

Abstract
In this study we investigated the mechanisms responsible for MAP kinase ERK1/2 activation following agonist activation of endogenous mu opioid receptors (MOR) normally expressed in cultured striatal neurons. Treatment with the MOR agonist fentanyl caused significant activation of ERK1/2 in neurons derived from wild type mice. Fentanyl effects were blocked by the opioid antagonist naloxone and were not evident in neurons derived from MOR knock-out (−/−) mice. In contrast, ERK1/2 activation by fentanyl was not evident in neurons from GRK3−/− mice or neurons pretreated with small inhibitory RNA for arrestin3. Consistent with this observation, treatment with the opiate morphine (which is less able to activate arrestin) did not elicit ERK1/2 activation in wild type neurons; however, transfection of arrestin3-(R170E) (a dominant positive form of arrestin that does not require receptor phosphorylation for activation) enabled morphine activation of ERK1/2. In addition, activation of ERK1/2 by fentanyl and morphine was rescued in GRK3−/− neurons following transfection with dominant positive arrestin3-(R170E). The activation of ERK1/2 appeared to be selective as p38 MAP kinase activation was not increased by either fentanyl or morphine treatment in neurons from wild type, MOR−/−, or GRK3−/− mice. In addition, U0126 (a selective inhibitor of MEK kinase responsible for ERK phosphorylation) blocked ERK1/2 activation by fentanyl. These results support the hypothesis that MOR activation of ERK1/2 requires opioid receptor phosphorylation by GRK3 and association of arrestin3 to initiate the cascade resulting in ERK1/2 phosphorylation in striatal neurons.
Opioid receptor activation results in both acute and long-lasting changes in neuronal physiology. The mechanisms responsible for the acute changes include Gαi/o and Gβγ protein activation that increases potassium conductance, decreases calcium conductance, and results in presynaptic inhibition (1–3). The mechanisms responsible for the long lasting effects of opioids are less clear but may include changes in adenylyl cyclase activity and activation of mitogen activating protein kinase (MAPK)2 pathways (4, 5). In this study we used primary cultured neurons isolated from mouse striata to address the mechanisms linking mu opioid receptor (MOR) activation to the phosphorylation and activation of the extracellular signal-related kinases 1 and 2 (ERK1/2) members of the MAPK family.
Phosphorylation of ERK1/2 by GPCRs involves growth factor receptor transactivation (4). As demonstrated for the β-adrenergic receptor, participation of arrestin is an integral part of GPCR signaling through the ERK1/2 signaling pathway in transfected HEK293 cells (6). Following agonist activation of MOR, the receptor becomes phosphorylated by G-protein receptor kinase (GRK), which initiates an arrestin-dependent desensitization process that involves clathrin-mediated endocytosis (7). The opioid activation of ERK1/2 by MOR agonist (D-Ala2,Me-Phe4,Gly-ol5) (DAMGO) was demonstrated in MOR-transfected cells (8), and ERK1/2 was activated in the pons and medulla of morphine-treated mice (9). However, the mechanism of ERK1/2 activation by opioids is not clear.
To assess whether GRK and arrestin were required for MOR activation of ERK1/2, we compared the activation of ERK1/2 in striatal neurons harvested from wild type, MOR knock-out mice (MOR−/−), and GRK 3 knock-out mice (GRK3−/−). Using siRNA to reduce arrestin3 expression and the dominant positive form of arrestin 3, arrestin-(R170E) (10), we further characterized the role of arrestin in the activation of ERK1/2 by MOR agonists. The results suggest that MOR agonists in striatal neurons produced selective activation of ERK1/2, the selective activation of ERK1/2 was rescued by the dominant positive arrestin, and this activation of ERK1/2 was GRK3 and arrestin3 dependent.
EXPERIMENTAL PROCEDURES
Materials
Culture media, serum, anisomycin, and the MEK inhibitor U0126 were purchased from Sigma. Morphine and fentanyl were provided by the National Institute on Drug Abuse. Norbinaltorphimine HCl and (−)U50,488 were obtained from Tocris (Ellisville, MO). Antibodies used include: rabbit anti-phospho-ERK1/2 (1/1000, Cell Signaling, Beverly, MA), rabbit anti-phospho-p38 (1/1000, Cell Signaling), mouse anti-arrestin3 (1/300 dilution, sc-13140 from Santa Cruz Biotechnology, Santa Cruz, CA), guinea pig anti-mu opioid receptor (1/1000 dilution, Neuromics, Minneapolis, MN), and rabbit anti-β-actin (1/10,000 dilution, ab8227, AbCam, Cambridge, MA). Secondary antibodies for confocal microscopy were purchased from Invitrogen and secondary antibodies for immunoblot analysis from Rockland (Gilbertsville, PA). The Li-Cor Blocking Buffer was purchased from Li-Cor Biosciences (Lincoln, NE). Pregnant C57Bl/6 mice were obtained from Charles River (Wilmington, MA). Transgenic mice lacking functional mu opioid receptors, MOR−/− (11), or G-protein receptor kinase 3, GRK3−/− (12) on a C57Bl/6 background, were kindly provided by Dr. John Pintar (R. W. Johnson School of Medicine) and Drs. Marc Caron and Robert Lefkowitz (Duke University), respectively, and bred within the University of Washington vivarium under specific pathogen-free conditions.
Striatal Neuronal Cultures and MOR-GFP AtT-20 Cells
This procedure was described previously (13) and adapted for these studies. The striatal region was dissected from 3–4-day-old C57BL/6 or transgenic mice and incubated in MEM containing 20 units/ml papain for 30 min at 37 °C. The tissue was then triturated using fire-polished Pasteur pipettes in MEM supplemented with 10% fetal bovine serum, 0.45% glucose, 0.5 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells were plated on poly-D-lysine-treated glass coverslips (BD Biosciences) at a density of 75,000 cells per coverslip for confocal microscopy, or in poly-D-lysine-coated 6-well plates for immunoblot analysis. Neuronal medium containing 50% MEM, 39% Ham’s F-12 medium, 10% horse serum, 1% fetal bovine serum, 0.45% glucose, 0.1 mg/ml apotransferrin, 0.5 mM kynurenic acid, 100 units/ml penicillin, and 100 μg/ml streptomycin was added 1 h after initial plating. Cultures were used 12–14 days after plating. MOR-GFP-transfected AtT-20 cells were previously described (14). Cells were grown in a humidified 5% CO2 incubator at 37 °C.
Transfection of Striatal Neuronal Cultures
For experiments using the dominant positive arrestin3-(R170E) (10), striatal cultures were grown in 6-well plates. Lipofectamine 2000 (Invitrogen) was added to MEM according to the manufacturer’s instructions, and 1.5 μg of cDNA coding for the dominant positive arrestin3-(R170E)-YFP or the empty vector YFP were prepared in Opti-MEM. DNA mixtures were added dropwise to the Lipofectamine 2000 mixture and incubated at room temperature for 30 min. After the incubation, the total mixture was added to cells in a 6-well tissue culture plate for 4 h. Cells were harvested on the second day following transfection for quantification of phospho-ERK1/2 and actin immunoreactivity as described. Treated cells transfected with dominant positive arrestin3-(R170E)-YFP or the empty vector YFP were compared with untreated cells transfected with the dominant positive arrestin3-(R170E)-YFP (basal). The dominant positive arrestin3-(R170E) was kindly provided by Dr. Vsevolod Gurevich (Vanderbilt, TN). BglII and KpnI restriction sites were added to the 5′ and 3′ ends of the cDNAs through polymerase chain reaction using the respective oligos: 5′-Arr3 BglI (5′-ATGCATAGATCTATGGGGGAGAAACCC) and 3′-Arr3 KpnI (5′-GGCCCGCGGTACCTAGCAGAACTGGTC). The resulting products were purified, digested, and then inserted into the multiple cloning site of the pEYFP-C1 vector (Clontech Laboratories). The 5′ oligo used to insert the BglII restriction site verified the coding sequence for arrestin3 remained in-frame with the EYFP sequence.
For transfection of the arrestin3 siRNA, chemically synthesized siRNA with 19-nucleotide duplex RNA and 2-nucleotide 3′ dTdT overhangs was purchased from (Invitrogen). The siRNA sequence targeting mouse arrestin3 was 5′-GGACCGGAAAGUGUUUGUG-3′. Lipofectamine 2000 (Invitrogen) was added to Opti-MEM according to the manufacturer’s instructions, whereas the RNA mixtures were prepared at a final concentration of 25 nM. RNA mixtures were added drop-wise to the Lipofectamine 2000 mixture and incubated at room temperature for 30 min. After the incubation, the total mixture was added to cells plated on coverslips for confocal microscopy, or to 6-well tissue culture plate for immunoblotting. Some wells received Lipofectamine 2000 in Opti-MEM only as a negative control (control). The mixture was removed after 4 h and fresh media was added to the cultures. Cells were harvested on the second day following transfection for quantification of p-ERK1/2, arrestin3, and actin immunoreactivity as described. Transfection efficiency was determined by transfecting 0.5 μg of the empty vector DsRed (Clontech, Palo Alto, CA) with the siRNA mixture as described above. Using confocal microscopy, DsRed positive cells were counted and taken over the total number of cells per × 63 field to calculate an average transfection efficiency at 28.5 ± 0.5%.
Quantitative Real-time Polymerase Chain Reaction (RT-PCR)
For further quantification of arrestin3 knockdown levels, RNA was harvested from striatal neuronal cultures prepared from 3– 4-day-old mouse pups using Qiashredder homogenizer columns and the RNeasy Mini Kit according to the manufacturer’s protocol (Qiagen, Valencia, CA). One step quantitative real-time PCR was performed on a Stratagene Mx3000 machine using the Brilliant SYBR Green QPCR Master Mix with StrataScript Reverse Transcriptase (Stratagene, La Jolla, CA). Arrestin2, arrestin3, and a control endogenous housekeeping gene, ribosomal protein ARBP, were amplified in separate reactions starting with 20 ng of total RNA for each sample. The oligo sequences used for amplification of mouse arrestin2 and arrestin3 were found through PrimerBank (15) and were as follows: arrestin2 (PrimerBank code 30089688a3), forward (GCCCCAATGGAAAGCTCACT), reverse (CCACGAGGTCAATGTGGTCC); and arrestin3 (PrimerBank code 21703856a3), forward (GGAGTAGACTTTGAGATTCGAGC), reverse (GGTCAGACATGAGGAAGTGGC). The oligos used for the housekeeping gene ARBP were forward (TGTTTGACAACGGCAGCATTT) and reverse (CCGAGGCAACAGTTGGGTA). Standard curves were run with serial dilutions of RNA and the PCR efficiencies were found to be similar for all oligo sets. Dissociation curves for the products of each oligo set had a single peak indicating specific amplification. For each sample, the Ct, or cycle at which the fluorescence measured reached the threshold to be above the ambient background signal, was determined for arrestin2, arrestin3, and ARBP. The ΔCt was calculated by subtracting the Ct for the endogenous control gene ARBP from the Ct of the gene of interest. Relative quantification was done using the ΔΔCt method (16). Relative quantity was then expressed as a -fold change by calculating 2−ΔΔCt. For absolute quantification of arrestin levels in the neuronal cultures, standard curves were generated from in vitro transcribed arrestin2 and arrestin3 RNA and the amount of arrestin RNA per 20 ng of total RNA harvested from the neuronal cultures was determined.
Confocal Microscopy
Striatal neurons grown on glass coverslips were treated for 30 min at 37 °C with fentanyl (100 nM) or morphine (10 μM). For antagonist studies, naloxone (10 μM) was added 30 min before agonist. Cells were fixed in 4% paraformaldehyde in phosphate-buffered saline (58 mM Na2HPO4, 17 mM NaH2PO4, 68 mM NaCl, pH 7.4) for 30 min. Coverslips were permeabilized and blocked with 5% goat serum, 3% bovine serum albumin, and 0.5% Triton X-100 for 1 h at room temperature. All cells were incubated with rabbit anti-phospho-ERK1/2, guinea pig anti-mu opioid receptor, and mouse anti-arrestin3, washed, and incubated for 1 h with goat anti-rabbit IgG 488 Alexa Fluor conjugate, goat anti-guinea pig IgG 633 Alexa Fluor conjugate, and goat anti-mouse IgG 555 at 1/500. Coverslips were washed, mounted onto a slide with Slowfade (Molecular Probes, Eugene, OR), and imaged with a Leica SL laser scanning confocal microscope. Phospho-ERK1/2 expression was quantified for siRNA experiments using Metamorph software (Molecular Devices, Sunnyvale, CA).
Immunoblotting
Striatal neurons or MOR-GFP AtT-20 cells were grown in 6-well plates were treated for 30 min at 37 °C with fentanyl (100 nM), morphine (10 μM), or U50,488 (10 μM). For antagonist and inhibitor studies naloxone (10 μM) and U0126 (1 μM) were added 30 min before agonist. 1 μM Norbinaltorphimine was added 60 min before agonist. Anisomycin (50 μM) was added for 30 min. Cells were serum-starved 24 h prior to drug treatment. After treatment, cells were resuspended in solubilization buffer (25 mM Tris, 150 mM NaCl with 1% CHAPS, pH 7.4), containing phosphatase inhibitor mixture Set 1 and protease inhibitor mixture Set 1 at a 1:100 dilution (Calbiochem, La Jolla, CA). Cultures or cells were scraped from plates and triturated, sonicated for 10 s, and centrifuged at 14,000 × g for 30 min at 4 °C. After determining protein concentrations for the whole cell lysate using the BCA Protein Assay kit (Pierce), 30 μg of protein/sample was used for quantification of phospho-ERK1/2, p-p38, and arrestin3. Proteins were suspended with an equal volume of Laemmli buffer, heated at 95 °C for 5 min, loaded, and separated on 10% bisacrylamide pre-cast gels (Invitrogen). The Benchmark pre-stained standard (Invitrogen) was loaded to confirm the appropriate molecular weight of each described protein. Gels were transferred to nitrocellulose (Whatman) and blocked in Tris-buffered saline, 5% bovine serum albumin for 1 h, incubated overnight at 4 °C with antibodies directed against phospho-ERK1/2, phospho-p38 MAPK, arrestin3, or β-actin. Membranes were washed in Tris-buffered saline, and then incubated with IRDye™ 800 conjugated affinity purified anti-rabbit IgG or anti-mouse IgG at a dilution of 1:10,000 in a 1:1 mixture of 5% milk/Tris-buffered saline and Li-Cor Blocking Buffer for 1 h at room temperature. Membranes were then washed in 1× Tris-buffered saline before scanning on the Odyssey Infrared Imaging System (Li-Cor Biosciencs). Membranes were re-probed with β-actin to confirm equal protein loading. Band pixel intensity was measured using the Odyssey software. Band intensities from treated groups were normalized to a percentage of control samples, and plotted using GraphPad (GraphPad Prism 4.0) software. Statistical significance was determined by an analysis of variance followed by Dunnett’s post hoc test, p < 0.05 or p < 0.01.
RESULTS
MOR Activation of ERK1/2 Phosphorylation Is Agonist Selective and GRK3 Dependent
The expression of phospho-ERK1/2 was assessed in agonist-treated neurons and compared with untreated striatal cells. Treatment with 100 nM fentanyl for 30 min significantly increased the activation of phosopho-ERK1/2 in the whole cell lysates prepared from wild type striatal neurons by 81 ± 13% above basal levels (p < 0.01, n = 7) (Fig. 1, A and B, left column, and Fig. 2). The increase in ERK1/2 phosphorylation was blocked by the MOR antagonist naloxone and was not evident in primary striatal neurons cultured from MOR−/− mice. Furthermore, the MEK inhibitor U0126 prevented the fentanyl-induced phosphorylation of ERK1/2 (p < 0.01, n = 4). Treatment with fentanyl (100 nM) for 2, 5, or 15 min did not significantly affect ERK1/2 phosphorylation (90 ± 6.5, 88 ± 14, and 88 ± 20%, respectively) compared with basal phospho-ERK1/2 levels in the whole cell lysate prepared from the wild type striatal neurons (Fig. 1D, n = 2–4). Lower doses of fentanyl were also used to treat neurons, and using immunoblot analysis the phosphorylation of ERK1/2 was not significantly increased from basal levels (data not shown, n = 3).
FIGURE 1. MOR activation of phospho-ERK1/2 is agonist selective and GRK3 dependent in striatal neurons.
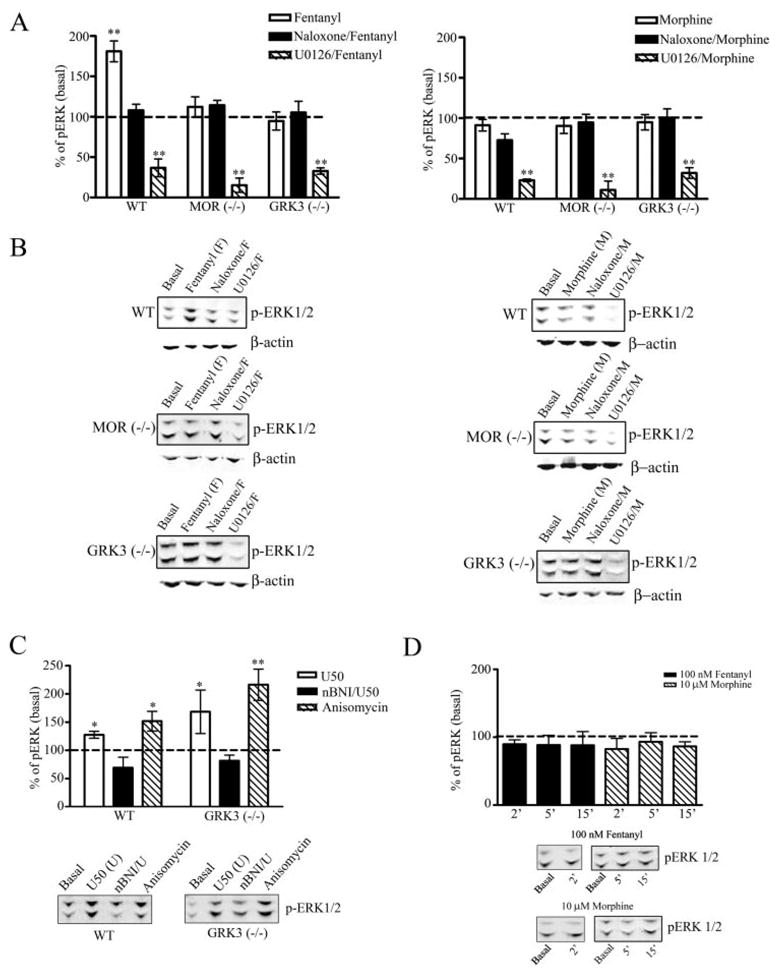
Agonist-induced activation of phospho-ERK1/2 by the endogenous MOR in wild type, MOR−/−, and GRK3−/− striatal neurons was assessed. Whole cell lysates prepared from neuronal cultures as described under “Experimental Procedures” were used to quantify the expression levels of phospho-ERK1/2. A, the results are the mean ± S.E., expressed as a percent of basal, untreated striatal neuronal cultures (**, p < 0.01 by Dunnett’s post hoc comparison, n = 4–7). B, representative immunoblots for the 44/42-kDa phospho-ERK1/2 protein are shown in which wild type (WT), MOR−/−, or GRK3−/− striatal neurons were vehicle treated (Basal) or treated for 30 min with fentanyl (100 nM) or morphine (10 μM) with or without pretreatment with naloxone (10 μM) or U0126 (1 μM). Representative immunoblots for β-actin demonstrates equal protein loading for each representative experiment. C, wild type and GRK3−/− neuronal cultures were treated with vehicle (Basal), 10 μM U50,488 (U50) (30 min), U50,488 in the presence of 1 μM norbinaltorphimine (nBNI/U, 1 h pretreatment), or 50 μM anisomycin. The results are the mean ± S.E., expressed as a percent of basal, untreated striatal neuronal cultures (*, p < 0.05; **, p < 0.01, by Dunnett’s post hoc comparison, n = 3–7). Representative immunoblots for the 44/42-kDa phospho-ERK1/2 protein are shown. D, the results are the mean ± S.E., expressed as a percent of basal, untreated striatal neuronal cultures (n = 2–4). Representative immunoblots for the 44/42-kDa phospho-ERK1/2 protein are shown in which cells were treated with vehicle (Basal), treated with fentanyl (100 nM) or morphine (10 μM) for 2 (2′), 5 (5′), or 15 min (15′).
FIGURE 2. Activation of phospho-ERK1/2 by MOR is agonist selective and results in arrestin3 colocalization in striatal neurons.
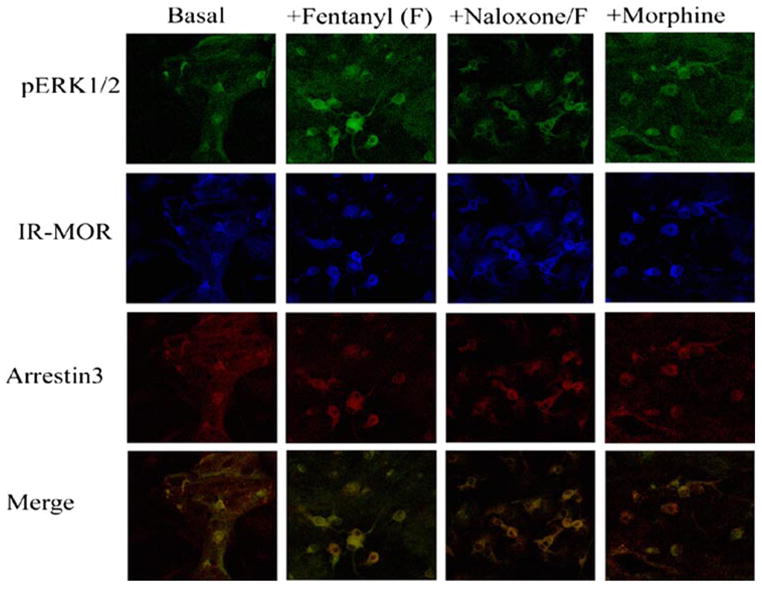
Agonist-induced activation of phospho-ERK1/2 by the endogenous MOR in wild type striatal neurons was assessed. Neuronal cultures prepared as described under “Experimental Procedures” were used to quantify the expression levels of phospho-ERK1/2. Representative confocal images are shown of pERK, MOR, and arrestin3 immunoreactivity in wild type striatal neurons that were vehicle treated (Basal), or treated for 30 min with fentanyl (100 nM), fentanyl with naloxone (10 μM) pretreatment, or morphine (10 μM). The merged images represent the colocalization of p-ERK and arrestin3 for each treatment described above.
In contrast to wild type neurons, fentanyl (100 nM) treatment of striatal neurons from GRK3−/− mice for 5, 15, or 30 min did not cause a significant activation of ERK1/2 (data for the 30-min time pointareshown, Fig. 1, leftcolumn). TheresultssuggestthatGRK3 was required for the fentanyl-induced activation of ERK1/2.
Treatment with 10 μM morphine for 30 min did not significantly increase the activation of phospho-ERK1/2 in whole cell lysates prepared from wild type, MOR−/−, or GRK3−/− striatal neurons compared with basal levels of phospho-ERK1/2 (Fig. 1, right column, and Fig. 2, n = 7). Furthermore, treatment with 10 μM morphine for 2, 5, or 15 min did not significantly increase phosphorylation of ERK1/2 (82.5 ± 15, 93 ± 13, and 86 ± 7, respectively) compared with basal levels (Fig. 1D, n = 2–6). This agonist specific difference in ERK1/2 activation in striatial neuronal cultures was also found in AtT-20 cells expressing a GFP-tagged MOR. Treatment with 10 μM morphine for 30 min did not significantly increase phospho-ERK levels (84.5 ± 8.2%) compared with basal levels (n = 6, data not shown), whereas treatment with 100 nM fentanyl produced a significant increase in the levels of phospho-ERK, 25.8 ± 7% above basal levels (p < 0.05, n = 6) (data not shown).
The lack of response of GRK3−/− cells to fentanyl was not due to an inability of these neurons to show receptor-activated ERK1/2 phosphorylation. The MEK inhibitor U0126 significantly reduced the basal activation of phospho-ERK1/2 in wild type, MOR−/−, and GRK3−/− striatal neurons (p < 0.01, n = 7), indicating the cultured neurons from the transgenic mice were capable of ERK1/2 phosphorylation. As a positive control, treatment with the kappa opioid receptor agonist U50,488 (10 μM) significantly increased phospho-ERK1/2 levels in both wild type and GRK3−/− striatal neurons by 27.6 ± 6.3 and 68.5 ± 38.5%, respectively (p < 0.05, n = 3–7) (Fig. 1C). In addition, treatment of both wild type and GRK3−/− neurons with the MAPK activator anisomycin (50 μM) significantly increased phospho-ERK1/2 levels 51.7 ± 17.5 and 116 ± 27.8%, respectively (p < 0.05 and p < 0.01, respectively, n = 3–7) (Fig. 1C). These data show that the GRK3−/− neurons were capable of ERK1/2 activation and further support the conclusion that GRK3 was specifically required for the mu opioid-induced activation of ERK1/2.
MOR Activation Specifically Induced Phosphorylation of ERK1/2, not p38 MAPK
Treatment with 100 nM fentanyl for 5, 15, or 30 min did not significantly increase phospho-p38 in whole cell lysates prepared from cultured striatal neurons derived from wild type, MOR−/−, or GRK3−/− mice (Fig. 3, n = 7). Furthermore, treatment with 10 μM morphine for 5, 15, or 30 min did not increase phosphorylated p38 in neurons cultured from wild type, MOR−/−, or GRK3−/− mice (Fig. 3). The expression of phosphorylated p38 was determined following treatment with fentanyl (100 nM) or morphine (10 μM) for 30 min and compared with untreated cells. Pretreatment of striatal neurons with naloxone did not significantly affect the phosphorylation of p38. Pretreatment with U0126, a selective inhibitor of ERK1/2 activation, did not significantly decrease the basal activation of phospho-p38 in wild type, MOR−/−, or GRK3−/− striatal neurons.
FIGURE 3. The MAP kinase phospho-p38 is not activated by MOR agonists in striatal neurons.
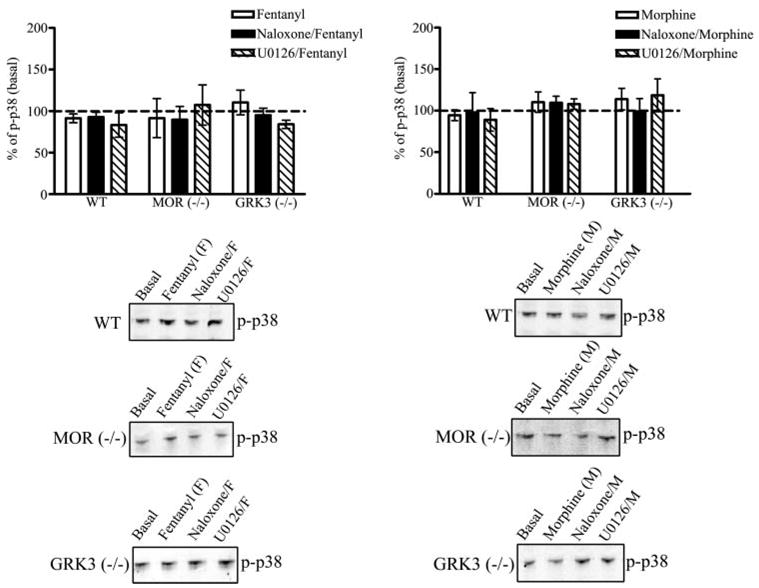
Agonist-induced activation of phospho-p38 by the endogenous MOR receptor in wild type, MOR−/−, and GRK3−/− striatal neurons was assessed. Whole cell lysates prepared from neuronal cultures as described under “Experimental Procedures” were used to quantify the expression levels of phospho-p38. Top, the results are the mean ± S.E., expressed as a percent of basal, untreated striatal neuronal cultures. Treatment of wild type, MOR−/−, or GRK3−/− striatal neurons with 100 nM fentanyl or 10 μM morphine for 30 min did not significantly increase the immunoreactivity of phospho-p38 (n = 7). The MOR antagonist, 10 μM naloxone, did not significantly increase phospho-p38 activation in fentanyl- or morphine-treated wild type, MOR−/−, or GRK3−/− striatal neurons (n = 7). Furthermore, 1 μM U0126, the phospho-ERK1/2 inhibitor, did not significantly decrease the immunoreactivity of phospho-p38 compared with basal levels in wild type, MOR−/−, and GRK3−/− striatal neurons treated with fentanyl or morphine (n = 4). Bottom, representative immunoblots for the 38-kDa phos-pho-p38 protein are shown in which cells were untreated (Basal), treated with fentanyl, treated with morphine, pretreated with naloxone, or pretreated with U0126 for wild type (WT), MOR−/−, or GRK3−/− striatal neurons.
MOR Activation of ERK1/2 Phosphorylation Was Arrestin3 Dependent
The requirement for GRK3 suggests that arrestin may also be involved in the mu receptor-induced phosphorylation of ERK1/2. Treatment of wild type striatal cultures with the siRNA directed against arrestin3 decreased the abundance of arrestin3 by 34% as detected by immunoblotting, without affecting actin-IR (Fig. 4A, inset). The expression of arrestin3 was 66 ± 7% of untreated neurons (p < 0.01, n = 4) (Fig. 4A). Neurons treated with the transfection reagent alone showed a fentanyl-induced increase in phospho-ERK1/2 (Fig. 4B). Whereas, the siRNA-induced depletion of arrestin3 significantly attenuated the fentanyl-induced phosphorylation of ERK1/2 in striatal neurons to 66 ± 9% of the basal level of phospho-ERK1/2 (p < 0.01, n = 4) (Fig. 4B).
FIGURE 4. Inhibition of the fentanyl-mediated activation of phospho-ERK1/2 by siRNA-induced knockdown of endogenous arrestin3.
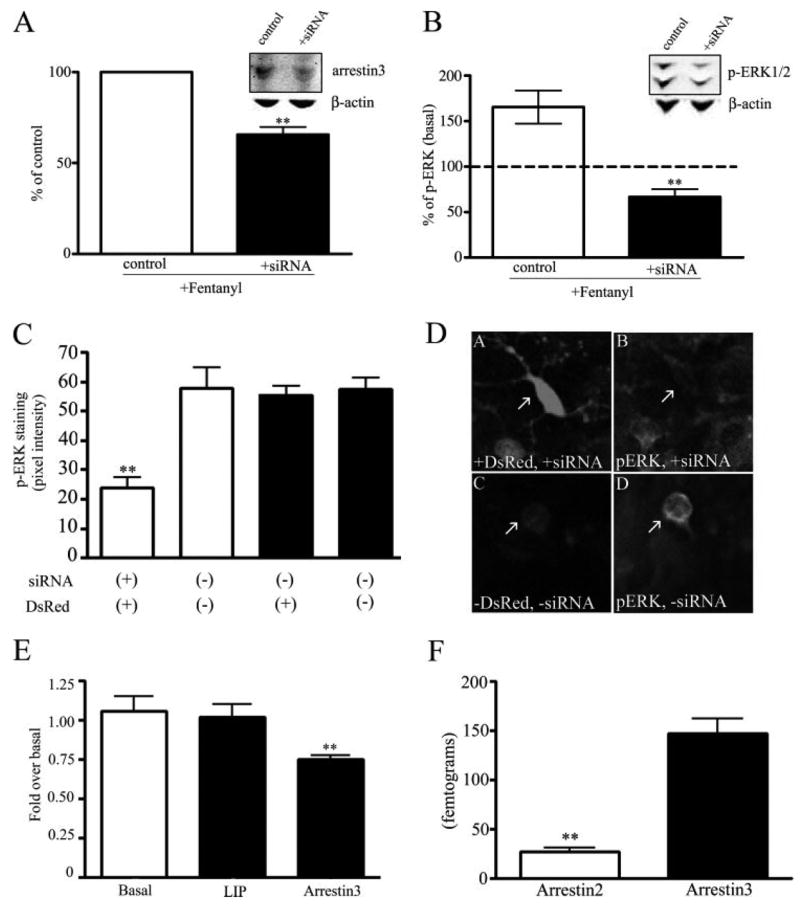
Fentanyl-induced activation of phospho-ERK1/2 by the endogenous MOR in siRNA arrestin3-treated wild type striatal neurons. A, immunoreactivity of arrestin3 (55 kDa) was determined in cells transfected with siRNA specific to arrestin3 and cells treated with transfection reagent only as described under “Experimental Procedures. ” Cells were treated with 100 nM fentanyl for 30 min (**, p < 0.01, by Student’s t test). A, inset, a representative immunoblot of arrestin3 (50 μg of protein) in cells transfected with the transfection reagent only (control) or arrestin3 siRNA (+siRNA) is shown. To control for equal loading of protein, membranes were reblotted with β-actin. B, phospho-ERK1/2 immunoreactivity (44/42 kDa) of cells transfected with transfection reagent only or siRNA-treated neurons was determined as described under “Experimental Procedures ” (**, p < 0.01, by Student’s t test). B, inset, representative immunoblot of phospho-ERK1/2 (30 μg of protein) in cells transfected with the transfection reagent only (control) or arrestin3 siRNA (+siRNA) is shown. C, the pixel intensity of the immunoreactivity of phospho-ERK1/2 was calculated using Metamorph software. Phospho-ERK1/2 was quantified in striatal neurons co-transfected with siRNA for arrestin3 and the empty vector DsRed (**, p < 0.01 by Dunnett’s post hoc comparison, n = 3) and compared with neurons without DsRed expression within the same field (open bars), neurons transfected with DsRed only, and neurons within the field of DsRed transfection only without DsRed expression (solid bars). In experiments, all cells were treated with the transfection reagent, Lipofectamine 2000. D, panel A, DsRed and arrestin3 siRNA co-transfected in striatal neurons, arrow points to transfected neuron; panel B, phospho-ERK1/2 immunoreactivity in the presence of arrestin3 siRNA, arrow points to the same transfected neuron as in panel A; panel C, no DsRed expressed, arrow points to untransfected neuron; panel D, phospho-ERK1/2 immunoreactivity in the absence of arrestin3 siRNA, arrow points to the same untransfected neuron in panel C. E, RNA was harvested from untreated and siRNA-treated neurons and quantitative RT-PCR was run. Arrestin3, arrestin2, and a control endogenous housekeeping gene ARBP were amplified in separate reactions from 20 ng of total RNA harvested from untreated neurons (basal), transfection reagent-treated neurons (Lip), and arrestin3 siRNA-treated neurons (arrestin3). Results are expressed as the -fold change over the untreated neurons (Basal) (**, p < 0.01, by Dunnett’s post hoc comparison). F, RNA was harvested from neurons and quantitative RT-PCR was run. Arrestin3 and arrestin2 were amplified in separate reactions from 20 ng of total RNA harvested from neurons. Standard curves were generated from in vitro transcribed arrestins to determine the absolute quantity of arrestin mRNA in the cultured neurons. Levels of arrestin2 and arrestin3 are expressed as femtograms of RNA. The abundance of arrestin2 was significantly lower than arrestin3 (**, p < 0.01, by Student’s t test).
Confocal imaging of transfected cells confirmed the results obtained by Western blot analysis. Following fentanyl treatment, the phosphorylated ERK1/2-IR intensity was 24 ± 3 average pixels per cell in striatal neurons co-transfected with siRNA for arrestin3 and DsRed compared with untransfected neurons in the same field (57 ± 7 average pixels per cell). Neurons transfected with DsRed but not arrestin3 siRNA responded to fentanyl treatment (100 nM, 30 min) with an increase in phosphorylated ERK1/2-IR intensity to the same extent (56 ± 3 average pixels per cell) as untransfected neurons (58 ± 4 average pixels per cell) (Fig. 4, C and D). Thus, arrestin3 siRNA expression reduced the fentanyl response by 59% as assessed with confocal microscopy. Primers were designed to detect arrestin3 message levels, and quantitative RT-PCR analysis showed that arrestin3 abundance was decreased by 25% (p < 0.01) compared with untreated neurons (Fig. 4E). The arrestin3 level for neurons treated with the transfection reagent alone was 1.09 ± 0.06-fold of basal, and the arrestin3 level for neurons treated with arrestin3 siRNA was 0.751 ± 0.02-fold of basal. Treatment with arrestin3 siRNA was selective as arrestin2 message levels were not affected. The arrestin2 level for neurons treated with arrestin3 siRNA was 1.04 ± 0.07-fold of basal.
A specific requirement for arrestin3 in the fentanyl-induced response was supported by assessing the absolute levels of mRNA expressed in cultured striatal neurons. Using primers designed to arrestin2 and arrestin3, quantitative RT-PCR showed that arrestin2 was expressed at 27.2 ± 4.1 fg in striatal neurons and arrestin3 was expressed at 147 ± 17.2 fg/20 ng of total RNA (Fig. 4F). Endogenous arrestin3 levels were significantly higher than endogenous arrestin2 levels in these cultured striatal neurons.
Dominant Positive Arrestin3-(R170E) Rescues Morphine-mediated Activation of Phospho-ERK1/2
Arrestin3-(R170E) is a dominant positive arrestin that eliminates the requirement of receptor phosphorylation in GPCR desensitization (10). Transfection of wild type striatal cultures with the dominant positive arrestin3-(R170E)-YFP significantly increased the morphine-induced phosphorylation of ERK1/2 compared with basal levels by 70 ± 18% (p < 0.01, Fig. 5B). YFP-transfected neurons treated with 10 μM morphine did not significantly increase phospho-ERK1/2 compared with basal levels. Treatment with 100 nM fentanyl of R170E-YFP-transfected striatal neurons increased phospho-ERK1/2 to 99 ± 28% compared with basal levels (p < 0.05, Fig. 5A). YFP-transfected striatal neurons treated with fentanyl significantly increased the phosphorylation of ERK1/2 by 95 ± 26% compared with basal levels (p < 0.05, Fig. 5A). The increase in fentanyl-induced phosphorylation of ERK1/2 was slightly higher than experiments in Fig. 1. However, this difference was not significant. The rescue by arrestin-(R170E) suggests that the inability of morphine to induce ERK1/2 phosphorylation was caused by its low efficiency in recruiting GRK3 and arrestin3.
FIGURE 5. Dominant positive arrestin rescues morphine-mediated and GRK-mediated activation of phospho-ERK1/2.
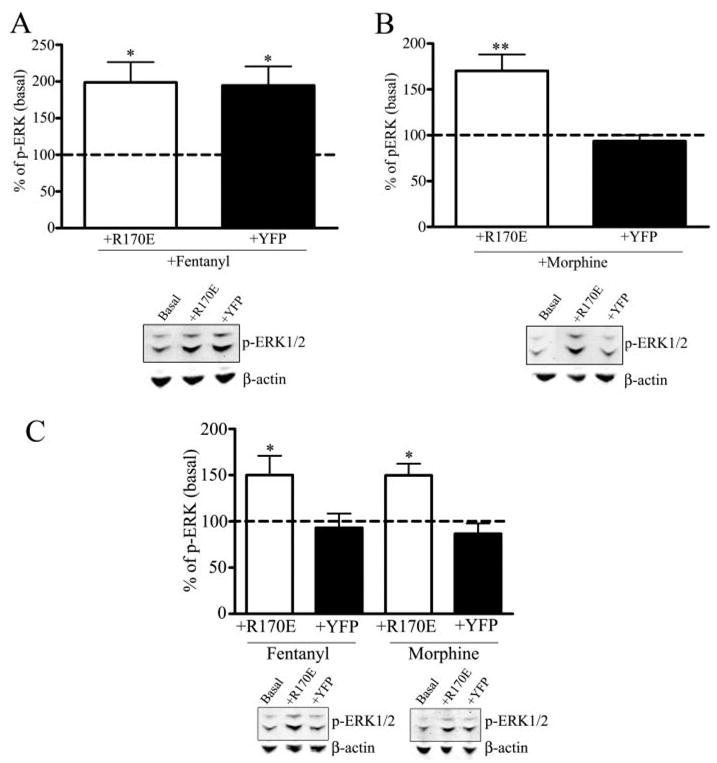
Morphine-induced activation of phospho-ERK1/2 by transfection of the dominant positive arrestin R170E in wild type and GRK3−/− striatal neurons. A, upper panel, striatal cultures were transfected with the dominant positive arrestin3-(R170E)-YFP or empty vector YFP and treated with 100 nM fentanyl. The intensity of the phospho-ERK1/2 immunoreactivity in agonist-treated transfected neurons was compared with untreated striatal neurons transfected with the dominant positive arrestin3-(R170E)-YFP (Basal). Fentanyl treatment increased the activation of phospho-ERK1/2 compared with basal levels (*, p < 0.05 by Dunnett’s post hoc comparison, n = 4). Fentanyl-treated YFP-transfected neurons increased the activation of phospho-ERK1/2 compared with basal levels (*, p < 0.05 by Dunnett’s post hoc comparison, n = 4). A, lower panel, a representative immunoblot for the 44/42-kDa phospho-ERK1/2 protein is shown in which cells were untreated in the presence of the dominant positive arrestin-(R170E) (Basal), treated with fentanyl in the presence of the dominant positive arrestin-(R170E) (+R170E) or YFP (+YFP). Representative immunoblots for β-actin demonstrates equal protein loading for each representative experiment. B, upper panel, striatal cultures were transfected with the dominant positive arrestin3-(R170E)-YFP or empty vector YFP and treated with 10 μM morphine for 30 min. The intensity of the phospho-ERK1/2 immunoreactivity in agonist-treated transfected neurons was compared with untreated striatal neurons transfected with the dominant positive arrestin3-(R170E)-YFP (Basal). Morphine treatment increased the activation of phospho-ERK1/2 compared with basal levels (**, p < 0.01 by Dunnett’s post hoc comparison, n = 4). B, lower panel, a representative immunoblot for the 44/42-kDa phospho-ERK1/2 protein is shown in which cells were untreated in the presence of the dominant positive arrestin-(R170E) (Basal), treated with morphine in the presence of the dominant positive arrestin-(R170E) (+R170E) or YFP (+YFP). Representative immunoblots for β-actin demonstrates equal protein loading for each representative experiment. C, GRK3−/− striatal cultures were transfected with the dominant positive arrestin3-(R170E)-YFP or empty vector YFP and treated with 100 nM fentanyl or 10 μM morphine for 30 min. The intensity of the phospho-ERK1/2 immunoreactivity in agonist-treated transfected neurons was compared with untreated striatal neurons transfected with the dominant positive arrestin3-(R170E)-YFP (basal). Fentanyl treatment increased the activation of phospho-ERK1/2 compared with basal levels (*, p < 0.05 by Dunnett’s post hoc comparison, n = 4). Morphine treatment increased the activation of phospho-ERK1/2 compared with basal levels (*, p < 0.05 by Dunnett’s post hoc comparison, n = 4). C, lower panel, a representative immunoblot for the 44/42-kDa phospho-ERK1/2 protein is shown in which cells were untreated in the presence of the dominant positive arrestin-(R170E) (Basal), treated with fentanyl in the presence of the dominant positive arrestin R170E (+R170E) or YFP (+YFP). Representative immunoblots for β-actin demonstrates equal protein loading for each representative experiment. C, lower panel, a representative immunoblot for the 44/42-kDa phospho-ERK1/2 protein is shown in which cells were untreated in the presence of the dominant positive arrestin-(R170E) (Basal), treated with morphine in the presence of the dominant positive arrestin-(R170E) (+R170E) or YFP (+YFP). Representative immunoblots for β-actin demonstrates equal protein loading for each representative experiment.
Dominant Positive Arrestin3-(R170E) Rescues Activation of Phospho-ERK1/2 Independent of GRK3
Transfection of GRK3−/− striatal cultures with the dominant positive arrestin3-(R170E)-YFP significantly increased the phosphorylation of ERK1/2 following fentanyl or morphine treatment compared with basal levels by 50 ± 20 and 50 ± 13%, respectively (p < 0.05, Fig. 5C). YFP-transfected neurons treated with 100 nM fentanyl or 10 μM morphine did not significantly increase phospho-ERK1/2 activation compared with basal levels (Fig. 5C). Although there was a significant increase of phosphorylated ERK1/2 in GRK3−/− striatal cultures compared with basal levels in wild type neurons, this increase was less than the fentanyl-induced activation of ERK1/2 in wild type cultures and fentanyl- or morphine-induced activation in R170E-YFP-transfected wild type striatal neurons. Again, the rescue of the MOR-induced phosphorylation of ERK1/2 by the dominant positive arrestin suggests the GRK3−/− neurons were specifically impaired in MOR phosphorylation and subsequent arrestin3 recruitment.
DISCUSSION
The novel finding of this study was that in mouse striatal neurons mu opioid receptor activation induced the phosphorylation of ERK1/2 MAPK by a mechanism that required agonist-induced mu receptor phosphorylation by GRK3 followed by arrestin3 recruitment. These results suggest that arrestin association mediates both homologous desensitization of MOR and enables MOR to participate in signal transduction pathways involving MAPK activation.
Desensitization of the mu opioid receptor by morphine has been characterized extensively with disparate results. However, two recent studies provided evidence that desensitization and endocytosis of MOR occurred with morphine treatment (17, 18). Activation of ERK1/2 by the mu opioid agonist DAMGO has been demonstrated in MOR-transfected cells and in neurons from morphine-treated mice (8, 9). However, the activation of ERK1/2 within the striatum and possible differences in the phosphorylation of ERK1/2 induced by MOR agonists has not been demonstrated. In this study we found that phosphorylation of ERK1/2 was significantly increased by fentanyl, a potent and efficacious MOR agonist, compared with basal levels of ERK1/2 phosphorylation. The effect was shown to be mediated by mu opioid receptors because naloxone, a MOR antagonist, blocked the fentanyl effect and similarly the effect was not seen in neurons isolated from MOR−/− mice. The fentanyl-induced increase was also blocked by the MEK inhibitor U0126.
However, when activation of ERK1/2 was assessed following morphine treatment in wild type striatal neurons, there was no significant increase in ERK1/2 phosphorylation compared with basal levels. This agonist selective activation of ERK1/2 in striatal neuronal cultures was also evident in a heterologous gene expression system similar to those in which GPCR-mediated ERK1/2 activation has been previously characterized (6). In AtT-20 cells expressing a GFP-tagged MOR, fentanyl but not morphine produced an increase in ERK1/2 phosphorylation. Although recent evidence has demonstrated that morphine treatment does cause rapid desensitization of the MOR under certain expression conditions (18), our data suggest that morphine was less effective than fentanyl at activating ERK1/2, which is consistent with its lower efficacy (14).
Because the activation of ERK1/2 by other GPCRs has been described as biphasic, we examined whether the fentanyl-induced increase in ERK1/2 phosphorylation was also biphasic. Previous reports have indicated that there is a G-protein-dependent early peak of activation, followed by a slower arrestin-dependent activation of ERK1/2 (6, 19). Both studies that investigated the biphasic activation of ERK1/2 used a transfected cell line that over-expressed the receptors and arrestin to characterize ERK1/2 phosphorylation. In whole cell lysates of striatal neurons containing the cytosolic and nuclear fractions, the phosphorylation of ERK1/2 was assessed at the endogenous levels of MOR and arrestin expression. Under these conditions, we did not observe a significant phosphorylation of ERK1/2 at the early time points suggesting an absence of the early, arrestin-independent activation of ERK1/2.
To determine whether the activation of ERK1/2 by fentanyl was specific or if MOR agonists might activate additional MAPK pathways, we also examined the stress-induced p38 MAPK. The p38 MAPK pathway has been demonstrated to play a major role in environmental stress and inflammatory signals, including cytokine activation (20). In addition, it has been demonstrated in rat dorsal root ganglion neurons, that chronic morphine treatment increased the activation of the MAPK pathways, ERK1/2, and p38 (21). The phosphorylation of the p38 MAPK pathway was not induced by fentanyl or morphine. Pretreatment with naloxone or the ERK1/2 inhibitor U0126 did not significantly increase the phosphorylation of p38. The results do not exclude a link between MOR and p38 in other systems, but they show that in these cultured neurons fentanyl specifically activated ERK1/2. The differences between p38 and ERK1/2 signaling observed support the concept that the pathways linking MOR activation to these MAP kinases are distinguishable.
To begin to elucidate the mechanisms underlying the specific activation of ERK1/2 by fentanyl, we investigated the role of GRK- and arrestin-dependent pathways. Following GRK phosphorylation of the receptor, arrestin binds and terminates the signaling of the receptor (22). In addition, the binding of arrestin targets the receptor to clathrin-coated pits for internalization and either degradation or resensitization (23, 24). Furthermore, it has been demonstrated that arrestin acts as a scaffolding protein, and promotes the stable association of signaling proteins with the receptor to activate MAPK pathways (19, 25). Arrestin scaffolding to MAPK has also been implicated in behavioral consequences using in vivo animal models (26). Therefore, to first determine whether GRK was involved in the fentanyl-mediated increase in ERK1/2 phosphorylation, we used striatal neuronal cultures derived from GRK3−/− mice. These mice have previously been shown to develop reduced behavioral tolerance to fentanyl (27). The fentanyl-induced activation of ERK1/2 was attenuated in GRK3−/− neurons indicating that GRK3 was specifically required for the MOR-mediated increase in phosphorylation of ERK1/2.
The lack of fentanyl effect on ERK1/2 phosphorylation in neurons derived from MOR−/− and GRK3−/− mice was not due to a gross inability of these cultures to show ERK1/2 phosphorylation. The MEK inhibitor U0126 dramatically decreased the phosphorylation of ERK1/2 compared with basal levels in wild type, MOR−/−, and GRK3−/− neurons. Furthermore, treatment with the MAPK activator anisomycin significantly increased levels of phosphorylated ERK1/2 in both wild type and GRK3−/− cultures. These data suggest that the mechanisms underlying both basal and induced ERK1/2 phosphorylation were still intact in these neurons. As an additional positive control for GPCR-induced phosphorylation of ERK1/2, activation of the kappa opioid receptor by U50,488 also induced phosphorylation of ERK1/2 in both wild type and GRK3−/− cultures. Previous work in AtT-20 cells has suggested that the kappa-mediated activation of ERK1/2 was GRK and arrestin independent, although kappa-induced phosphorylation of p38 required both GRK3 and arrestin3 (28). Therefore, although both the MOR−/− and GRK3−/− neurons could demonstrate ERK1/2 activation, they were specifically unable to increase ERK1/2 phosphorylation in response to fentanyl. These results suggest a specific role for GRK3 in the fentanyl-induced phosphorylation of ERK1/2.
Next, we examined the role of arrestin3 in the phosphorylation of ERK1/2 induced by fentanyl in striatal neurons. Using both immunoblot analysis and confocal microscopy, a significant decrease in the phosphorylation of ERK1/2 by fentanyl in siRNA-treated neurons was evident. The complete attenuation of MOR-mediated activation of ERK1/2 caused by a partial knockdown of arrestin was surprising. This may suggest that at endogenous levels of MOR expression, arrestin levels are limiting. The attenuation of ERK1/2 phosphorylation that was observed with the knockdown of arrestin3, defines the necessity of arrestin3 in the MOR-mediated activation of ERK1/2. Furthermore, our quantitative RT-PCR data supports a specific role for arrestin3 because it was the isoform of arrestin most abundantly expressed in the cultured striatal neurons.
As discussed earlier, the differential ability of fentanyl, but not morphine, to lead to increased levels of phosphorylated ERK1/2 may result from the lower efficacy of morphine. It is possible that morphine-activated MOR does not recruit arrestin readily compared with other more efficacious MOR agonists or the coupling of arrestin to MOR is less efficient with morphine activation. Consistent with this, numerous studies have suggested that morphine does not efficiently promote MOR regulation by GRK and arrestin. Morphine has been shown either to be unable to induce MOR phosphorylation (29, 30) or to induce less phosphorylation of MOR compared with higher efficacy agonists such as DAMGO (31, 32). Additionally, morphine was unable to promote internalization of MOR in heterologous cell systems (7), although more recently morphine was shown to produce internalization in striatal neurons when MOR- and GFP-tagged arrestin were overexpressed (18). Although we did not directly examine receptor internalization or arrestin binding in the present study, the inability of morphine to activate ERK1/2 compared with the higher efficacy agonist fentanyl is consistent with decreased coupling of morphine-activated receptors to arrestin.
Because the stability of the arrestin-receptor interaction likely depends on the phosphorylation state of the receptor, we used a dominant positive arrestin to eliminate the requirement of receptor phosphorylation. The dominant positive arrestin3-(R170E) has previously been reported to bind to agonist-occupied receptors and mediate desensitization independent of receptor phosphorylation (10, 33). When the arrestin3-(R170E) mutant and a phosphorylation insensitive delta opioid receptor were expressed, desensitization of the receptor was similar to the wild type (33). As expected, morphine produced a significant increase in levels of phosphorylated ERK1/2 in neurons transfected with arrestin3-(R170E). These data suggest that bypassing the requirement for receptor phosphorylation with the dominant positive arrestin enables morphine-activated receptors to more efficiently couple to arrestin to facilitate activation of ERK1/2. Because transfection of arrestin-(R170E) might have activated ERK1/2 independently of agonist treatment, we also examined untreated transfected neurons. However, phosphorylated ERK1/2 levels were not increased in arrestin3-(R170E) transfected, untreated neurons. Thus, the ability of the dominant positive arrestin to promote morphine-induced activation of ERK1/2 supports the concept that morphine is less efficient at recruiting arrestin.
In the usual mode of GPCR desensitization, GRK phosphorylation of the receptor is presumed to be necessary. In our study using GRK3−/− neurons, neither fentanyl nor morphine induced significant ERK1/2 phosphoryation. Again, when we transfected the dominant positive arrestin into neurons, thus removing the requirement of receptor phosphorylation, we were able to rescue the phosphorylation of ERK1/2 in the GRK3−/− striatal neurons. Interestingly, the phosphorylation of ERK1/2 by both MOR agonists was similar, suggesting a similar mechanism was involved. The rescue of MOR-mediated ERK1/2 activation in GRK3−/− neurons by arrestin3-(R170E) again demonstrates that these neurons were not globally deficient in their ability to activate ERK1/2, but rather that GRK3 was essential for MOR-induced phosphorylation of ERK1/2.
In this study, we were able to determine that the activation of MOR was agonist dependent in striatal neurons. The phosphorylation of ERK1/2 was dependent on both GRK3 and arrestin3 expression. Furthermore, when the requirement of MOR phosphorylation was eliminated by the transfection of arrestin3-(R170E), morphine-mediated activation of ERK1/2 was significantly increased. These data suggest that the addition of the dominant positive arrestin rescues the ability for the less efficacious agonist morphine to promote the arrestin-receptor interaction and mediate activation of ERK1/2. This study suggests the coupling of MOR to arrestin3 can alter the signaling properties of MOR. Increasing the strength of the association of the signaling proteins increases the efficiency of MOR to activate ERK1/2. This study helps further elucidate the signaling mechanisms of mu opioid receptors in brain.
Acknowledgments
We thank John Pintar for the MOR−/− mice; Marc Caron and Robert Lefkowitz for the GRK3−/− mice; Vsevold Gurevich for the bovine arrestin3-(R170E) mutant. We also thank Andrea Francois, Michael Soskis, and Greg Martin for technical assistance.
Footnotes
This work was supported by United States Public Health Service Grants RO1DA11672 and T32NS07332.
The abbreviations used are: MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; GPCR, G-protein coupled receptor; GRK3, G-protein coupled receptor kinase 3; IR, immunoreactivity; MEM, minimal essential media; MOR, Mu opioid receptor; p-p38, phospho-p38 MAPK; R170E, dominant positive arrestin3; RT, reverse transcriptase; siRNA, small inhibitory ribonucleic acid; YFP, yellow fluorescent protein; GFP, green fluorescent protein; DAMGO, D-Ala2,Me-Phe4,Gly-ol5; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid; dsRED, red fluorescent protein isolated from the Indo Pacific reef coral Discoma species.
References
- 1.Kovoor A, Henry DJ, Chavkin C. J Biol Chem. 1995;270:589–595. doi: 10.1074/jbc.270.2.589. [DOI] [PubMed] [Google Scholar]
- 2.Kovoor A, Nappey V, Kieffer BL, Chavkin C. J Biol Chem. 1997;272:27605–27611. doi: 10.1074/jbc.272.44.27605. [DOI] [PubMed] [Google Scholar]
- 3.Kovoor A, Celver JP, Wu A, Chavkin C. Mol Pharmacol. 1998;54:704–711. [PubMed] [Google Scholar]
- 4.Schulz R, Eisinger DA, Wehmeyer A. Eur J Pharmacol. 2004;500:487–497. doi: 10.1016/j.ejphar.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 5.Clark MJ, Neubig RR, Traynor JR. J Pharmacol Exp Ther. 2004;310:215–222. doi: 10.1124/jpet.103.064824. [DOI] [PubMed] [Google Scholar]
- 6.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 7.Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, Evans CJ, von Zastrow M. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- 8.Belcheva MM, Vogel Z, Ignatova E, Avidor-Reiss T, Zippel R, Levy R, Young EC, Barg J, Coscia CJ. J Neurochem. 1998;70:635–645. doi: 10.1046/j.1471-4159.1998.70020635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narita M, Ioka M, Suzuki M, Suzuki T. Neurosci Lett. 2002;324:97–100. doi: 10.1016/s0304-3940(02)00141-6. [DOI] [PubMed] [Google Scholar]
- 10.Kovoor A, Celver J, Abdryashitov RI, Chavkin C, Gurevich VV. J Biol Chem. 1999;274:6831–6834. doi: 10.1074/jbc.274.11.6831. [DOI] [PubMed] [Google Scholar]
- 11.Schuller AG, King MA, Zhang J, Bolan E, Pan YX, Morgan DJ, Chang A, Czick ME, Unterwald EM, Pasternak GW, Pintar JE. Nat Neurosci. 1999;2:151–156. doi: 10.1038/5706. [DOI] [PubMed] [Google Scholar]
- 12.Peppel K, Boekhoff I, McDonald P, Breer H, Caron MG, Lefkowitz RJ. J Biol Chem. 1997;272:25425–25428. doi: 10.1074/jbc.272.41.25425. [DOI] [PubMed] [Google Scholar]
- 13.Macey TA, Gurevich VV, Neve KA. Mol Pharmacol. 2004;66:1635–1642. doi: 10.1124/mol.104.001495. [DOI] [PubMed] [Google Scholar]
- 14.Celver J, Xu M, Jin W, Lowe J, Chavkin C. Mol Pharmacol. 2004;65:528–537. doi: 10.1124/mol.65.3.528. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Seed B. Nucleic Acids Res. 2003;31:e154. doi: 10.1093/nar/gng154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dheda K, Huggett JF, Bustin SA, Johnson MA, Rook G, Zumla A. BioTechniques. 2004;37:112–119. doi: 10.2144/04371RR03. [DOI] [PubMed] [Google Scholar]
- 17.Dang VC, Williams JT. Mol Pharmacol. 2005;68:1127–1132. doi: 10.1124/mol.105.013185. [DOI] [PubMed] [Google Scholar]
- 18.Haberstock-Debic H, Kim KA, Yu YJ, von Zastrow M. J Neurosci. 2005;25:7847–7857. doi: 10.1523/JNEUROSCI.5045-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDonald PH, Lefkowitz RJ. Cell Signal. 2001;13:683–689. doi: 10.1016/s0898-6568(01)00203-0. [DOI] [PubMed] [Google Scholar]
- 20.Tibbles LA, Woodgett JR. Cell Mol Life Sci. 1999;55:1230–1254. doi: 10.1007/s000180050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma W, Zheng WH, Powell K, Jhamandas K, Quirion R. Eur J Neurosci. 2001;14:1091–1104. doi: 10.1046/j.0953-816x.2001.01731.x. [DOI] [PubMed] [Google Scholar]
- 22.Bohn LM, Dykstra LA, Lefkowitz RJ, Caron MG, Barak LS. Mol Pharmacol. 2004;66:106–112. doi: 10.1124/mol.66.1.106. [DOI] [PubMed] [Google Scholar]
- 23.Pippig S, Andexinger S, Lohse MJ. Mol Pharmacol. 1995;47:666–676. [PubMed] [Google Scholar]
- 24.Tsao PI, von Zastrow M. J Biol Chem. 2000;275:11130–11140. doi: 10.1074/jbc.275.15.11130. [DOI] [PubMed] [Google Scholar]
- 25.Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. Proc Natl Acad Sci U S A. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 27.Terman GW, Jin W, Cheong YP, Lowe J, Caron MG, Lefkowitz RJ, Chavkin C. Br J Pharmacol. 2004;141:55–64. doi: 10.1038/sj.bjp.0705595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruchas MR, Macey TA, Lowe JD, Chavkin C. J Biol Chem. 2006;281:18081–18089. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, Caron MG. Proc Natl Acad Sci U S A. 1998;95:7157–7162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Neuron. 1999;23:737–746. doi: 10.1016/s0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- 31.Yu Y, Zhang L, Yin X, Sun H, Uhl GR, Wang JB. J Biol Chem. 1997;272:28869–28874. doi: 10.1074/jbc.272.46.28869. [DOI] [PubMed] [Google Scholar]
- 32.Koch T, Schulz S, Pfeiffer M, Klutzny M, Schroder H, Kahl E, Hollt V. J Biol Chem. 2001;276:31408–31414. doi: 10.1074/jbc.M100305200. [DOI] [PubMed] [Google Scholar]
- 33.Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. J Biol Chem. 2002;277:9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]