

Abstract
Arginine kinase belongs to the family of enzymes, including creatine kinase, that catalyze the buffering of ATP in cells with fluctuating energy requirements and that has been a paradigm for classical enzymological studies. The 1.86-Å resolution structure of its transition-state analog complex, reported here, reveals its active site and offers direct evidence for the importance of precise substrate alignment in the catalysis of bimolecular reactions, in contrast to the unimolecular reactions studied previously. In the transition-state analog complex studied here, a nitrate mimics the planar γ-phosphoryl during associative in-line transfer between ATP and arginine. The active site is unperturbed, and the reactants are not constrained covalently as in a bisubstrate complex, so it is possible to measure how precisely they are pre-aligned by the enzyme. Alignment is exquisite. Entropic effects may contribute to catalysis, but the lone-pair orbitals are also aligned close enough to their optimal trajectories for orbital steering to be a factor during nucleophilic attack. The structure suggests that polarization, strain toward the transition state, and acid-base catalysis also contribute, but, in contrast to unimolecular enzyme reactions, their role appears to be secondary to substrate alignment in this bimolecular reaction.
A combination of kinetic and structural studies over the last 30 years has given us an understanding of how enzymes can achieve their catalysis. Acid-base catalysis, enzyme-induced strain, or binding favoring the transition state have become textbook mechanisms (1), the result of extensive studies of mostly unimolecular (single substrate) enzymes. In bimolecular systems, the potential importance of entropic effects (2) (the alignment of substrates) is much greater. Basic questions such as the relative importance of entropic and other effects, and whether enzymes precisely align orbitals (3, 4) or merely guide substrates to the correct vicinity, remain open. Recently, the kinetic effects of distorting the geometry of the hydride transfer in isocitrate dedydrogenase (5) indicated that the donor-acceptor distance was not the only factor affecting kinetic rate. This finding was cited as affirmation of the importance of precise orientational alignment. Unfortunately, there have been no atomic structures that have shown directly how precisely multiple reactants are oriented relative to one another in an enzyme active site, prior to a multimolecular reaction.
Understanding of bimolecular reactions has lagged behind unimolecular reactions because of technical difficulties in direct visualization. In studies of bisubstrate transition-state analogues (6, 7) the reactants have been covalently linked to obtain a stable complex. The positions of the reactants are constrained by the covalent bond between them and offer only indirect insights regarding their alignment prior to the reaction. Other prior studies of substrate analogs or mutants with slow enough catalysis to be visualized usually have had active site or substrate stereochemistry that is at least slightly distorted, precluding comments about precise alignment (8–10). What is required are systems that can be studied at high resolution, in which the changes required to make a stable complex that can be visualized have had minimal impact on substrate alignment. Here is reported the 1.86-Å structure of a phosphagen kinase dead-end transition-state analog complex in which a nitrogen substitutes for a phosphorus atom as a nitrate mimics the planar γ-phosphoryl during an associative in-line transfer between ATP and the phosphoryl acceptor (11–13). In such a case, the positions of the reactants in the system reported here are constrained only by natural interactions with the enzyme, offering an opportunity to measure how well substrates can be aligned as part of an enzyme mechanism.
Phosphagen (guanidino) kinases catalyze the reversible transfer of a phosphoryl group between ATP and an “energy storage” phosphagen such as creatine in the human enzyme—creatine kinase (CK), or arginine for the enzyme (EC 2.7.3.3) studied here:
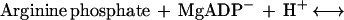 |
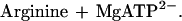 |
It is through this buffering reaction that cells can support bursts of nerve or muscle activity that would otherwise drain ATP to levels that would not sustain other essential functions (14). Phosphagen kinases have a mechanism that is one of the most thoroughly studied by classical methods (15), but full understanding has been hindered through lack of an atomic structure.
After many unsuccessful attempts, an ≈3-Å structure of the octameric mitochondrial CK [MibCK, (16)] was determined, revealing a two-domain subunit structure. Although this work was very informative and critical to the success of the work reported here, it was not possible to come to firm conclusions about the catalytic mechanism because, in the absence of creatine, much of the active site was disordered. In fact, the structure initiated debate about aspects of the mechanism that had previously seemed settled. A histidine had been expected to act as an acid-base catalyst (17), but none was close enough to the active site to be an obvious candidate (16). Perhaps a conformational change is required for activity (16), or alternatively, acid-base catalysis is not involved, and CK acts as a “conzyme,” achieving catalysis through (merely) bringing the substrates into close proximity (15). These questions are settled by the arginine kinase (AK) structure reported here.
Differences were to be expected between the published structure of MibCK and that of the AK reported here. The enzymes share only ≈40% sequence identity, have different quaternary structures, have different cellular locations, and differ in that MibCK is membrane associated, but AK is not. Furthermore, the structures have been determined in different catalytic states. The MibCK structure was determined in the presence of the nucleotide, but not creatine. Slight differences between the structures of different subunits of the octamer, regions of disorder, and a comparison of the structure with spectroscopic data (18) suggested to the authors that it was the structure of a flexible inactive “open” form of the enzyme that had been determined (16). This finding was consistent with evidence for a conformational change that occurs when all substrates have been bound to either AK or CK (19, 20). By contrast, the AK was expected to be in the “closed” reactive conformation because it was crystallized in the presence of the complete set of transition-state analog components (21).
This report focuses on the active site of AK. It examines whether the substrates are aligned precisely enough for entropy reduction (2) or orbital steering (3, 4) to be significant factors in the catalysis of this paradigm for bimolecular reactions. It also identifies the amino acids of the active site that might be critical to these and other aspects of catalysis.
METHODS
Diffraction Data Collection.
AK from horseshoe crab (Limulus polyphemus) was cloned, expressed, complexed with analog components (ADP, nitrate, and arginine), as described (21, 22). Diffraction data were collected with an R-Axis II image plate detector (Molecular Structure) and a rotating anode x-ray source for the previously reported crystals and a second crystal form crystallizing under identical conditions (Table 1). Data used for an initial structure determination (native and two derivatives) were collected at about 298 K, whereas 1.86-Å data for refinement were collected at 100 K after serial transfer of crystals to a 25% glycerol cryo-protectant.
Table 1.
Crystallographic and refinement statistics
| Data collection | Native | Native | Native | K2PtCl4 | Thiomersal |
|---|---|---|---|---|---|
| Crystal form | 1: P212121, | 2: P212121, | 2: P212121, | 2: P212121, | 2: P212121, |
| a = 64.2 Å, | a = 66.4 Å, | a = 65.4 Å, | a = 66.7 Å, | a = 66.8 Å, | |
| b = 66.0 Å, | b = 72.5 Å, | b = 70.9 Å, | b = 72.8 Å, | b = 72.6 Å, | |
| c = 86.7 Å | c = 80.4 Å | c = 80.4 Å | c = 80.3 Å | c = 80.4 Å | |
| Temperature, K | 298 | 298 | 100 | 298 | 298 |
| Resolution limit, Å | 2.25 | 2.25 | 1.86 | 3.0 | 3.0 |
| Completeness | 94.6 | 94.3 | 98.9 | 98.9 | 84.5 |
| (at resolution limit), % | (88.3) | (76.5) | (98.5) | (97.3) | (77.5) |
| Redundancy* | 4.5 | 7.4 | 6.7 | 6.6 | 5.9 |
| Rsym† = Σ|Ih−〈Ih〉|/Σ|Ih|, % | 7.0 | 6.0 | 4.7 | 3.8 | 7.5 |
| Number of heavy atom sites | 0 | 0 | 0 | 3 | 3 |
| Riso and phasing power‡ | 18%, 1.0 | 17%, 1.2 | |||
| Model | 356 of 357 amino acids (missing Met-1) + 298 water molecules + arginine, ADP, Mg2+, and two NO3− | ||||
| Refinement | Rcryst1 = 0.196 | 〈B〉overall = 16 Å2 | Cross-validated | ||
| Resolution = 5−1.86 Å | Rfree = 0.224 | 〈B〉substrate analogs = 9 Å2 | Luzzatti error (27) | 0.24 Å | |
Average number of observation of each reflection.
Rsym = Σ|Ih − 〈Ih〉|/Σ|Ih|, where 〈Ih〉 is the average intensity of symmetry equivalent observations.
Riso = Σ|FPH − Fp|/Σ|Fp|, or the mean fraction isomorphous difference, and phasing power = fH/E where fH is the heavy atom structure amplitude and E is the lack of closure error.
Rcryst = Σ|Fo − Fc|/Σ|Fo|, Rfree was assessed with 877 (3%) reflections omitted from the refinement. Refinement and Rcryst used 28346 reflections for which F/σ(F) > 2.0.
Phase Determination.
Molecular replacement was attempted by superimposing a subunit of MibCK (16) onto the AK crystal by using the amore and x-plor implementations (23, 24) of the rotation and translation functions, and allowing the small and large domains to have independent orientations. Molecular replacement solutions that were consistent between the crystal forms were found for the large domain of an MibCK subunit, but not the small domain. Molecular replacement and multiple isomorphous replacement (MIR) phases were combined at 3-Å resolution and improved by intercrystal form averaging by using DMMulti (25). The starting model was based on the MibCK large domain and the molecular replacement solution was marginal, having near-random agreement with the diffraction amplitudes (R = 0). The starting map, based on poor phases, was of poor quality, enabling only parts of the AK sequence of the large domain to be modeled (26), but the small domain was not yet visible. Some rigid-body improvement of the two-domain molecular replacement solution was possible with the remodeled large domain, but the model was outside the convergence radius of conventional atomic refinement with an Rfree (27) of 0.54. Local real-space refinement (28, 29) was critical in starting atomic refinement without overfitting the partial model and in bringing Rfree below 0.5, after which conventional refinement was possible. The model was improved through several iterations of rebuilding, refinement by a combination of real (28) and reciprocal (30) space methods, and map improvement. Maps were calculated by using phases calculated by Fourier-inversion of an omit-map (31) calculated from the latest model, combination with MIR phases, and improvement by averaging between crystal forms (25).
Model, Refinement, and Quality.
After completion of the protein model, refinement was extended to 1.86-Å resolution data by using the data collected at cryo-temperatures and reciprocal-space refinement. To be sure of avoiding bias, the transition-state components were modeled only late in refinement (Rfree = 0.3) when the electron density was unambiguous. The entire protein is visible except for the N-terminal methionine that mass spectrometry indicates is present in about 50% of our sample. In addition to the protein, 298 ordered water molecules, MgADP−, and two nitrates were modeled. Refinement yielded Rcryst = 0.196 and Rfree = 0.224 (27) for 2 σ data between 5- and 1.86-Å resolution and a cross-validated estimate of overall rms positional error (27) of 0.24 Å. This report discusses the alignment of nucleophiles’ valence orbitals with neighboring nonbonded electrophiles. This result can be determined implicitly from the angles at a nucleophilic atom between its covalent bonds and the direction to the electrophile. The 0.24-Å overall estimate of error indicates that such angles can be measured with an average precision of about ±9°. However, the low average B-factor for the substrate analogs compared with the protein (9 cf. 16 Å2) indicates greater precision than average in the active site.
This report also compares the consistency of the observed configuration of the analog components with a likely structure of the actual transition state that cannot be observed crystallographically. The transition-state structure, most consistent with the data for the analogs, was generated by least-squares refinement as follows: (i) the nitrate nitrogen was replaced with a phosphorus, (ii) additional phosphorus to Oβ and Nη distance restraints were calculated according to the putative 20% covalent bond formation (32, 33), (iii) bond angle restraints were added for the phosphorus and its neighbors as if the analog components were joined in one covalent complex, and (iv) the structure was refined against the supplemented stereochemical restraints for the actual transition state and the available diffraction data for the analog complex.
The observed analog structure is consistent with prior spectroscopic results. For example, the glycosidic ADP torsion angle (46°) is consistent with the 50 ± 5° (34) determined by NMR, and the structure confirms octahedral coordination of the Mg2+ with single ligands from oxygens of each of the α, β, and γ ATP (35, 36) phosphates.
RESULTS AND DISCUSSION
Overview of Structure, Similarities, and Differences with MibCK.
As expected of 40% sequence identity, AK shares the same subunit topology as CK (16). A small α-helical N-terminal domain is followed by a larger C-terminal domain (residues 112–357) that is similar to the C-terminal domain of glutamine synthetase (37, 38)—an 8-stranded antiparallel β-sheet is flanked by 7 α-helices (Fig. 1). It was suggested that the ATP of the binary MibCK complex was not in the exact position required for catalysis (16), and, indeed the ADP of the complete transition-state analog (TSA) complex is shifted an rms of 2.4 Å in AK relative to ATP in the binary CK complex. The bridging of the substrates between the two domains (Fig. 1) and the 4-Å rms shift of the small domain [relative to the MibCK structure, (16)] offers a molecular explanation for the substantial changes seen with X-ray solution scattering of both AK and CK upon addition of all (but not individual) TSA components (20). Relative to the inactive open structure seen with MibCK, the closed active structure seen here shows substantial backbone changes (up to 15 Å) for active site residues, primarily in loops that were partially disordered in the MibCK structure (16). Following the example of hexokinase (39), generation of the active conformation by induced fit after binding of all substrates may prevent wasteful hydrolysis of ATP, should it bind in the absence of the guanidino phosphoryl acceptor.
Figure 1.

Structure of AK. The N-terminal domain is shown in yellow and the C-terminal domain in green. The TSA complex ligands, shown in ball-and-stick, lie left to right in the order ADP, Mg2+ (light blue), nitrate, and arginine with arginine bridging between the large domain and a region of the small domain (highlighted in red) that is likely involved with substrate specificity and movement of the small domain. Two loops (63–68, red; and 309–318, blue) that were disordered in the MibCK structure are found in the AK TSA complex in substantially different conformation.
Configuration of the Substrate Analog Components.
Clear electron density (Fig. 2a) and the high resolution refinement (Rfree = 0.22 for 5- to 1.86-Å resolution) allow detailed analysis of the active site. Studies of the reaction in small molecule models had previously suggested a preassociative concerted phosphoryl transfer. The transition state was thought to approximate a hybrid of a dissociated metaphosphate intermediate and a pentavalent γ-phosphorus in which there is ≈20% simultaneous apical covalent bonding to the β-phosphoryl oxygen and guanidino nitrogen (32, 33). Although there are no covalent bonds to constrain them, the positions and geometry of the substrate analog components are remarkably consistent with the presumptive transition state. For example, the observed distance between guanidino N and Oβ through the analog nitrate is 5.9 Å compared with 6.0 Å expected of the transition state. Similarly, unlike the γ-phosphoryl of either the substrate complex or the presumptive transition state, the position of the nitrate of the analog is not constrained by (partial) covalent bonding to the β phosphoryl or guanidinium, yet is positioned nearly exactly where the γ-phosphoryl would be expected (Fig. 2a) in the transition state. Such consistency, combined with the low B-factors of the substrate analogs that are one-half the average (Table 1) indicates that the enzyme has a role in restricting the freedom of the reactive groups and could bring an entropic component to catalysis (2).
Figure 2.

Details of the active site. For clarity only atoms in the immediate neighborhood are shown with carbon-colored black, oxygen red, nitrogen dark blue, magnesium light blue, sulfur yellow and phosphorus gray. Distances are shown in Å. (a) Stereo diagram comparing part of the experimental analog structure with omit-map electron density and the structure of the presumptive transition state (gray atoms). Small molecule model systems suggest a preassociative concerted phosphoryl transfer and a pentavalent γ-phosphorus transition state with about 20% covalent bonding to both the β-phosphoryl oxygen and guanidino nitrogen (32, 33). The transition-state coordinates were derived from the experimental coordinates by replacing the nitrate with a phosphoryl group, and refining with additional distance and angle restraints appropriate for the estimated 20% partial covalent bonding. (b–e) Details of the enzyme-substrate analog interactions: (b) α and β phosphoryl groups of the ADP are held in place by extensive hydrogen bonds/salt bridges with four highly conserved arginines; (c) the nitrate (mimicking a planar phosphoryl group during transfer) is sandwiched between two conserved arginines and the Mg2+ ion whose position is constrained by ligands from the α and β phosphoryl groups of the ADP; (d) the guanidinium of the substrate arginine is clamped with salt bridges/hydrogen bonds to two carboxylates and a conserved cysteine that likely exists as a thiolate (54); and (e) interactions of the substrate amino and carboxylate groups with loop residues 63–68 of the enzyme. The carboxylate-to-backbone interactions might be conserved between all phosphagens and their kinases. The amino groups are present in arginine and lombricine but absent from creatine and glycocyamine. The tyrosine interacting with the amino group is conserved among AKs, but is a valine in all other phosphagen kinases. Immediately preceding residue 61 (and interactions with the carboxylate) is an insertion in other sequences whose size inversely correlates with the size of substrate (42).
The dense network of interactions that hold the substrates in place is shown in Fig. 2. It is a concentration of five conserved arginines and the bound Mg2+ that hold the ATP phosphoryl groups in place. The arginines are conserved in all known phosphagen kinase sequences, except Drosophila in which residues 280 and 309 are leucine and alanine, respectively. Of the carboxylates that hold the substrate guanidinium in place, one (Glu-225) is completely conserved, and the other (Glu-314) is conserved, except in CK. With minor adjustment to prior sequence alignments (40–42), MibCK Asp-321 can be aligned with AK Glu-314. Then CK would have a shorter carboxylate to compensate for the bulk of a methyl group at the Nɛ substrate position, compared with the other phosphagen kinases (arginine, lombricine, and glycocyamine) in which a glutamate interacts with the smaller hydrogen at the Nɛ position.
Substrate Alignment and Its Mechanistic Implications.
Substrates are positioned not only in close proximity but with orbitals aligned close to optimally. Consistent with in-line transfer, the donor and acceptor atoms (ADP Oβ, and guanidinyl Nη) are positioned to form bonds within 0.2° of orthogonal to the phosphoryl (nitrate) plane. With respect to the guanidinyl Nη, the optimal direction for nucleophilic attack by its lone pair is at an angle of ≈110° to the Nη—Cγ bond in a plane perpendicular to the guanidinium plane. The nitrate nitrogen, mimicking the Pγ, is at 115°, close to ideal. For the reaction in the opposite direction, extended Hückel calculation of phosphate valence electron density suggests optimal direction of nucleophilic attack by the oxygen at an angle of 100° with respect to the Pβ—Oβ bond. The observed Pβ—Oβ—N(Pγ) angle is within 1° of optimal. Thus, for the reactions in both directions, the enzyme is steering the orbitals on trajectories within 5° of optimal, precision estimated to enhance catalysis by up to 105 (4).
Other Catalytic Effects.
Supplementing entropic and orbital steering effects, strain toward the transition state and other effects may contribute to catalysis. Unlike the other guanadinyl nitrogens, the reactive Nη2 does not hydrogen bond with sp2-consistent geometry. This result would require ≈0.4-Å shifts of the Nη2 and acceptor Oɛ of glutamates 225 and 314 to the edge of the experimental electron density. The observed configuration is consistent with an Nη2 intermediate between the sp2 substrate and presumed sp3 configuration on the reaction pathway. Caution is needed in interpreting such subtleties, even at 1.9-Å resolution. However, as in the serine proteases, catalysis is perhaps facilitated by an improvement of hydrogen-bonding configuration as the reaction proceeds, channeling binding energy into strain and lowering of the activation barrier (43, 44). The catalytic effect is likely smaller than in the serine proteases because it is the angle at the hydrogen bond acceptor that is improved, not the hydrogen bond distance, on which the energy is more dependent.
Acid-Base Catalysis?
A histidine was long implicated in acid-base catalysis of phosphagen kinases (17, 45) and thought to assist in the abstraction of a proton from the Nη. With no histidines close to the presumed reactive site, Fritz-Wolf et al. (16) suggested that conformational changes from their MibCK structure would be required for activity, whereas Stroud (15) suggested that entropic effects alone might be sufficient for catalysis. The current structure resolves this enigma. One histidine, previously shown by mutagenesis to be important (46) in CK, forms a base-stacking interaction with the adenine. Nucleotide-binding appears to be the role for the important histidine not proton abstraction from the Nη, for which glutamates 225 and 314 are the only candidates (Fig. 2d). Although an ionized carboxylate had been implicated in an undetermined role for CK (17), one could be forgiven for not expecting a glutamate to act as a base, even though there is precedent in Staphylococcal nuclease (47, 48). The counter-intuitive role of a glutamate as a base, that we hypothesize here, can be rationalized at two levels: (i) acid-base catalysis may be secondary to the role of glutamate in positioning the guanidinium, and (ii) in the reactive state the pKs may be more optimally matched for isoergonic proton transfer (49) than between free glutamate and arginine. Proton elimination would have to occur as or after the Nη—Pγ bond is formed, and the transitional Nη+ cation would be acidic. Also, the pKs of glutamates 225 and 314 are likely raised by the close proximity of the γ-phosphate of ATP or arginine phosphate anion. Whether the proposed base is Glu-225 or Glu-314 is not indicated by the crystallographic structure.
As in several enzymes, such as Staphylococcal nuclease, positive charges (arginines 229 and 309 and Mg2+) pull electrons toward the phosphate oxygens from the phosphorus, preparing it for nucleophilic attack in either direction of the reaction. Similarly the interactions of arginines 126 and 280 (Fig. 2b) with the ATP O3β appear to stabilize the partial charge on O3β, after ATP hydrolysis in the forward reaction or in preparation for O3β nucleophilic attack on Pγ in the reverse reaction.
The “essential” cysteine (271 in AK, 278 in MibCK) took its name from the observation that covalent modifications resulted in inactive enzyme (50–52). The effect of gross modifications can be rationalized by the structure in terms of steric conflict with the substrate guanidinium. More subtle effects of conservative mutations (22, 53, 54) have led to several hypotheses that the cysteine may not be catalytically important, but may be involved in synergism between the binding of the two substrates or in a hinge movement required for the enzyme to become active. The AK structure (Fig. 2d), in which the cysteine interacts with the nonreactive guanidinyl Nη1, suggests that, after all, it is likely to have at least an accessory role in catalysis and is consistent with the finding that CK is most active with the cysteine as a thiolate ion (54). The cysteine, even if not essential, could enhance the catalytic activity by (i) further constraining the position of the substrate guanidinium, and (ii) drawing partial positive charge away from the reactive nucleophilic Nη2. It may accept a hydrogen bond from the substrate and may contribute to the observed pH activity profile (17).
Substrate Specificity.
Phosphagen kinases show broad diversity in the substrates for which the enzyme from particular species are specific. All substrates have guanidinium reactive groups, but they can be either amino acids (e.g., lombricine and arginine) or carboxylic acids (creatine and glycocyamine) and have a wide range of molecular masses: 117–271 Da. In AK, the carboxylate (common to all substrates) accepts three hydrogen bonds from the backbone amino groups of residues 63–5 (Fig. 2e). Immediately preceding these interactions, at residue 61, there is a 5-amino acid insertion in the sequences of enzymes with the shorter substrates (42). From the sequence alignments, it had been argued that the size of the insertion might control substrate specificity (42). With the insertion point being close to the arginine carboxylate, the AK structure is consistent with the loop being one of the factors that might control specificity. It corresponds to a flexible loop identified in the MibCK structure (16) that likely only adopts the ordered conformation seen in the AK structure upon substrate binding. The AK structure suggests that this loop is key to the small domain movement (Fig. 1). Therefore, specificity might be mediated not only in steric interactions, but through the ability of substrates only of appropriate length to bridge the active site and, through interactions at each end, initiate the conformational changes required for activity.
CONCLUSION
The structure of AK presented in this communication substantially advances our mechanistic understanding of a reaction central to cellular energy transactions. It has also provided an opportunity to examine a bimolecular transition-state analog system that satisfied three criteria that have been difficult to meet simultaneously in the past: (i) that changes made to block reactivity have minimal impact on the structure, (ii) that there be no additional constraints upon molecular positions than present during the reaction; and (iii) that the structure determination be of sufficient precision. The results of this study are fully consistent with the proposals that restraining the freedom of the reactants (2) and steering the substrates so that their orbitals are within 5 or 10° of the optimal trajectories (3, 4) would be used to achieve catalysis in bimolecular enzyme reactions.
The roles of active-site residues in the catalytic mechanism are summarized in Fig. 3. In addition to the steering and positioning effects, roles seen in the more thoroughly studied, unimolecular systems are also seen in this bimolecular system: effecting transfer of partial charge, strain toward the transition state, and acid-base catalysis. It appears that all possible enhancements of catalysis are employed. In judging their relative importance, it is noteworthy that carboxylates have been selected by evolution at positions where catalytic bases are required, presumably because precise positioning of substrates claimed precedence over optimization of the proposed role of the carboxylates in acid-base catalysis.
Figure 3.

Roles of neighboring amino acids in the catalytic mechanism of AK: (a) the forward reaction and (b) the reverse reaction. In this schematic representation, only the reactive groups of participants are shown. ∗, The structure does not indicate whether it is Glu-225 or Glu-314 that acts as the proposed acid/base catalyst.
Acknowledgments
We thank W. Kabsch for providing MibCK coordinates prior to general release, and M. Schwartz, A. Steigman and D. Caspar for helpful discussions. This work was supported by grants from the American Heart Association, Florida Affiliate (to M.S.C.), National Institutes of Health Grant GM 55837 (to M.S.C.), National Science Foundation Grants IBN 96–31907 (to W.R.E.) and BIR 94-18741 (to M.S.C. for refinement methods), and the Lucille P. Markey Charitable Trust (Structural Biology Program). Cryo-data collection apparatus was purchased with National Cancer Institute Grant CA 47439 to Don Caspar.
ABBREVIATIONS
- AK
arginine kinase
- CK
creatine kinase
- Mi
mitochondrial
- MIR
multiple isomorphous replacement
- TSA
transition-state analog
Footnotes
Data deposition: The atomic coordinates and x-ray amplitudes have been deposited in the Protein Data Bank, Biology Department, Brookhaven National Laboratory, Upton, NY 11973 (PDB ID code 1bg0).
References
- 1.Fersht A. Enzyme Structure and Function. New York: Freeman; 1985. [Google Scholar]
- 2.Page M I, Jencks W P. Proc Natl Acad Sci USA. 1971;68:1678–1683. doi: 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Storm D R, Koshland D E., Jr Proc Natl Acad Sci USA. 1970;66:445–452. doi: 10.1073/pnas.66.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dafforn A, Koshland D E., Jr Proc Natl Acad Sci USA. 1971;68:2463–2467. doi: 10.1073/pnas.68.10.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mesecar A D, Stoddard B L, Koshland D E., Jr Science. 1997;277:202–206. doi: 10.1126/science.277.5323.202. [DOI] [PubMed] [Google Scholar]
- 6.Kantrowitz E R, Lipscomb W N. Science. 1988;241:669–674. doi: 10.1126/science.3041592. [DOI] [PubMed] [Google Scholar]
- 7.Schneider G, Lindqvist Y, Brändén C-I. Annu Rev Biophys Biomol Struct. 1992;21:119–143. doi: 10.1146/annurev.bb.21.060192.001003. [DOI] [PubMed] [Google Scholar]
- 8.Gouaux J E, Lipscomb J E. Proc Natl Acad Sci USA. 1988;85:4205–4208. doi: 10.1073/pnas.85.12.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karpusas M, Branchaud B, Remmington S J. Biochemistry. 1990;29:2213–2219. [PubMed] [Google Scholar]
- 10.Bolduc J M, Dyer D H, Scott W G, Singer P, Sweet R M, Koshland D E, Stoddard B L. Science. 1995;268:1312–1318. doi: 10.1126/science.7761851. [DOI] [PubMed] [Google Scholar]
- 11.Hansen D E, Knowles J R. J Biol Chem. 1981;256:5967–5969. [PubMed] [Google Scholar]
- 12.Milner-White E J, Watts D C. Biochem J. 1971;122:727–740. doi: 10.1042/bj1220727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McLaughlin A C, Cohn M. J Biol Chem. 1972;247:4382–4388. [PubMed] [Google Scholar]
- 14.McGilvery R W. Biochemistry, A Functional Approach. Philadelphia: Saunders; 1979. [Google Scholar]
- 15.Stroud R M. Nat Struct Biol. 1996;3:567–569. doi: 10.1038/nsb0796-567. [DOI] [PubMed] [Google Scholar]
- 16.Fritz-Wolf K, Schnyder T, Wallimann T, Kabsch W. Nature (London) 1996;381:341–345. doi: 10.1038/381341a0. [DOI] [PubMed] [Google Scholar]
- 17.Cook P F, Kenyon G L, Cleland W W. Biochemistry. 1981;20:1204–1210. doi: 10.1021/bi00508a023. [DOI] [PubMed] [Google Scholar]
- 18.Vasák M, Nagayamne K, Wüthrich K, Mertens M L, Kägi J H R. Biochemistry. 1979;18:5050–5055. doi: 10.1021/bi00590a004. [DOI] [PubMed] [Google Scholar]
- 19.Reed G H, Cohn M. J Biol Chem. 1972;247:3073–3081. [PubMed] [Google Scholar]
- 20.Forstner M, Kriechbaum M, Laggner M P, Wallimann T. J Mol Struct. 1996;383:217–227. [Google Scholar]
- 21.Zhou G, Parthasarathy G, Somasundaram T, Ables A, Roy L, Strong S J, Ellington W R, Chapman M S. Protein Sci. 1997;6:444–449. doi: 10.1002/pro.5560060222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strong S J, Ellington W R. Comp Biochem Physiol B. 1996;113:809–816. doi: 10.1016/0305-0491(95)02104-3. [DOI] [PubMed] [Google Scholar]
- 23.Navaza J, Saludjian P. Methods Enzymol. 1997;277:581–594. doi: 10.1016/S0076-6879(97)76079-8. [DOI] [PubMed] [Google Scholar]
- 24.Brünger A T. Methods Enzymol. 1997;276:558–580. doi: 10.1016/S0076-6879(97)76078-6. [DOI] [PubMed] [Google Scholar]
- 25.Cowtan K D, Main P. Acta Crystallogr D. 1993;49:148–157. doi: 10.1107/S0907444992007698. [DOI] [PubMed] [Google Scholar]
- 26.Kleywegt G J, Jones T A. Methods Enzymol. 1997;277:208–230. doi: 10.1016/s0076-6879(97)77013-7. [DOI] [PubMed] [Google Scholar]
- 27.Brünger A T. Methods Enzymol. 1997;277:366–396. doi: 10.1016/s0076-6879(97)77021-6. [DOI] [PubMed] [Google Scholar]
- 28.Chapman M S. Acta Crystallogr A. 1995;51:69–80. [Google Scholar]
- 29.Chapman M S, Blanc E. Acta Crystallogr D. 1997;53:203–206. doi: 10.1107/S0907444996012280. [DOI] [PubMed] [Google Scholar]
- 30.Brünger A T. x-plor Version 3.1: A System for X-ray Crystallography and NMR. New Haven, CT: Yale Univ. Press; 1992. [Google Scholar]
- 31.Bhat T N. J Appl Crystallogr. 1988;21:279–281. [Google Scholar]
- 32.Buchwald S L, Friedman J M, Knowles J R. J Am Chem Soc. 1984;106:4911–4916. [Google Scholar]
- 33.Skoog M T, Jencks W P. J Am Chem Soc. 1984;106:7597–7606. [Google Scholar]
- 34.Murali N, Joaori G K, Rao B D N. Biochemistry. 1994;33:14227–14236. doi: 10.1021/bi00251a035. [DOI] [PubMed] [Google Scholar]
- 35.Leyh T S, Goodhart P J, Nguyen A C, Kenyon G L, Reed G H. Biochemistry. 1985;24:308–316. doi: 10.1021/bi00323a011. [DOI] [PubMed] [Google Scholar]
- 36.Cohn M, Shih N, Nick J. J Biol Chem. 1982;257:7646–7649. [PubMed] [Google Scholar]
- 37.Almassy R J, Janson C A, Hamlin R, Xuong N-H, Eisenberg D S. Nature (London) 1986;323:304–309. doi: 10.1038/323304a0. [DOI] [PubMed] [Google Scholar]
- 38.Kabsch W, Fritz-Wolf K. Curr Opin Struct Biol. 1997;7:811–818. doi: 10.1016/s0959-440x(97)80151-0. [DOI] [PubMed] [Google Scholar]
- 39.Bennett W S, Jr, Steitz T A. Proc Natl Acad Sci USA. 1978;75:4848–4852. doi: 10.1073/pnas.75.10.4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dumas C, Camonis J. J Biol Chem. 1993;268:21599–21606. [PubMed] [Google Scholar]
- 41.Strong S J, Ellington W R. Biochim Biophys Acta. 1995;1246:197–200. doi: 10.1016/0167-4838(94)00218-6. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki T, Kawasaki Y, Furukohri T, Ellington W R. Biochim Biophys Acta. 1997;1343:152–159. doi: 10.1016/s0167-4838(97)00128-3. [DOI] [PubMed] [Google Scholar]
- 43.Fersht A R, Blow D M, Fastrez J. Biochemistry. 1973;12:2035–2041. doi: 10.1021/bi00735a002. [DOI] [PubMed] [Google Scholar]
- 44.Robertus J D, Kraut J, Alden R A, Birktoft J J. Biochemistry. 1972;11:4293–4303. doi: 10.1021/bi00773a016. [DOI] [PubMed] [Google Scholar]
- 45.Rosevear P R, Desmeules P, Kenyon G L, Mildvan A S. Biochemistry. 1981;20:6155–6164. doi: 10.1021/bi00524a038. [DOI] [PubMed] [Google Scholar]
- 46.Chen L H, Borders C L, Jr, Vásquez J R, Kenyon G L. Biochemistry. 1996;35:7895–7902. doi: 10.1021/bi952798i. [DOI] [PubMed] [Google Scholar]
- 47.Dunn B M, Di Bello C, Anfinsen C B. J Biol Chem. 1973;248:4769–4774. [PubMed] [Google Scholar]
- 48.Cotton F A, Hazen E E, Legg M J. Proc Natl Acad Sci USA. 1979;76:2551–2555. doi: 10.1073/pnas.76.6.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kyte J. Mechanism in Protein Chemistry. New York: Garland; 1995. [Google Scholar]
- 50.Virden R, Watts D C. Biochem J. 1966;99:162–172. doi: 10.1042/bj0990162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Watts D C, Rabin B R. Biochem J. 1962;85:507–516. doi: 10.1042/bj0850507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu H, Yao Q-Z, Tsou C-L. Biochim Biophys Acta. 1989;997:78–82. doi: 10.1016/0167-4838(89)90137-4. [DOI] [PubMed] [Google Scholar]
- 53.Lin L, Perrymann M B, Friedman D, Roberts R, Ma T S. Biochim Biophys Acta. 1994;1206:97–104. doi: 10.1016/0167-4838(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 54.Furter R, Furter-Graves E M, Wallimann T. Biochemistry. 1993;32:7022–7029. doi: 10.1021/bi00078a030. [DOI] [PubMed] [Google Scholar]