gray matter junctions where microcirculatory flow is poor (2, 3). In addition to the sequential progression from cerebritis to necrosis during brain abscess evolution, the activation of resident glial cells and influx of peripheral leukocytes demonstrate temporal patterns (1). Specifically, microglial and astrocyte activation is evident immediately following the entry of bacteria into the CNS parenchyma and persists throughout abscess evolution (4, 5). Neutrophils are the initial leukocyte subset to infiltrate developing abscesses and are observed as early as 12 h following bacterial exposure (5, 6). Macrophages and T cells are associated with lesions as they progress, with infiltrates generally peaking around days 3 and 7, respectively (5, 7). Beginning around 7 to 10 days postinfection, a highly vascularized fibrotic wall forms around the necrotic milieu, effectively forming a barrier to contain the infection (5, 8). It is important to note that the kinetics of cellular activation, influx, and bordering functions represent general time frames and there is likely overlap between each of these processes during brain abscess evolution. Despite recent advances made in detection and therapy, brain abscess remains a serious CNS infectious disease that can lead to long-term complications including seizures, loss of mental acuity, and focal neurological defects that are lesion site-dependent (9, 10). Common etiologic agents of brain abscess are the Streptococcal strains and Staphylococcus aureus (9, 10). Based upon its prevalence in human CNS infection, our laboratory has used S. aureus to establish an experimental brain abscess model in the mouse that accurately reflects the course of disease progression in humans, providing an excellent model system to identify critical molecules responsible for the establishment of CNS antibacterial immunity (1, 6, 11).
Identifying the signals, effectors, and sequence of innate immune responses in the experimental brain abscess model has been a focus of our laboratory over recent years (1, 5, 6, 11–13). A pivotal component of innate immunity is the intracellular protein MyD88, which represents the central adapter protein for the majority of the TLRs (1) identified to date (with the exception of TLR3) in addition to transducing activation signals emanating from the IL-1 and IL-18 receptors (14 –16). Previous studies have demonstrated that MyD88 knockout (KO)3 mice exhibit dramatic defects in antibacterial immunity in a variety of infectious disease models, highlighting the importance of this adapter in influencing a wide array of host responses (17–21).
In considering the host immune responses that ensue during brain abscess development, MyD88-dependent signals are likely to impact a number of effector pathways. First, conserved structural motifs of S. aureus termed pathogen-associated molecular patterns likely serve as triggers for multiple TLR family members (22, 23). For example, TLR2 has been shown to mediate the recognition of Gram-positive peptidoglycan and bacterial lipoproteins and has well-described effects on mediating host innate immune responses to numerous Gram-positive species (20, 24 –29). In addition, other TLRs that may participate in bacterial recognition in concert with TLR2 during brain abscess evolution include TLR1 and TLR6, which form functional heterodimers with TLR2 (30 –33), in addition to TLR9, which recognizes bacterial DNA (34, 35). A total of 13 TLRs have been identified to date, and almost every TLR has been found to be expressed in the CNS or in purified glia (i.e., microglia and astrocytes), suggesting that this family of pattern recognition receptors plays an important role in CNS innate immune responses (36 – 44).
In addition to TLRs, MyD88 is an essential component of signaling via the IL-1 and IL-18 receptors (16, 17, 45, 46). Previous studies from our laboratory have demonstrated that IL-1 is pivotal during the acute phase of brain abscess development to control bacterial burdens and ensure host survival (12). Therefore, any effects observed following the loss of MyD88 could conceivably originate from defects i TLR, IL-1, or IL-18 signaling either alone or in combination. We have not yet examined the functional importance of IL-18 in the experimental brain abscess model; therefore, any potential contributions from this pathway emanating from MyD88-dependent activation remain unknown.
Although one report has recently described an important role for MyD88-dependent signals in bacterial meningitis (47), the functional significance of this central adaptor in regulating the host innate immune response to brain abscess has not yet been examined and may differ from that for meningitis based upon the highly focal nature of lesions in the former and the cell types available to immediately respond to infection. Therefore, to determine the functional importance of MyD88-dependent pathways in the CNS response to S. aureus, we evaluated brain abscess pathogenesis in MyD88 KO and wild-type (WT) mice. The results presented demonstrate that MyD88-dependent mechanisms play an essential role in eliciting a protective innate immune response during the acute phase of brain abscess development, whereas alternative pathway(s) are responsible for pathogen containment.
Materials and Methods
Mice
MyD88 gene KO mice (originally from Dr. S. Akira, Osaka University, Osaka, Japan) (17) were originally purchased from the Centre de La Recherche Scientifique (Paris, France) and have been previously backcrossed with C57BL/6 mice for over 10 generations (18, 48). Age- and sex-matched C57BL/6 mice (Harlan Sprague Dawley) were used as WT controls. For all brain abscess studies, MyD88 KO and WT mice were used between 6 and 8 wk of age.
Generation of experimental brain abscess
Live S. aureus (strain RN6390, provided by Dr. A. Cheung, Dartmouth Medical School, Hanover, NH) were encapsulated in agarose beads before implantation in the brain as previously described (6). Earlier studies from our laboratory have established that the introduction of sterile agarose beads does not induce detectable inflammation or peripheral immune cell infiltrates (5, 6). To induce brain abscesses, mice were anesthetized with 2.5% Avertin i.p. and a 1-cm longitudinal incision was made along the vertex of the skull extending from the ear to the eye. A rodent stereotaxic apparatus equipped with a Cunningham mouse adaptor (Stoelting) was used to implant S. aureus-encapsulated beads into the caudate putamen using the following coordinates relative to bregma: plus 1.0 mm rostral, plus 2.0 mm lateral, and minus 3.0 mm deep from the surface of the brain. A burr hole was made and a 5-μl Hamilton syringe fitted with a 26-gauge needle was used to slowly deliver 2-μl beads (104 CFU) into the brain parenchyma. The needle remained in place for 2.5 min following injection to minimize bead efflux and potential leakage into the meninges. The burr hole was sealed with bone wax and the incision closed using surgical glue. The animal use protocol, approved by the University of Arkansas for Medical Sciences Institutional Animal Care and Use Committee (Little Rock, AR), is in accord with the National Institutes of Health guidelines for the use of rodents. Our extensive experience with the experimental brain abscess model has established that bacterial burdens expand rapidly during the first 24 h following infection (by 1 to 2 log compared with the initial inoculum) and reach peak levels by ~3–5 days postinfection (5, 12). We routinely deliver equivalent bacterial burdens between individual animals as evident by the fact that many of our previous studies, including the present one, demonstrate no alterations in bacterial replication between many of the WT or KO strains we have tested in the experimental brain abscess model (11) (T. Kielian, unpublished observations).
Preparation of brain abscess homogenates
To prepare brain abscess homogenates for downstream protein analysis, lesion sites were visualized by the stab wound created during injections and sectioned within 1 to 2 mm on all sides. Upon recovery, brain abscesses were homogenized in 500 μl of PBS supplemented with a Complete protease inhibitor cocktail tablet (Roche) and 160 U/ml RNase inhibitor (Promega) using a Polytron homogenizer (Brinkmann Instruments). At this point, a 20-μl aliquot of abscess homogenate was removed for a quantitative culture of viable bacteria as described below. Subsequently, homogenates were centrifuged at 14,000 rpm for 15 min at 4°C to pellet membrane material and supernatants were removed and stored at −70°C until ELISA and multiplex cytokine microbead array analysis as described below.
Quantitation of viable bacteria from brain abscesses
To quantitate the numbers of viable bacteria associated with brain abscesses, serial 10-fold dilutions of abscess homogenates were plated onto blood agar plates (BD Biosciences). Titers were calculated by enumerating colony growth and are expressed as CFU per milliliter of homogenate.
ELISA
Protein levels of murine MIP-2 were quantified in brain abscess homogenates using an ELISA kit (DuoSet; R&D Systems) according to the manufacturer’s instructions (level of sensitivity = 15.6 pg/ml). Results were normalized to the amount of total protein extracted from tissues to correct for differences in sampling size as previously described (5, 12).
Use of a multianalyte array to detect proinflammatory mediator expression
To expand the analysis of inflammatory mediators differentially expressed between MyD88 KO and WT mice, a mouse 20-plex cytokine microbead array system was used according to the manufacturer’s instructions (BioSource International). This microbead array allows for the simultaneous detection of 20 individual inflammatory molecules in a single 75-μl brain abscess homogenate sample including IL-1α, IL-1β, TNF-α, IFN-γ, IL-2, IL-3, IL-4, IL-5, IL-6, IL-10, IL-12 p40/p70, IL-13, IL-17, IFN-g-induced protein 10 (IP-10), monokine induced by IFN-γ, MCP-1, KC, GM-CSF, vascular endothelial growth factor, and basic fibroblast growth factor. Results were analyzed using a Bio-Plex workstation (Bio-Rad) and adjusted based on the amount of total protein extracted from abscess tissues for normalization. The level of sensitivity for each microbead cytokine standard curve ranged from 1 to 35 pg/ml.
Quantitation of cellular infiltrates in brain abscesses of MyD88 KO and WT mice by FACS analysis
To determine whether MyD88-dependent signals regulated the influx of neutrophils, macrophages, and/or microglia into brain abscesses, abscess-associated cells were quantitated by FACS analysis as previously described with minor modifications (49 –51). Briefly, mice were perfused to eliminate leukocytes from the vasculature, whereupon the entire infected hemisphere was collected to recover abscess-associated cells. This approach ensured that equivalent tissue regions were procured from both MyD88 KO and WT mice for downstream comparisons of leukocyte infiltrates. Following vascular perfusion, tissues were minced in HBSS (Mediatech) supplemented with 10% FBS and filtered through a 70-μm nylon mesh cell strainer using a rubber policeman. The resulting slurry was digested for 30 min at 37°C in HBSS supplemented with 2 mg/ml collagenase type I (Sigma-Aldrich) and 5000 U/ml DNase I (Invitrogen Life Technologies) to obtain a single-cell suspension. Following enzyme neutralization, cells were layered onto a discontinuous Percoll gradient (1.03–1.088 g/ml) and centrifuged at 2,400 rpm for 20 min at room temperature in a swinging bucket rotor. After centrifugation, myelin debris was carefully aspirated and the cell interface collected. Following extensive washing, cells were stained with directly conjugated Abs (Gr-1-allophycocyanin, CD11b-Alexa 488, and CD45-PE, all from BD Pharmingen) to detect neutrophils (Gr-1+, CD11b+, and CD45high), macrophages (Gr-1−, CD11b+, and CD45high), and microglia (Gr-1−, CD11b+, and CD45low-int, where int is intermediate). Cells were analyzed using a BD FACSAria (BD Biosciences) cytometer with a compensation set based on the staining of each individual fluorochrome alone and correction for autofluorescence with unstained cells. Controls included cells stained with directly conjugated isotype control Abs to assess the degree of nonspecific staining.
For reporting the differences in cellular influx between MyD88 KO and WT mice, we normalized (i.e., divided) the numbers of FACS-purified neutrophils, macrophages, or microglia recovered from MyD88 WT mice by those collected from MyD88 KO animals to express the latter as a percentage of WT (set to 100%). This approach was required because it is difficult to achieve identical bacterial burdens in mice between independent brain abscess experiments. As a result, the absolute numbers of infiltrating cells within brain abscesses of MyD88 WT and KO mice differed between individual experiments, requiring us to normalize our data within each independent replicate. Therefore, this method enabled us to pool the results from three independent experiments because the relative ratio of each cell type between MyD88 KO and WT mice remained consistent throughout our independent replicates.
Quantitation of brain abscess size
At the indicated time points postinfection (i.e., 24 – 42 h), MyD88 KO and WT mice were perfused transcardially to eliminate leukocytes from the vasculature, whereupon brains were removed and immediately flash frozen on dry ice. Before cryostat sectioning, brain tissues were embedded in optimal cutting temperature medium and serial 10-μm sections were made throughout the entire lesioned tissue and stained with H&E to demarcate abscess margins. The evaluation of serial sections throughout the affected brain parenchyma ensured that the largest abscess cross-sectional area would be identified for comparisons of lesion size. Abscess area (reported as mm2) was calculated using the MetaMorph image analysis program (Universal Imaging). Pathological changes associated with brain abscesses from MyD88 KO and WT mice were assessed by a neuropathologist (Dr. R. Mrak, Department of Pathology, University of Arkansas for Medical Sciences, Little Rock, AR) who was blinded to treatment groups.
Statistics
Significant differences between experimental groups were determined using the unpaired Student’s t test at the 95% confidence interval with Sigma Stat (SPSS Science). This analysis was determined to be most appropriate because, although we are evaluating changes in proinflammatory mediator expression over time, repeated measurements are not made on the same animal (mice are sacrificed to collect abscess homogenates at each time point), precluding ANOVA and post hoc analysis of the data.
Results
MyD88-dependent signals are essential for the widespread induction of proinflammatory mediators during the acute phase of brain abscess development
One of the main pathogens associated with brain abscesses in humans is the Gram-positive bacterium S. aureus (1, 9, 10). We have previously demonstrated that MyD88 plays an important role in enabling microglia to recognize S. aureus and respond with the robust production of numerous cytokines and chemokines (37). However, the functional importance of this central adapter molecule for TLR, IL-1R, and IL-18R signaling during brain abscess development remains to be demonstrated. To address this question, we compared brain abscess pathogenesis in MyD88 KO and WT mice.
MyD88-dependent signal(s) were essential in affording survival during the first 24 h following intracerebral S. aureus injections, as revealed by a significant decrease in survival in MyD88 KO mice compared with WT animals (18.3 ± 6.6 vs 58.9 ± 2.6%, respectively, p < 0.05; n = 33 KO and 38 WT mice). Throughout the course of our studies, only a total of 4 of 42 MyD88 KO mice survived to 42– 48 h following S. aureus exposure, with most animals succumbing to infection at earlier time points (i.e., 24 –36 h), even when the bacterial inoculum was reduced. Because most infected MyD88 KO mice were moribund before this 42– 48 h interval they were sacrificed, which precluded us from determining long-term survival data and statistical analysis. Based on this finding, the studies reported here were conducted primarily during the first 24 h following S. aureus infection.
The dramatic sensitivity of MyD88 KO mice to intracerebral S. aureus led us to hypothesize that these animals may have defects in inducing a protective antibacterial inflammatory response in the CNS parenchyma. To examine this possibility, we initially examined the expression of several proinflammatory mediators previously determined to play a pivotal role in the host response during the acute phase of brain abscess development, namely IL-1β, MIP-2, and MCP-1 (6, 12). As shown in Fig. 1, the expression of all three mediators was significantly attenuated in brain abscesses of MyD88 KO mice at 24 h following bacterial exposure. Of these, IL-1β was the most significantly affected with levels in MyD88 KO mice ~5-fold lower compared with WT animals (Fig. 1). Based on the dramatic reduction in proinflammatory mediator expression coupled with increased mortality rates in MyD88 KO mice, it was envisioned that these differences would correlate with enhanced bacterial burdens in KO animals. Surprisingly, this was not the case because the number of viable S. aureus organisms within brain abscesses was similar between MyD88 KO and WT mice at 24 h following bacterial exposure (1.48 × 106 ± 3.44 × 105 vs 1.28 × 106 ± 3.39 × 105, respectively, p = 0.542; n = 3–5 mice per group in each of four independent experiments). This finding indicates that MyD88-dependent signal(s) do not play an important role in dictating pathogen burdens in brain abscesses, at least at the early time points examined in this study.
FIGURE 1.

MyD88 KO mice exhibit defects in the induction of several proinflammatory mediators. MyD88 KO and WT mice (n = 4 or 5 per group) were sacrificed 1 day following S. aureus infection, whereupon IL-1β, MIP-2, and MCP-1 protein levels were quantitated by ELISA (mean ± SD). Results were normalized to the amount of total protein recovered to correct for differences in abscess sampling size. Significant differences are denoted by asterisks (*, p < 0.05; **, p < 0.001). Results are representative of three independent experiments.
To delineate the kinetics of attenuated mediator expression in MyD88 KO mice during the acute stage of brain abscess development, cytokine and chemokine levels were quantitated in MyD88 WT and KO mice at 6, 12, and 24 h following bacterial exposure using a multiplex cytokine microbead array capable of simultaneously detecting 20 individual mediators in a single abscess homogenate sample. The results demonstrated a time-dependent induction of proinflammatory mediators following the intra-cerebral inoculation of S. aureus. For example, IL-12 p70 and the neutrophil chemokines MIP-2/CXCL2 and KC/CXCL2 were induced as early as 6 h postinfection, whereas numerous mediators including IL-1α and β, IL-6, IL-10, MCP-1/CCL2, and MIP-1α/CCL3 were not detected until 12 h following bacterial exposure (Figs. 2 and 3). Importantly, the majority of these proteins were significantly reduced in brain abscesses of MyD88 KO mice compared with WT animals (Figs. 2 and 3), although a few differences were observed relative to the timing of when the alterations in protein levels between WT and KO mice were first evident. For example, MIP-2 expression was not significantly attenuated in brain abscesses of MyD88 KO mice until 12–24 h postinfection (Fig. 3A). In addition, some mediators appeared to be more robustly affected by the loss of MyD88 including IL-1β, IL-12 p70, IL-6, KC, and MIP-1α, whereas others (i.e., MCP-1 and MIP-2) were not as dramatically reduced in MyD88 KO animals. Importantly, IL-10 and basic fibroblast growth factor expression within brain abscesses of MyD88 WT and KO mice were equivalent at all time points examined (Fig. 2E and data not shown), indicating that the loss of MyD88-dependent signals targets the expression of a select subset of genes. The relatively low protein concentrations detected using this highly sensitive fluorescence-based array can be attributed to the inability of mediators to accumulate to sufficiently high levels at the early time points examined and the fact that abscesses were homogenized in a sizeable buffer volume (500 μl) to facilitate efficient homogenization. Collectively, these results indicate that MyD88-dependent signals play a pivotal role in the establishment of proinflammatory mediator expression in the CNS parenchyma during the acute stage of brain abscess development.
FIGURE 2.

MyD88-dependent signals control the expression of select cytokines during the early stage of brain abscess development. MyD88 KO and WT mice (n = 4 to 5 per group) were infected with S. aureus intracerebrally as described in Materials and Methods. Animals were sacrificed at 6, 12, or 24 h following bacterial exposure, whereupon multiplex microbead array analysis was performed on abscess homogenates to allow for the simultaneous quantitation of 20 distinct mediators at the protein level. Results are shown for abscess-associated IL-1β (A), IL-1α (B), IL-12 p70 (C), IL-6 (D), and IL-10 (E) (mean ± SD) and were normalized based on the amount of total protein recovered from abscesses to correct for differences in tissue sampling size. Significant differences in cytokine expression between brain abscesses in MyD88 KO and WT mice are indicated by asterisks (*, p < 0.05; **, p < 0.001). Results are representative of two independent experiments.
FIGURE 3.

Chemokine induction during acute brain abscess development is highly dependent on MyD88. MyD88 KO and WT mice (n = 4 or 5 per group) were infected with S. aureus intracerebrally as described in Materials and Methods. Animals were sacrificed at 6, 12, or 24 h following bacterial exposure, whereupon multiplex microbead array analysis was performed on abscess homogenates to allow the simultaneous quantitation of 20 distinct mediators at the protein level. Results are shown for abscess-associated MIP-2 (A), KC (B), MIP-1α (C), and MCP-1 (D) (mean ± SD) and were normalized based on the amount of total protein recovered from abscesses to correct for differences in tissue sampling size. MIP-2 levels were determined using a standard sandwich ELISA approach because this chemokine was not included in the multiplex array. Significant differences in chemokine expression between brain abscesses of MyD88 KO and WT mice are indicated by asterisks (*, p < 0.05; **, p < 0.001). Results are representative of two independent experiments.
Neutrophil and macrophage influx into developing brain abscesses is significantly attenuated in MyD88 KO mice
The rather broad-scale reduction in chemokine expression in brain abscesses of MyD88 KO mice led us to examine whether this finding extended to alterations in the degree of peripheral immune cell influx into lesions. To quantitate the potential differences in neutrophils, macrophages, and microglia between MyD88 KO and WT mice, three-color FACS analysis was used. As shown in Fig. 4, MyD88 KO mice demonstrated a dramatic reduction in neutrophil infiltrates into evolving brain abscesses, with an approximate 8-fold decrease in the relative percentage of neutrophils compared with that in WT animals. Macrophage infiltrates were also significantly lower in MyD88 KO mice; however, this difference was not as dramatic compared with that for neutrophils (Fig. 4). The relative percentage of microglia detected in MyD88 KO mice was significantly higher compared with WT animals. This result is likely due to the fact that both neutrophil and macrophage infiltrates were reduced in MyD88 KO mice, leading to an apparent increase in the proportion of microglia detected rather than an absolute increase in microglial numbers.
FIGURE 4.

Neutrophil and macrophage influx into brain abscesses is significantly impaired in MyD88 KO mice. MyD88 KO and WT mice (n = 10 per group) were infected with S. aureus intracerebrally and sacrificed at 12 h following bacterial exposure, whereupon abscess-associated neutrophils (Gr-1+, CD11b+, and CD45high), macrophages (Gr-1−, CD11b+, and CD45high), and microglia (Gr-1−, CD11b+, and CD45low) were quantitated by FACS analysis. Results are expressed as the percentage of cells normalized to WT values (set at 100%) and represent the mean ± SEM from three independent experiments. Significant differences in abscess-associated cells recovered from MyD88 KO vs WT animals are denoted by asterisks (*, p < 0.05; **, p < 0.001).
To determine whether abscess-associated neutrophils, macrophages, or microglia from MyD88 KO or WT mice displayed any differences in cytokine expression profiles immediately ex vivo, we performed multiplex microbead arrays on conditioned supernatants from each cell type following a 24-h incubation period in vitro without bacterial restimulation. Neutrophils recovered from the abscesses of MyD88 KO mice demonstrated defects in the production of numerous proinflammatory mediators, including IL-6, MIP-1α, and IP-10, compared with WT cells (Fig. 5A). This pattern of reactivity mimicked what was observed with abscess homogenates. Fewer significant changes were observed between abscess-associated macrophages recovered from MyD88 KO and WT mice, with only IL-6 significantly attenuated in the former (Fig. 5B). Although MyD88 KO abscess-associated macrophages also expressed less TNF-α and MIP-1α compared with WT cells, these differences did not approach statistical significance (Fig. 5B). Interestingly, IL-10 expression did not significantly differ in both neutrophils and macrophages recovered from the abscesses of MyD88 KO and WT mice (Fig. 5, A and B), which is in agreement with the finding that IL-10 levels were not significantly altered in brain abscess homogenates (Fig. 2E). Microglia isolated from abscesses of MyD88 KO mice revealed the most extensive deficits in proinflammatory mediator production. Specifically, all mediators tested were reduced in abscess-associated microglia from MyD88 KO mice; however, only a subset (i.e., IL-6, TNF-α, and IL-10) reached statistical significance (Fig. 5C). The relatively low levels of mediator expression detected in abscess-associated cells by the fluorescence-based microbead array may be explained by the fact that cells were recovered relatively early following S. aureus infection in the brain (i.e., 12 h) and we elected not to restimulate the various cell types with S. aureus during the subsequent 24 h in vitro culture period in an attempt to reflect their activation state in situ. Together, these results demonstrate that MyD88-dependent signal(s) are important for neutrophil and macrophage recruitment into the infected CNS and their subsequent activation in response to parenchymal S. aureus infection. Microglia also use MyD88 to trigger the expression of numerous proinflammatory mediators, which is likely a key step toward initiating the CNS innate immune response to bacterial infection.
FIGURE 5.

The expression of numerous cytokines and chemokines is attenuated in abscess-associated cells from MyD88 KO mice. Abscess-associated neutrophils (A), macrophages (B), and microglia (C) were recovered from the lesions of MyD88 KO and WT mice at 12 h following S. aureus infection as described in Materials and Methods. Cells were cultured overnight without additional stimulation and conditioned supernatants were analyzed for mediator expression by multiplex microbead array assays. The amount of each cytokine/chemokine was normalized based on total cell numbers and represents the mean ± SEM from three independent experiments. Significant differences in mediator expression between the various cell types recovered from MyD88 KO vs WT mice are denoted with asterisks (*, p < 0.05; **, p < 0.001).
Brain abscess size is exacerbated in MyD88 KO mice
The finding that proinflammatory mediator expression and cellular influx was significantly attenuated in the brain abscesses of MyD88 KO mice led us to consider that the degree of tissue injury may be more severe in these animals. The extent of brain abscess involvement was determined by performing H&E stains on serial tissue sections prepared throughout the entire lesion to identify the largest abscess cross-sectional area, and pathological changes were evaluated by a neuropathologist who was blinded to the various experimental groups. As shown in Fig. 6, abscesses were significantly more expansive in MyD88 KO mice compared with WT animals. Another striking difference was the fact that the infected hemisphere in MyD88 KO mice had lost any resemblance of normal parenchyma and was grossly edematous, distended, and displayed more extensive necrosis compared with WT tissues. Whereas focal, well-demarcated abscesses were evident in WT animals with the characteristic accumulation of neutrophils in the necrotic center as evident by H & E staining, lesions were ill-defined in MyD88 KO mice and lacked the focal concentration of infiltrating leukocytes (Fig. 6A). The quantitation of lesion sizes demonstrated that abscess size was significantly increased in MyD88 KO mice at all time points examined (Fig. 6B). We were not able to evaluate lesions later than 42 h postinfection because very few MyD88 KO mice survived past this time interval. Collectively, these results suggest that MyD88-dependent signals play a pivotal role in restricting the extent of parenchymal damage during brain abscess evolution.
FIGURE 6.
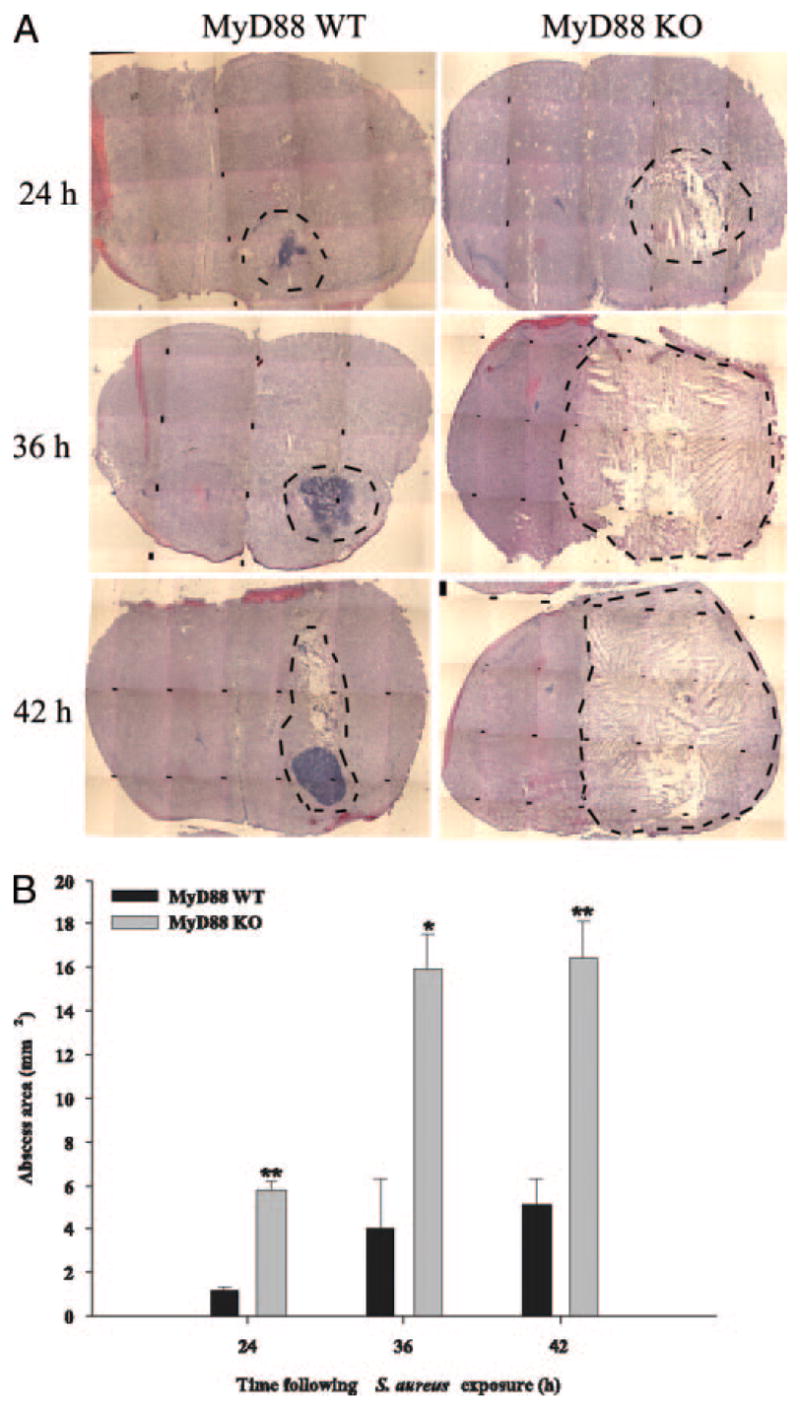
Brain abscess size is significantly greater in MyD88 KO mice. MyD88 KO and WT mice (n = 3 or 4 per group per time point) were injected intracerebrally with S. aureus and sacrificed at 24, 36, and 42 h following bacterial exposure, whereupon brain tissues were flash frozen on dry ice for subsequent cryostat sectioning. Serial sections were prepared throughout the entire abscess to ensure that the maximal cross-sectional area was identified and stained with H&E to demarcate the extent of tissue damage. A, A series of microscopic images (original magnification, ×4) were assembled for each individual MyD88 KO or WT animal to demonstrate the extent of abscess formation and edema/necrosis. Abscess margins (including cerebritis) are demarcated with dotted lines. B, Abscess area (mm2; mean ± SD) was quantitated using the MetaMorph image analysis program by measuring the largest lesion size for each tissue specimen. Significant differences in brain abscess size between MyD88 KO vs WT mice are denoted with asterisks (*, p < 0.05; **, p < 0.001).
Discussion
This study has demonstrated an essential role for MyD88-dependent signals during the early phase of brain abscess development. In particular, the loss of MyD88 led to a significant decrease in survival, which correlated with the complete disruption of normal brain tissue architecture in the infected parenchyma. Concomitant with these outcomes was the inability to recruit significant numbers of neutrophils and macrophages into the brain parenchyma of MyD88 KO animals, the former representing a leukocyte population that we had previously demonstrated to be essential during the acute stage of brain abscess formation (6). Finally, MyD88 KO mice displayed significant decreases in the expression of numerous proinflammatory mediators, which likely contributed, in part, to the impaired leukocyte recruitment observed in KO animals.
One striking finding of this study was the important contribution of MyD88-dependent signals in regulating the extent of parenchymal damage during the acute stage of brain abscess formation. Indeed, brain abscesses were grossly disseminated in MyD88 KO mice and often encompassed the entire injected hemisphere, whereas lesions were contained and well-demarcated in WT animals at all time points examined. In addition, MyD88-dependent signals were pivotal for regulating the extent of cerebral edema and necrosis because both were dramatically exaggerated in the brain abscesses of MyD88 KO mice; however, the identity of the downstream effectors of MyD88 responsible for these phenomena remain to be characterized. Currently, we do not have a good understanding of the pathological mechanisms that dictate whether animals succumb to infection during brain abscess development. However, interpretation of available studies suggests a multifactorial etiology in which both elevated cerebral edema and necrosis play a role (2, 3, 6). Cerebral edema is generally divided into two types, vasogenic and cytotoxic, depending on the mechanism of edema formation (52). Cytotoxic edema originates from the disruption of normal osmotic gradients across the cell membrane and the resultant influx of water into the cell and eventual cell swelling/lysis. In contrast, vasogenic edema occurs because of brain-blood barrier compromise, resulting in extracellular fluid accumulation. Both forms of cerebral edema can contribute to an increase in intracranial pressure; however, vasogenic edema is thought to represent the predominant response observed in brain abscess (2, 3). This pathogenic fluid dynamic in the CNS is likely one causative agent for the ensuing tissue necrosis observed in brain abscesses through the physical compression of cerebral vasculature and subsequent ischemia. Work by others has demonstrated that aquaporin 4 (AQP4), a key astrocytic water channel that is critical in controlling edematous responses in the CNS parenchyma, is up-regulated along brain abscess margins during disease evolution (2). The functional importance of AQP4 in regulating cerebral edema during brain abscess development was demonstrated by the finding that AQP4 KO mice displayed significantly elevated vasogenic edema and intracranial pressure compared with WT mice; however, AQP4 had no impact on brain abscess size or bacterial burdens (2). Importantly, the kinetics of increased brain water content in AQP4 KO mice was not observed until day 3 postinfection, which is well beyond the survival of infected MyD88 KO mice in our current study. It remains to be determined whether AQP4 is down-regulated in MyD88 KO mice, which would effectively result in increased cerebral edema as we have observed in these animals.
In addition to the exaggerated edematous response in brain abscesses of MyD88 KO mice, these lesions were also typified by extensive necrosis, which we propose is one mechanism that leads these animals to succumb to infection more rapidly. A recent study by Lehnardt et al. (53) has demonstrated that group B streptococci and their secreted bacterial factors induce neuronal cell death via a TLR2/MyD88-dependent pathway that is mediated by activated microglia. However, it is important to note that this study used heat-inactivated group B streptococci, which differs from our study investigating the CNS response to live organisms. Therefore, there is a clear distinction between the necrotic cell death that is associated with brain abscesses in the CNS parenchyma vs responses to heat-inactivated organisms that have been reported to induce the apoptotic death of neurons in vitro. In fact, although we have not yet examined the extent of apoptosis in brain abscesses, work by others has demonstrated that apoptosis represents a relatively minor component of parenchymal cell death in the brain abscess model (7). The necrotic cell death characteristic of brain abscesses differs from that described for bacterial meningitis, where apoptosis is implicated as one of the main pathophysiological effector mechanisms (54, 55). Another possibility to account for the accelerated mortality rate of MyD88 KO mice is that the animals may become septic due to the dramatic destruction of brain parenchyma and failure to contain lesions; however, this remains speculative at the present time. We do not routinely examine animals for evidence of sepsis because our extensive work with this model has demonstrated that bacteria are normally efficiently sequestered in the brain parenchyma of WT animals (T. Kielian, unpublished observations). We would like to reiterate the fact that bacterial burdens per se are not the sole deciding factor in dictating the extent of tissue injury and/or mortality during brain abscess development. This is supported by our histological findings depicting severe edema and necrosis in the brains of MyD88 KO mice at the later time points examined (i.e., 24 and 42 h postinfection) despite similar S. aureus levels between WT and KO mice. Cytokines and chemokines elaborated at high levels can also exert direct neurotoxic effects as demonstrated by others (56, 57), and this remains another potential mechanism of necrotic cell death in the brain abscess model. In addition, a reduction in NF-κB signaling, presumably through the loss of MyD88, might lead to a reduction in prosurvival signals (58), culminating in increased cell death. Currently, we do not know the exact molecular mechanisms responsible for necrotic cell death in the brain during abscess evolution, although several possibilities can be proposed such as depletion of cellular energy sources, direct toxic effects by bacterial virulence factors, and/or ischemia/hypoxia due to the mass effect of the edematous response to infection.
Neutrophil and macrophage accumulation in brain abscesses was significantly attenuated in MyD88 KO mice compared with WT animals, which correlated with a dramatic decrease in the production of numerous chemokines in the former. The number of abscess-associated neutrophils in MyD88 KO mice was more significantly affected compared with macrophages, which agreed with the finding that the expression of neutrophil chemokines (i.e., MIP-2 and KC) was more attenuated in MyD88 KO animals compared with macrophage chemoattractants (i.e., MCP-1 and MIP-1α). We have previously demonstrated that neutrophils play an essential role in bacterial containment in the brain abscess model (6). However, it is important to note that in this earlier study significant differences in bacterial burdens in mice with impaired neutrophil influx into the infected CNS were not observed until days 3 and 5 postinfection (6). Because our current experiments with MyD88 KO mice were limited by rapid mortality rates, it is likely that compensatory responses were sufficient to counteract the defects in neutrophil accumulation in these animals or that the low numbers of neutrophils reaching the abscess were sufficient to initially contain S. aureus infection in MyD88 KO mice early during the disease process. Unlike the majority of the inflammatory mediators examined, MIP-2 demonstrated a distinct temporal course of expression. Previous studies from our laboratory (6) have established that MIP-2 is one of the key chemokines responsible for dictating neutrophil entry into the brain parenchyma during brain abscess development (along with KC). Interestingly, MIP-2 levels were equivalent between MyD88 KO and WT mice at 6 h postinfection and did not become significantly attenuated in the former until 12 h following S. aureus exposure. The reason for this delay in MIP-2 attenuation in MyD88 KO mice may be explained by the presence of a MyD88-independent mechanism for chemokine production early following infection that is not potentiated due to the lack of a MyD88-dependent feedback loop, because numerous other proinflammatory mediators are also inhibited in MyD88 KO mice. An example may be IL-1α, which has been shown to enhance CXC chemokine mRNA stability (59). Therefore, one could postulate that the reduction in IL-1α expression observed in MyD88 KO mice may reduce the half-life of MIP-2; however, this response takes time to develop, effectively leading to the observed lag in chemokine inhibition. The question that remains is what signals are responsible for this initial apparent MyD88-independent increase in MIP-2 expression immediately following infection, an issue that currently remains unresolved.
In addition to regulating the induction of inflammatory gene transcription, MyD88-dependent signals have recently been shown to stabilize cytokine mRNA expression (60). Specifically, IFN-γ treatment of MyD88 KO macrophages led to significant decreases in TNF-α and IP-10 mRNA expression, which was a consequence of reduced mRNA half-life (60). We have recently found that CD4+ T cells recovered from brain abscesses produce significant levels of IFN-γ (T. Kielian, unpublished observations), indicating that this cytokine is a component of the abscess milieu and, by extension, that the lack of MyD88-dependent signaling may contribute to a decrease in TNF-α and IP-10 expression by reducing mRNA stability. Another interesting finding relates to the fact that IL-1 has also been reported to stabilize KC mRNA expression, whereas IL-10 decreases KC mRNA stability (59, 61). Because IL-1 expression is attenuated in brain abscesses of MyD88 KO mice whereas IL-10 levels are not affected, the net balance of these cytokines could account for the dramatic reduction in KC levels in lesions of MyD88 KO mice, in addition to reduced transcriptional activation due to diminished NF-κB-dependent induction, because KC and numerous other cytokine/chemokine genes are driven by this important transcription factor (62, 63).
Not only were the numbers of abscess-associated neutrophils and macrophages reduced in brain abscesses of MyD88 KO mice, but their activation status was also impaired. This was reflected by the lower expression levels of several cytokines and chemokines in MyD88 KO cells including IL-6, TNF-α, MIP-1α, and IP-10. In contrast to peripheral leukocyte influx, the relative percentage of microglia was increased in the brain abscesses of MyD88 KO mice. This finding may be explained by the fact that the high frequency of infiltrating neutrophils and macrophages into WT animals effectively reduced the percentage of abscess-associated microglia detected. In contrast, because neutrophil and macrophage influx is dramatically reduced in MyD88 KO animals, it allowed resident microglia to be more easily detected, effectively increasing the relative percentage of these cells. The more global attenuation of proinflammatory mediator release by abscess-associated microglia recovered from MyD88 KO mice was similar to findings in our previous report documenting the impaired responsiveness of MyD88 KO microglia to S. aureus in vitro (37). One issue that remains relates to whether the observed reduction in cytokine and chemokine production by neutrophils and microglia from MyD88 KO mice was due to increased cell death in culture. This is an important point, because the survival time of neutrophils in culture is relatively short and is compounded by the fact that in our study neutrophils were presumably activated because they were recovered from an infectious milieu. We normalized our cytokine/chemokine expression data to the number of cells plated per well because it was difficult to accurately enumerate cells after the overnight culture period using a hemocytomer due to limiting input numbers. Therefore, although we assume that neutrophil viability is minimal following this period, we cannot definitively make this statement. However, it should be noted that not all of the mediators examined were differentially expressed between purified MyD88 KO and WT cells. For example, IL-10 and TNF-α levels produced by MyD88 KO and WT abscess-associated neutrophils were equivalent, suggesting that the reduction of select mediators in MyD88 KO cells was not a result of cell death and the subsequent nondiscriminate release of intracellular cytokines. Similarly, the production of IL-10 and IP-10 was not altered between MyD88 KO and WT macrophages and MIP-1α release was not significantly affected in WT and KO microglia. Collectively, these findings also argue against cell death-dependent effects of mediator release, although this possibility cannot be completely discounted.
One intriguing and unexpected finding that surfaced during the course of these studies was the fact that bacterial burdens were equivalent in MyD88 KO and WT mice despite the dramatic inhibition of CNS innate immune responses in the former. Several mechanisms have been shown to contribute to S. aureus killing, including complement, anti-microbial peptides (i.e., defensins), lysozyme, phagocytosis, and the generation of reactive oxygen/nitrogen intermediates, among others (64). In our initial studies we quantitated the number of abscess-associated bacteria in MyD88 KO and WT mice that survived to 24 h following infection. Because no differences in bacterial burdens were evident, we reasoned that perhaps bacterial replication was enhanced in those MyD88 KO mice that had died before the initial 24 h time point examined. To investigate this possibility, we sacrificed MyD88 KO and WT animals at 6, 12, and 24 h such that bacterial burdens could be determined in the hours leading up to the time interval in which MyD88 KO mice typically succumbed to infection. Surprisingly, there was no evidence of altered bacterial replication in MyD88 KO mice at any of the time points examined, despite the dramatic inhibition of proinflammatory mediator expression and the influx of professional phagocytes into lesions. Previous studies from our laboratory have demonstrated that IL-1 is pivotal for bacterial containment during the acute phase of brain abscess pathogenesis (12). However, similar to what was observed in the current study with MyD88 KO mice, bacterial burdens were equivalent in IL-1 KO and WT mice at 24 h postinfection and did not become significantly elevated in the former until day 3 following bacterial exposure. In addition, the relative bacterial burdens reported for MyD88 KO and IL-1 KO mice at 24 h following S. aureus infection were equivalent between this study and our previous publication, respectively (i.e., ~ 106 CFU). Because the majority of MyD88 KO mice did not survive beyond 48 h postinfection, it was not possible to evaluate bacterial burdens at later time points. We would like to emphasize that a trend toward elevated bacterial replication was observed in MyD88 KO mice; however, this difference did not reach statistical significance. Therefore, we predict that if MyD88 KO mice had lived for longer periods of time following intracerebral S. aureus infection the observed trend toward increased bacterial burdens in MyD88 KO animals may have become more dramatic and approached statistical significance, although this possibility remains speculative. Collectively, these findings suggest that MyD88-independent mechanisms are responsible for controlling bacterial replication in the CNS parenchyma. Indeed, because TLRs and the IL-1 and IL-18 receptors are not phagocytic receptors, it may be expected that alternative phagocytic pattern recognition receptors are responsible for pathogen uptake and neutralization (65, 66). Some candidate receptors that may participate in regulating S. aureus burdens in the CNS parenchyma include members of the scavenger receptor family as well as opsonic receptors including Fc and complement receptors (67, 68). Alternatively, another potential explanation to account for the similarity in bacterial burdens between MyD88 KO and WT mice is that perhaps bacterial burdens cannot increase further in MyD88 KO mice because of the extensive necrosis associated with the infected tissue, which would be expected to compromise blood flow and oxygenation of the infected parenchyma. This may effectively retard bacterial expansion due to limited nutrient and oxygen bioavailability. Our findings demonstrating no alterations in pathogen burdens in brain abscesses of MyD88 KO mice differ from those described by Koedel et al. in a mouse model of pneumococcal meningitis (47). In this report, the authors demonstrated that bacterial burdens were significantly elevated in MyD88 KO mice, which correlated with the impaired expression of several proinflammatory mediators, the latter of which is in agreement with our present study. Importantly, these findings highlight the fact that disparate mechanisms are responsible for bacterial containment between brain infections in the CNS parenchyma (i.e., abscesses) vs the meninges.
In summary, our results establish an essential role for MyD88-dependent signals in the induction of host immune responses in brain abscesses and in sequestering lesions from expanding significantly into the surrounding normal brain parenchyma. Therefore, the dramatic phenotype observed during the early stage of brain abscess formation in MyD88 KO mice is likely influenced by the loss of multiple TLRs as well as signaling via the IL-1R. The relative role of IL-18 signaling is currently not known, because we have not yet investigated the importance of this cytokine in the experimental brain abscess model. Subsequent studies can begin to delineate the specific repertoire of MyD88-dependent pathways required for these phenomena by examining brain abscess pathogenesis in double receptor KO mice, selecting candidate receptors that are known to be MyD88 dependent.
Acknowledgments
We thank Dr. Robert E. Mrak for histopathological analysis of brain abscesses and Shuliang Liu, Gail Wagoner, and Napoleon Phillips for excellent technical assistance.
Footnotes
This work was supported by National Institutes of Health, National Institute of Mental Health Grant R01 MH65297, and National Institute of Neurological Disorders and Stroke Grant P30 NS047546 (to the Core Facility at University of Arkansas for Medical Sciences, Little Rock, AR).
Abbreviations used in this paper: KO, knockout; AQP4, aquaporin 4; IP-10, IFN-γ-induced protein 10; WT, wild type.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Kielian T. Immunopathogenesis of brain abscess. J Neuroinflammation. 2004;1:16. doi: 10.1186/1742-2094-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bloch O, Papadopoulos MC, Manley GT, Verkman AS. Aquaporin-4 gene deletion in mice increases focal edema associated with staphylococcal brain abscess. J Neurochem. 2005;95:254–262. doi: 10.1111/j.1471-4159.2005.03362.x. [DOI] [PubMed] [Google Scholar]
- 3.Lo WD, Wolny A, Boesel C. Blood-brain barrier permeability in staphylococcal cerebritis and early brain abscess. J Neurosurg. 1994;80:897–905. doi: 10.3171/jns.1994.80.5.0897. [DOI] [PubMed] [Google Scholar]
- 4.Kielian T, Hickey WF. Proinflammatory cytokine, chemokine, and cellular adhesion molecule expression during the acute phase of experimental brain abscess development. Am J Pathol. 2000;157:647–658. doi: 10.1016/S0002-9440(10)64575-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baldwin AC, Kielian T. Persistent immune activation associated with a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuroimmunol. 2004;151:24–32. doi: 10.1016/j.jneuroim.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Kielian T, Barry B, Hickey WF. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J Immunol. 2001;166:4634–4643. doi: 10.4049/jimmunol.166.7.4634. [DOI] [PubMed] [Google Scholar]
- 7.Stenzel W, Soltek S, Miletic H, Hermann MM, Korner H, Sedgwick JD, Schluter D, Deckert M. An essential role for tumor necrosis factor in the formation of experimental murine Staphylococcus aureus-induced brain abscess and clearance. J Neuropathol Exp Neurol. 2005;64:27–36. doi: 10.1093/jnen/64.1.27. [DOI] [PubMed] [Google Scholar]
- 8.Flaris NA, Hickey WF. Development and characterization of an experimental model of brain abscess in the rat. Am J Pathol. 1992;141:1299–1307. [PMC free article] [PubMed] [Google Scholar]
- 9.Mathisen GE, Johnson JP. Brain abscess. Clin Infect Dis. 1997;25:763–779. doi: 10.1086/515541. [DOI] [PubMed] [Google Scholar]
- 10.Townsend GC, Scheld WM. Infections of the central nervous system. Adv Intern Med. 1998;43:403–447. [PubMed] [Google Scholar]
- 11.Kielian T, Haney A, Mayes PM, Garg S, Esen N. Toll-like receptor 2 modulates the proinflammatory milieu in Staphylococcus aureus-induced brain abscess. Infect Immun. 2005;73:7428–7435. doi: 10.1128/IAI.73.11.7428-7435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kielian T, Bearden ED, Baldwin AC, Esen N. IL-1 and TNF-α play a pivotal role in the host immune response in a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuropathol Exp Neurol. 2004;63:381–396. doi: 10.1093/jnen/63.4.381. [DOI] [PubMed] [Google Scholar]
- 13.Kielian T, Cheung A, Hickey WF. Diminished virulence of an α-toxin mutant of Staphylococcus aureus in experimental brain abscesses. Infect Immun. 2001;69:6902–6911. doi: 10.1128/IAI.69.11.6902-6911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akira S. TLR signaling. Curr Top Microbiol Immunol. 2006;311:1–16. doi: 10.1007/3-540-32636-7_1. [DOI] [PubMed] [Google Scholar]
- 15.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. PG - 253– 8. Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 16.Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- 17.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 18.Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, Ryffel B. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J Clin Invest. 2004;114:1790–1799. doi: 10.1172/JCI21027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, Medzhitov R, Sher A. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol. 2002;168:5997–6001. doi: 10.4049/jimmunol.168.12.5997. [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 21.Bafica A, Santiago HC, Goldszmid R, Ropert C, Gazzinelli RT, Sher A. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol. 2006;177:3515–3519. doi: 10.4049/jimmunol.177.6.3515. [DOI] [PubMed] [Google Scholar]
- 22.Kaisho T, Akira S. Pleiotropic function of Toll-like receptors. Microbes Infect. 2004;6:1388–1394. doi: 10.1016/j.micinf.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 23.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 24.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 25.Echchannaoui H, Frei K, Schnell C, Leib SL, Zimmerli W, Landmann R. Toll-like receptor 2-deficient mice are highly susceptible to Streptococcus pneumoniae meningitis because of reduced bacterial clearing and enhanced inflammation. J Infect Dis. 2002;186:798–806. doi: 10.1086/342845. [DOI] [PubMed] [Google Scholar]
- 26.Knapp S, Wieland CW, van ‘t Veer C, Takeuchi O, Akira S, Florquin S, van der Poll T. Toll-like receptor 2 plays a role in the early inflammatory response to murine pneumococcal pneumonia but does not contribute to antibacterial defense. J Immunol. 2004;172:3132–3138. doi: 10.4049/jimmunol.172.5.3132. [DOI] [PubMed] [Google Scholar]
- 27.Koedel U, Angele B, Rupprecht T, Wagner H, Roggenkamp A, Pfister HW, Kirschning CJ. Toll-like receptor 2 participates in mediation of immune response in experimental pneumococcal meningitis. J Immunol. 2003;170:438–444. doi: 10.4049/jimmunol.170.1.438. [DOI] [PubMed] [Google Scholar]
- 28.Dziarski R, Gupta D. Staphylococcus aureus peptidoglycan is a toll-like receptor 2 activator: a reevaluation. Infect Immun. 2005;73:5212–5216. doi: 10.1128/IAI.73.8.5212-5216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirschfeld M, Kirschning CJ, Schwandner R, Wesche H, Weis JH, Wooten RM, Weis JJ. Cutting edge: inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by toll-like receptor 2. J Immunol. 1999;163:2382–2386. [PubMed] [Google Scholar]
- 30.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci USA. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S. Cutting edge: role of toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169:10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 32.Bulut Y, Faure E, Thomas L, Equils O, Arditi M. Cooperation of Toll-like receptor 2 and 6 for cellular activation by soluble tuberculosis factor and Borrelia burgdorferi outer surface protein A lipoprotein: role of Toll-interacting protein and IL-1 receptor signaling molecules in Toll-like receptor 2 signaling. J Immunol. 2001;167:987–994. doi: 10.4049/jimmunol.167.2.987. [DOI] [PubMed] [Google Scholar]
- 33.Hajjar AM, O’Mahony DS, Ozinsky A, Underhill DM, Aderem A, Klebanoff SJ, Wilson CB. Cutting edge: functional interactions between toll-like receptor (TLR) 2 and TLR1 or TLR6 in response to phenol-soluble modulin. J Immunol. 2001;166:15–19. doi: 10.4049/jimmunol.166.1.15. [DOI] [PubMed] [Google Scholar]
- 34.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 35.Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, Wagner H, Lipford GB. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci USA. 2001;98:9237–9242. doi: 10.1073/pnas.161293498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esen N, Tanga FY, DeLeo JA, Kielian T. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the Gram-positive bacterium Staphylococcus aureus. J Neurochem. 2004;88:746–758. doi: 10.1046/j.1471-4159.2003.02202.x. [DOI] [PubMed] [Google Scholar]
- 37.Esen N, Kielian T. Central role for MyD88 in the responses of microglia to pathogen-associated molecular patterns. J Immunol. 2006;176:6802–6811. doi: 10.4049/jimmunol.176.11.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83:711–730. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun. 2003;17:13–19. doi: 10.1016/s0889-1591(02)00055-7. [DOI] [PubMed] [Google Scholar]
- 40.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 41.Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005;49:360–374. doi: 10.1002/glia.20117. [DOI] [PubMed] [Google Scholar]
- 42.Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 43.Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer JL, Di Marco F, French L, Tschopp J. MyD88, an adapter protein involved in interleukin-1 signaling. J Biol Chem. 1998;273:12203–12209. doi: 10.1074/jbc.273.20.12203. [DOI] [PubMed] [Google Scholar]
- 46.Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science. 1997;278:1612–1615. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- 47.Koedel U, Rupprecht T, Angele B, Heesemann J, Wagner H, Pfister HW, Kirschning CJ. MyD88 is required for mounting a robust host immune response to Streptococcus pneumoniae in the CNS. Brain. 2004;127:1437–1445. doi: 10.1093/brain/awh171. [DOI] [PubMed] [Google Scholar]
- 48.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 49.Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 50.Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting: phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- 51.Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T. TNF-α expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis: regulation by Th1 cytokines. J Immunol. 1995;154:944–953. [PubMed] [Google Scholar]
- 52.Kimelberg HK. Water homeostasis in the brain: basic concepts. Neuroscience. 2004;129:851–860. doi: 10.1016/j.neuroscience.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 53.Lehnardt S, Henneke P, Lien E, Kasper DL, Volpe JJ, Bechmann I, Nitsch R, Weber JR, Golenbock DT, Vartanian T. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol. 2006;177:583–592. doi: 10.4049/jimmunol.177.1.583. [DOI] [PubMed] [Google Scholar]
- 54.Koedel U, Scheld WM, Pfister HW. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis. 2002;2:721–736. doi: 10.1016/s1473-3099(02)00450-4. [DOI] [PubMed] [Google Scholar]
- 55.Nau R, Bruck W. Neuronal injury in bacterial meningitis: mechanisms and implications for therapy. Trends Neurosci. 2002;25:38–45. doi: 10.1016/s0166-2236(00)02024-5. [DOI] [PubMed] [Google Scholar]
- 56.Thornton P, Pinteaux E, Gibson RM, Allan SM, Rothwell NJ. Interleukin-1-induced neurotoxicity is mediated by glia and requires caspase activation and free radical release. J Neurochem. 2006;98:258–266. doi: 10.1111/j.1471-4159.2006.03872.x. [DOI] [PubMed] [Google Scholar]
- 57.Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. Tumor necrosis factor-α induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–21368. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- 58.Shishodia S, Aggarwal BB. Nuclear factor-κB activation: a question of life or death. J Biochem Mol Biol. 2002;35:28–40. doi: 10.5483/bmbrep.2002.35.1.028. [DOI] [PubMed] [Google Scholar]
- 59.Tebo JM, Datta S, Kishore R, Kolosov M, Major JA, Ohmori Y, Hamilton TA. Interleukin-1-mediated stabilization of mouse KC mRNA depends on sequences in both 5′- and 3′-untranslated regions. J Biol Chem. 2000;275:12987–12993. doi: 10.1074/jbc.275.17.12987. [DOI] [PubMed] [Google Scholar]
- 60.Sun D, Ding A. MyD88-mediated stabilization of interferon-γ-induced cytokine and chemokine mRNA. Nat Immunol. 2006;7:375–381. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]
- 61.Kim HS, Armstrong D, Hamilton TA, Tebo JM. IL-10 suppresses LPS-induced KC mRNA expression via a translation-dependent decrease in mRNA stability. J Leukocyte Biol. 1998;64:33–39. doi: 10.1002/jlb.64.1.33. [DOI] [PubMed] [Google Scholar]
- 62.Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002;13:413–421. doi: 10.1016/s1359-6101(02)00026-6. [DOI] [PubMed] [Google Scholar]
- 63.Ghosh S. Regulation of inducible gene expression by the transcription factor NF-κB. Immunol Res. 1999;19:183–189. doi: 10.1007/BF02786486. [DOI] [PubMed] [Google Scholar]
- 64.Komatsuzawa H, Ouhara K, Yamada S, Fujiwara T, Sayama K, Hashimoto K, Sugai M. Innate defences against methicillin-resistant Staphylococcus aureus (MRSA) infection. J Pathol. 2006;208:249–260. doi: 10.1002/path.1898. [DOI] [PubMed] [Google Scholar]
- 65.Henneke P, Takeuchi O, Malley R, Lien E, Ingalls RR, Freeman MW, Mayadas T, Nizet V, Akira S, Kasper DL, Golenbock DT. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J Immunol. 2002;169:3970–3977. doi: 10.4049/jimmunol.169.7.3970. [DOI] [PubMed] [Google Scholar]
- 66.Underhill DM, Gantner B. Integration of Toll-like receptor and phagocytic signaling for tailored immunity. Microbes Infect. 2004;6:1368–1373. doi: 10.1016/j.micinf.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 67.Peiser L, Mukhopadhyay S, Gordon S. Scavenger receptors in innate immunity. Curr Opin Immunol. 2002;14:123–128. doi: 10.1016/s0952-7915(01)00307-7. [DOI] [PubMed] [Google Scholar]
- 68.Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC. Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia. 2002;40:195–205. doi: 10.1002/glia.10148. [DOI] [PubMed] [Google Scholar]