

Abstract
In K/BxN mice, arthritis is induced by autoantibodies against glucose-6-phosphate-isomerase (GPI). To investigate B cell tolerance to GPI in nonautoimmune mice, we increased the GPI-reactive B cell frequency using a low affinity anti-GPI H chain transgene. Surprisingly, anti-GPI B cells were not tolerant to this ubiquitously expressed and circulating autoantigen. Instead, they were found in two functionally distinct compartments: an activated population in the splenic marginal zone (MZ) and an antigenically ignorant one in the recirculating follicular/lymph node (LN) pool. This difference in activation was due to increased autoantigen availability in the MZ. Importantly, the LN anti-GPI B cells remained functionally competent and could be induced to secrete autoantibodies in response to cognate T cell help in vitro and in vivo. Therefore, our study of low affinity autoreactive B cells reveals two distinct but potentially concurrent mechanisms for their activation, of which one is T cell dependent and the other is T cell independent.
In the B cell compartment, tolerance to many self proteins is actively maintained by either purging self-reactive B cells from the repertoire through clonal deletion and receptor editing or by functionally silencing them through the induction of anergy (1–5). However, these processes are clearly incomplete, as B cell–driven autoimmune responses still occur. A prime example is the K/BxN mouse model of rheumatoid arthritis in which mice spontaneously develop a T and B cell–dependent inflammatory joint disease by 4–5 wk of age (6). Autoantibodies are key mediators in arthritis induction and by themselves can transfer disease to most naive strains of mice (7). In this model, the autoantigen for both the KRN transgenic (tg) T cells and non-tg B cells has been identified as the glycolytic enzyme glucose-6-phosphate-isomerase (GPI) (8). It remains unclear how autoreactive B and T cells escape tolerance induction to this ubiquitously expressed autoantigen and initiate the pathogenic autoantibody response.
It is difficult to study the mechanisms of tolerance induction for autoreactive B cells in unmanipulated animals because of their low precursor frequency. The use of Ig tg models has been invaluable to increase the frequency of autoreactive B cells, allowing their fate to be tracked in both healthy and autoimmune mice. Initial studies used high affinity somatically mutated Ig transgenes directed toward model or disease-associated antigens (Ags) and defined several fates for autoreactive B cells in nonautoimmune mice, including clonal deletion, anergy, and receptor editing (1–5, 9–13). These models were extremely useful and identified the mechanisms used to induce tolerance in B cells with high affinity Igs. However, most B cells undergoing tolerance induction in the BM are not somatically mutated, and those that are able to escape tolerance induction most likely express Igs with low affinity for autoantigens (14–16). Studies using 10–1,000-fold lower affinity Ig B cell receptor transgenes demonstrate either only partial tolerance or antigenic ignorance in the autoreactive B cell compartment (17–19). Collectively, these studies demonstrate that B cells with low affinity Igs may be able to escape tolerance mechanisms normally induced in B cells with high affinity Igs. It is from these low affinity B cell populations that the autoimmune response is likely to initiate.
Mature peripheral B cells can bedivided into two main subsets based on surface phenotype: recirculating follicular B cells (B220+CD1dlow- CD21/35intCD23int) and marginal zone (MZ) B cells (B220+CD1dhighCD21/35highCD23low) (20). These populations are also physically segregated into different compartments. Follicular B cells are found in both the spleen and LN, whereas MZ B cells are localized exclusively to the spleen where they are separated from the B cell follicle by the marginal sinus (21). MZ B cells are a rare nonrecirculating B cell population that has been shown to be easily activated by low levels of antigen (Ag), to be potent APCs for naive T cells, and has been implicated in autoreactive B cell responses (22, 23). Engagement by self-Ag drives the selection of B cells into the MZ compartment; however, it remains controversial whether this is dependent on high or low affinity interactions (21, 22, 24, 25).
To understand how GPI-reactive B cells escape tolerance induction to the ubiquitously expressed autoantigen GPI, it was important at the outset to establish how their tolerance was induced/maintained in nonarthritic mice. To increase the frequency of GPI-reactive B cells and follow their fate in nonautoimmune mice, we generated low affinity anti-GPI Ig tg mice, termed VH147 tg mice. VH147 tg mice are H chain alone tg mice in which GPI-reactive B cells are identifiable by their ability to bind GPI in a flow cytometry–based assay, allowing for the simultaneous detection of multiple VH147 H chain/endogenous L chain pairs. Therefore, these mice offer the unique opportunity to follow the fate of an oligoclonal repertoire of low affinity GPI-reactive B cells in the context of a diverse nonautoimmune B cell repertoire.
In this study, we demonstrate that low affinity GPI-binding B cells populated both the spleen and the LN of nonautoimmune mice but were dramatically enriched in the MZ of the spleen. Surprisingly, although the anti-GPI B cells showed evidence of autoantigen encounter beginning in the BM, no imprint of tolerance was evident and the GPI-reactive B cells remained functionally competent to proliferate and secrete Ig. Importantly, only the anti-GPI B cells found in the expanded MZ population up-regulated activation markers and spontaneously differentiated into autoantibody-secreting cells in vivo. Although the follicular/LN anti-GPI B cells were ignorant to self-Ag in nonautoimmune mice, they were recruited into the autoimmune response by GPI-specific T cell help in vivo. Collectively, these data demonstrate that low affinity self-reactive B cells can participate in autoantibody responses in both T cell–dependent and –independent mechanisms.
RESULTS
Generation and characterization of KRN5-147 hybridoma and VH147 tg mice
We wished to examine how B cell tolerance to GPI was induced in nonautoimmune mice in a physiologically relevant context. Ig tg mice using only a H chain transgene have been used in past studies to increase the frequency of Ag-specific B cells while retaining a diverse B cell repertoire (12, 13, 22). In this study, we generated H chain alone tg mice with the aim of allowing the tg H chain to pair with endogenous L chains in the mouse to form both GPI- and non-GPI–binding B cells, thus maintaining a polyclonal B cell repertoire. To create a low affinity anti-GPI Ig tg mouse, candidate GPI-binding Igs were identified in hybridoma panels generated from K/BxN mice at the initiation of arthritis. The goal was to isolate spontaneously arising anti-GPI Igs that were present in the early phase of arthritis, thus hypothesizing that these would be from B cells responsible for the induction of the autoimmune response.
One hybridoma, KRN5-147 (anti-GPI IgG1), was selected based on its ability to bind an epitope on GPI that was prevalent in arthritic K/BxN serum (unpublished data). In addition, it had a relatively low affinity for GPI (KD∼2 × 10−6) by surface plasmon resonance and had an unmutated H chain V region (unpublished data). These features closely mimic the Igs of B cells that undergo selection in the BM and possibly escape tolerance induction to join the peripheral B cell pool (14). To generate the tg construct, the rearranged genomic VDJ fragment of the 147 hybridoma was amplified by PCR and cloned into an IgM H chain expression vector (1). Using flow cytometry and allotype-specific antibodies, the tg Ig protein was expressed and allelically excluded rearrangement of endogenous Ig (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20060701/DC1).
The majority of anti-GPI B cells display an activated MZ phenotype in the spleen
VH147 tg mice were examined for the presence of anti-GPI B cells and were found to contain a readily detectable population of GPI binders (Fig. 1 A). As one control, hen egg lysozyme Ig tg mice on an IgM-deficient background (HELμMT) were used as a population of non–GPI-binding B cells to define the GPI+ gate. VH147 tg mice contained populations of both anti-GPI and non-GPI B cells in the BM, spleen, LN, and peritoneal cavity with anti-GPI frequencies ranging from 6–13% of the total B cell population (Fig. 1 A and not depicted). This demonstrated that the VH147 transgene significantly increased the frequency of GPI-binding B cells compared with transgene negative (tg neg) mice (0.6–0.8% GPI+), allowing their fate to be followed in nonarthritic C57BL/6 mice. Analysis of total B cell numbers in VH147 tg mice revealed a decrease in the absolute number of B cells compared with tg neg littermates (6.8 ± 2.4 × 106 vs. 18.4 ± 6.2 × 106; n = 12). This reduction in B cell numbers was not caused by a transgene integration artifact because a second independently derived founder line showed a similar decrease in B cell numbers (7.2 ± 4.7 × 106; n = 5). Decreased B cell numbers have also been noted in other H chain Ig tg mice and could be caused by deletion of some GPI-binding B cells in the BM and/or inefficient pairing of the 147 H chain with many endogenous L chains (9, 10).
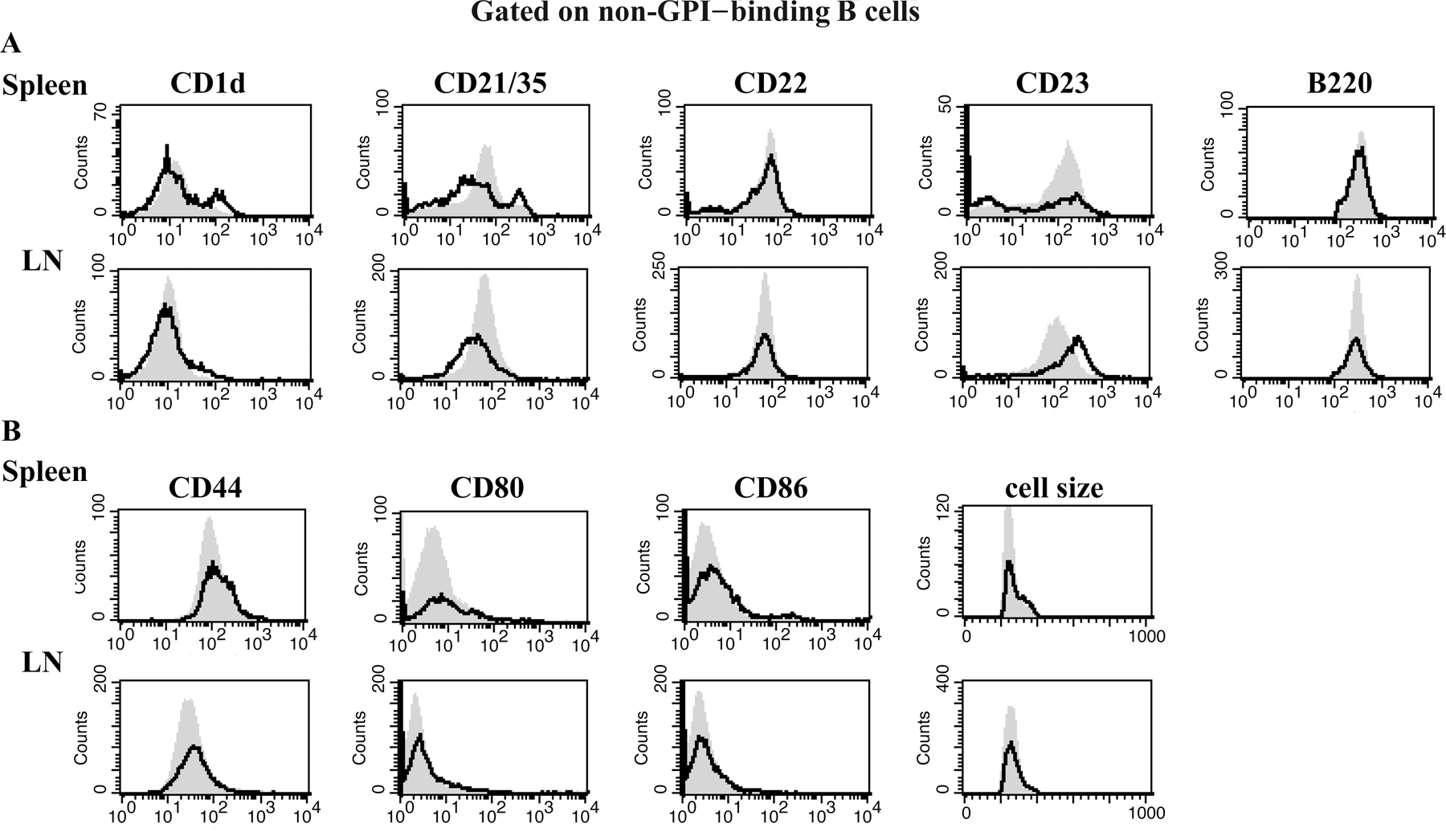
Figure 1.

Anti-GPI B cells in the spleen exhibit an activated MZ phenotype, whereas those in the LN are naive mature follicular cells. Spleen or LN cells from VH147 tg or tg neg B6.K mice were stained with anti-B220, GPI-his, and antibodies against phenotypic markers or activation markers and analyzed by flow cytometry. (A) The percentages of GPI-binding B cells out of total B220+ cells in the spleen are given for the boxed regions. The GPI+ gate was drawn based on the HELμMT, which contains a monoclonal population of non-GPI-binding B cells. (B) Histograms show surface marker expression for splenic or LN B220+GPI+ cells from VH147 tg mice (open histograms) overlaid tg neg B cells (shaded histograms). (C) Histograms show activation marker expression for B220+GPI+ MZ (CD21/35high), FO (CD21/35int), or LN VH147 tg (open histograms) overlaid unfractionated tg neg B cells (shaded histograms). (D) Histograms are gated on B220+CD21/35high MZ B cells from the VH147 tg GPI+ (open histograms) overlaid GPI− (shaded histograms) populations. n = 5 mice of each genotype.
To characterize the anti-GPI B cell population in VH147 tg mice, spleen and LN cells were stained with anti-B220, GPI-his protein, and a panel of cell surface markers used to define B cell subsets. Anti-GPI B cells in both the spleen and LN were found in the mature follicular compartment, B220highCD22highCD1dlowCD21/35intCD23int (Fig. 1 B). Intriguingly, the majority (59.5 ± 8.3%) of GPI-binding B cells in the VH147 tg spleen were not mature follicular B cells, but instead had the phenotype of MZ B cells, CD1dhighCD21/35highCD23low (Fig. 1 B). Consistent with an increase in MZ B cells, immunofluorescent staining of spleen sections revealed an expanded MZ in VH147 tg mice; however, we were unable to specifically identify the GPI-binding B cells in the tissue sections because of their low affinity for GPI (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20060701/DC1; and not depicted). Preferential MZ B cell development has been reported previously in mice with reduced B cell numbers (20, 26, 27). Because VH147 tg mice had a 3–4-fold reduction in B cell numbers, it was important to distinguish whether the enrichment of GPI-binding B cells in the MZ was simply caused by lymphopenia. To address this, mixed BM chimeras (VH147 tg + tg neg) with a more complete B cell compartment (13.9 ± 4.8 × 106 B cells; n = 4) were generated. The GPI-binding B cells were still enriched in the MZ compartment of the mixed BM chimeric mice (29.4 ± 2.3%) compared with tg neg chimeric mice (4.1 ± 0.9%), but not to the same extent as in nonchimeric VH147 tg mice. FACS analysis of non-GPI binders in both the VH147 spleen and LN, with the exception of an elevated level of CD23, closely matched tg neg B cells, demonstrating that the skewed MZ phenotype was specific to the GPI-binding B cells (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20060701/DC1). This indicated that reduced B cell numbers may explain part of the MZ enrichment, but as suggested by others, self-reactivity is likely to have also played a role in their phenotypic skewing (21, 22, 24, 25).
To determine if the GPI-binding B cells in VH147 tg mice showed evidence of having encountered GPI Ag, the VH147 CD21/35high MZ and CD21/35int follicular splenic subsets, as well as LN cells, were stained for activation markers and compared with naive tg neg B cells (Fig. 1 C). Interestingly, only the anti-GPI B cells in the MZ and not the follicle or LN of VH147 tg mice were activated, expressing elevated levels of CD44, CD69, CD80, and CD86 and an increase in cell size (Fig. 1 C). However, they had not formed germinal centers, as there was no evidence of an expanded germinal center B cell population in VH147 tg mice by immunofluorescence microscopy or flow cytometry (not depicted). MZ B cells have been reported to be a partially activated population of B cells, possibly as a result of positive selection by autoantigen (22). In VH147 tg mice, the GPI-binding MZ B cells expressed even higher levels of the activation markers CD44, CD69, CD80, and CD86 (1.5–3-fold elevated) compared with non-GPI binding VH147 or tg neg MZ B cells (Fig. 1 D and not depicted), indicating that their activation status was not simply a result of their MZ phenotype. Control non-GPI binders in both the spleen and LN did not express elevated levels of any of the activation markers tested (Fig. S3). Together, these data demonstrate that the majority of anti-GPI B cells in the spleen were activated MZ B cells, whereas a minority in the spleen and those in the LN remained naive mature follicular B cells.
MZ B cells exhibit the highest GPI autoantigen exposure in vivo
The differences in activation phenotype suggested that the GPI-reactive B cells in the splenic MZ, follicle, and LN have had different exposure to autoantigen. To address which population exhibited the highest exposure to GPI and where GPI-reactive B cells first encountered their autoantigen, bound GPI was quantitated on anti-GPI B cells isolated from the BM, peripheral blood, LN, and spleen, separating MZ and follicular B cells, using flow cytometry (Fig. 2, A and B). Not all B cells in VH147 tg mice are GPI-reactive; therefore, for this quantitation, we divided the percentage of cells bound to endogenous GPI by the percentage of total possible GPI binders. Endogenous GPI was bound to a percentage of anti-GPI B cells in all organs tested. This indicated that anti-GPI B cells were first exposed to their autoantigen in the BM and continued to encounter it in the peripheral blood, spleen, and LN. Consistent with their activated phenotype, the highest percentage of GPI-bound B cells was found in the MZ, where 43.4 ± 4.8% of the total possible GPI-binding B cells were bound to GPI (Fig. 2 B). Because MZ B cells have high levels of the complement receptors (Crs) CD21/35 on their surface, the GPI could be bound directly to the Ig receptor as well as indirectly in the form of immune complexes (20). To test whether the increased endogenous GPI detected on the MZ B cells was caused by their elevated level of CD21/35, we bred the VH147 transgene onto a Cr-deficient (Cr2−/−) background (28). In the absence of Crs, the MZ pool still contained the highest percentage of B cells bound to endogenous GPI (Fig. 2 C). The percentage of GPI-bound B cells was actually higher in the MZ of Cr2−/− mice (70.1 ± 6.1%), perhaps because of the lack of other Cr-expressing cells in the MZ binding to GPI/anti-GPI immune complexes, leaving them free to bind the anti-GPI B cells via their Ig receptors.
Figure 2.

MZ B cells exhibit the highest exposure to GPI autoantigen in vivo. (A) Cells from VH147 tg mice or HELμMT were stained with a pool of anti-GPI antibodies to determine the percentage of cells bound to endogenous GPI (top) or exogenous GPI followed by the pool of anti-GPI mAbs to determine the total percentage of possible GPI-binders (bottom). The plots are gated on CD21/35+ cells for the HELμMT spleen, the VH147 blood and LN, IgM+ cells for the BM, CD21/35int cells for the follicle, and CD21/35high cells for the MZ. The percentages are given for the boxed regions. The positive gate is drawn based on the control HELμMT spleen sample. (B) The percentage of anti-GPI B cells bound to endogenous GPI was determined by dividing the percentage of cells staining with anti-GPI antibodies alone by the total percentage of possible GPI binders. Each symbol represents an individual mouse for a total of n = 5 mice for each organ. The mean percentage of GPI-binding B cells is represented as a histogram. There is a significantly higher percentage of B cells bound to GPI in the MZ compared with the other organs (P < 0.0004) and a lower percentage bound in the BM (P < 0.025). The difference between the BM and peripheral blood is not statistically significant (P = 0.0687). BL, peripheral blood; FO, follicular. (C) The percentage of anti-GPI B cells bound to endogenous GPI in VH147 tg Cr2−/− mice. There is a significantly higher percentage of B cells bound to GPI in the MZ (P = 0.001; n = 3 mice).
The VH147 transgene is a H chain alone transgene. Therefore, it was possible that the activation differences we observed were the result of differential endogenous L chain usage by the anti-GPI B cells rather than increased Ag availability. To determine the L chains that generate GPI-binding Igs when paired with the VH147 H chain, L chains were cloned from anti-GPI B cells sorted from the spleen or LN. The majority of GPI binders in both the spleen (52%) and LN (84%) used the Vκ14-111 L chain (Fig. 3 A), the L chain used by the original KRN5-147 hybridoma. Sequences using other Vκ genes were also isolated, indicating that the VH147 H chain could pair with several different L chains to generate an anti-GPI specificity (Fig. 3 A). Overall, the spleen and LN GPI-binding populations used a similar set of endogenous L chains; however, two Vκ sequences (Vκ9-124 and Vκ4-52) were isolated only from the spleen and one Vκ sequence (Vκ17-121) only from the LN GPI-binders. To determine if there was any difference in the relative affinity of these VH147/Vκ pairs compared with those found in both the spleen and LN populations, we isolated hybridomas using these L chains from unmanipulated VH147 tg mice. There was no clear distinction in the relative GPI affinity between the L chains found exclusively in the spleen and those found in both the spleen and LN (Fig. 3 B). Therefore, the spleen and LN GPI-binding B cells used similar endogenous L chain repertoires with the same relative affinity for GPI.
Figure 3.

Spleen and LN B cells from VH147 tg mice use similar endogenous L chains. (A) L chain cDNA was amplified and cloned from sorted VH147 tg spleen or LN anti-GPI B cells as described in Materials and methods. L chain assignment was determined by using the Ig BLAST and IMGT/V programs. The percentage of sequences using the given L chains is depicted from a total of n = 25 sequences each from the spleen and LN. (B) Relative affinity ELISA of VH147 tg B cells paired with endogenous L chains. Purified Ig from VH147 tg hybridomas was plated on serially diluted GPI-his-biotin bound to neutravidin-coated wells. Igs with a higher relative affinity for GPI will bind to lower concentrations of GPI than Igs with lower relative affinities. The graph represents the mean OD ± SEM from triplicate wells, normalized as the fraction of maximal binding. This experiment was repeated twice using at least 2 hybridomas for each L chain, yielding similar results.
To show definitively that MZ anti-GPI B cells were exposed to more autoantigen in vivo and exclude minor receptor affinity differences not measurable in an ELISA assay, we used a H+L chain transgene (VH147+Vκ14-111) on a Rag−/− background (mk147 Rag−/−). This L chain was derived from the original KRN5-147 hybridoma. As expected, the B cells in mk147 Rag−/− mice strongly bound GPI (Fig. 4 A). Importantly, both CD21/35highCD23low MZ and CD21/35intCD23int follicular B cell subsets were present in mk147 tg mice (Fig. 4 A). CD21/35highCD23low MZ B cells were increased in frequency in mk147 Rag−/− mice compared with tg neg mice (24.4 ± 1.9% vs. 5.7 ± 1.3%; P = 0.004; n = 6); however, there was not the dramatic skewing observed in VH147 tg mice (59.5 ± 8.3%). This indicated that, in addition to Vκ14-111, other L chains must have also contributed to the GPI-binding MZ repertoire in VH147 tg mice. The CD21/35highCD23low MZ B cells in mk147 Rag−/− mice expressed a slightly higher level of CD23 and lower level of CD1d than MZ B cells in tg neg mice, the reason for which was not clear (Fig. 4 A). However, when analyzed by histology, these CD21/35highCD23low B cells localized to the MZ, confirming that they were MZ B cells and not a recently defined CD21/35highCD23+ MZ precursor population that localized instead to the splenic follicle (unpublished data) (27, 29).
Figure 4.

Monoclonal anti-GPI tg mice have an expanded MZ population with increased in vivo autoantigen exposure. (A) Spleens from tg neg or mk147 Rag−/− mice were stained for GPI binders (left), MZ (CD21/35highCD23low) or follicular (CD21/35int/CD23low) populations gated on B220+ cells, or CD1d levels on CD21/35high (open histograms) overlaid CD21/35int (shaded histograms) B cells. The percentages of follicular and MZ cells are given for the indicated regions. (B) Cells from mk147 Rag−/− mice (open histograms) or HELμMT (shaded histograms) were stained for endogenously bound GPI as described in Fig. 2. The percentage and mean fluorescence intensity are given for the indicated regions. The mean percentage(C) and mean fluorescence intensity (D) of GPI-binding B cells ± SEM are represented as histograms. There is a significantly higher percentage of B cells bound to GPI (P < 0.03) as well as intensity of GPI staining (P < 0.05) in the MZ compared with the other organs. BL, peripheral blood; FO, follicular. n = 5 mice of each genotype. MFI, mean fluorescence intensity.
The monoclonal B cell repertoire in mk147 Rag−/− mice allowed us to clearly test whether MZ and follicular anti-GPI B cells with identical B cell receptors still exhibited differences in autoantigen exposure (Fig. 4, B–D). Similar to the findings in VH147 tg mice, mk147 Rag−/− B cells from all organs tested had bound to GPI with the highest percentage of GPI-bound cells again found in the splenic MZ (40.9 ± 6.2%; Fig. 4 C). Additionally, the MZ B cells demonstrated the highest intensity of staining per cell (Fig. 4, B and D). Therefore, the B cells in the MZ, as a population, have engaged more GPI than their counterparts in other compartments. These data directly demonstrate that MZ B cells had greater access than follicular B cells to circulating autoantigens, as had been suggested in previous studies (20, 21). Thus, the exclusive activation of MZ anti-GPI B cells was because a higher level of GPI autoantigen was available to them in the MZ.
Self-Ag encounter does not render anti-GPI B cells tolerant
Autoreactive B cells that have encountered their self-Ag are often rendered tolerant, such that they are refractory to subsequent stimulation in vivo or in vitro (30). To determine whether the anti-GPI B cells were tolerant, spleen and LN cells were tested for their ability to proliferate and secrete Ig to in vitro stimuli. Anti-GPI B cells from both the VH147 tg spleen and LN proliferated robustly to LPS, anti-CD40 + IL-4, and cognate T cell help (KRN T cells) (Fig. 5 A). On day 3, anti-GPI Ig was detectable in the supernatants from VH147 tg spleen, but not LN, cells cultured with LPS, anti-CD40 + IL-4, or KRN T cells (Fig. 5 B). By day 7, both VH147 tg spleen and LN cells had secreted anti-GPI Ig in response to LPS and KRN T cells (Fig. 5 C). The early antibody response by VH147 tg spleen cells together with their Ig secretion in response to anti-CD40 + IL-4 alone was consistent with their MZ phenotype (22). None of the supernatants bound to plates coated with an irrelevant Ag, nor was anti-GPI Ig detected in the supernatants from tg neg B cells, confirming the specificity of the assay (Fig. 5, B and C; and not depicted). The tg neg spleen and LN B cells did exhibit a lower but detectable level of proliferation in response to KRN T cell help (Fig. 5 A). This low level of proliferation was most likely caused by a bystander response to factors such as CD40L and IL-4. In summary, despite their encounter with self-Ag, neither anti-GPI B cells in the spleen nor LN were rendered unresponsive to in vitro stimulation.
Figure 5.

Anti-GPI B cells are not tolerant but proliferate and secrete Ig to in vitro stimuli and have a slow turnover rate in vivo. (A–C) Purified B cells from tg neg spleen, tg neg LN, VH147 spleen, or VH147 LN were cultured in vitro in media alone, 25 μg/ml LPS, 10 μg/ml anti-CD40 + 100 U/ml IL-4, or with purified KRN T cells. (A) Proliferation was measured on day 3 as a dilution of CFSE intensity, gating on B220+GPI+ VH147 tg or B220+ tg neg cells by flow cytometry. Data is represented as the percentage of cells that divided ± SD (n = 3). A greater percentage of anti-GPI B cells proliferated to KRN T cells in the spleen (P = 0.0001) and LN (P = 0.0121) compared with tg neg B cells. Anti-GPI Ig secretion in the culture supernatants was measured on days 3 (B) and 7 (C) by ELISA. Data is presented from a representative experiment of triplicate wells ± SD from a minimum of 3 experiments per culture condition. VH147 tg spleen cells produce a greater amount of anti-GPI Ig than tg neg spleen cells to LPS, anti–CD40+IL-4, and KRN T cells on days 3 and 7 (P = 0.0001). VH147 tg LN cells produce more anti-GPI Ig than tg neg LN cells to LPS and KRN T cells on day 7 (P = 0.0001). (D) In vivo turnover rate. Mice were continuously labeled with BrdU for 15 d and followed for BrdU incorporation on days 6, 10, and 15 by flow cytometry. Histograms represent the mean percentage of incorporated BrdU ± SD for VH147 GPI+ B cells or tg neg B cells. Four VH147 tg and three tg neg mice were analyzed at each time point. BM chimeras are shown for day 10 of BrdU labeling. n = 4 mixed chimeric, and n = 3 100% chimeric mice.
B cells that have not been tolerized should have a normal turnover rate in vivo (31). To estimate the turnover rate of anti-GPI B cells, VH147 tg or tg neg mice were continuously labeled with BrdU for 15 d and analyzed by flow cytometry for the amount of incorporated BrdU in their spleens and LNs (Fig. 5 D). No difference in the percentage of BrdU+ cells was detected between the VH147 tg anti-GPI B cells and tg neg B cells in either the spleen or the LN, nor was there a difference when the cells were divided into CD21/35high MZ versus CD21/35low follicular populations (Fig. 5 D and not depicted). Previous experiments have demonstrated that some autoreactive B cells have an accelerated turnover rate only when in the presence of nonautoreactive B cells (32). Although VH147 tg mice contained populations of GPI-binding and non–GPI-binding B cells, to ensure that their normal turnover rate was not caused by the lack of competition from non-tg B cells we measured the turnover rate of anti-GPI B cells in mixed BM chimeras (Fig. 5 D). The percentage of BrdU-labeled anti-GPI B cells was the same whether they were in the presence of tg neg B cells (mixed) or only VH147 B cells (100% VH147). Therefore, the anti-GPI B cells in VH147 tg mice had a normal in vivo turnover rate. This indicates that, although only the MZ B cells in the spleen had up-regulated activation markers in vivo, none of the anti-GPI B cell populations had been rendered tolerant and all remained long-lived functionally competent populations of autoreactive B cells.
Anti-GPI B cells in the MZ spontaneously secrete Ig
VH147 tg mice contain populations of anti-GPI B cells that are fully capable of producing Ig upon in vitro stimulation. To determine if these B cells were actively secreting antibody in vivo, we measured anti-GPI Ig in the serum by ELISA and anti-GPI antibody-secreting cells (ASCs) directly ex vivo by ELISPOT (Fig. 6). All VH147 tg mice expressed low but detectable titers of anti-GPI Ig in the serum, whereas serum anti-GPI Ig was undetectable in tg neg littermate control mice (Fig. 6 A). This is consistent with GPI engagement on anti-GPI B cells not only up-regulating activation markers, but also resulting in the production of autoantibody. To quantitate which cells were secreting the autoantibody, ASCs were measured directly ex vivo in an ELISPOT assay. Anti-GPI ASCs were detected from the spleen, but not the LN of VH147 mice (Fig. 6 B). The fact that only the MZ GPI binders up-regulated activation markers in response to their exposure to GPI in vivo suggested that they might be the population responsible for this autoantibody production. To directly test this, the ELISPOT assay was repeated using VH147 tg spleen samples that contained either purified follicular B cells or an enriched population of MZ B cells (Fig. 6 C). ASCs were present within the enriched MZ samples. However, ASCs were not found in the purified follicle samples that had been depleted of MZ B cells. Therefore, only the MZ anti-GPI B cells had differentiated into ASCs in vivo. Collectively with Fig. 2, these data show that anti-GPI B cells in the MZ had encountered more GPI than their counterparts in the follicle or LN and in response became activated to secrete autoantibody.
Figure 6.

VH147 tg anti-GPI B cells in the splenic MZ but not follicle or LN spontaneously secrete Ig. (A) Serum anti-GPI Ig titers from VH147 tg or tg neg mice were measured by ELISA. Each symbol represents an individual mouse for a total of 19 VH147 and 16 tg neg mice. The mean serum titer is represented by a bar. VH147 tg serum has higher titers of anti-GPI Ig (P = 0.0001). (B) Spontaneous ASCs from the spleen or LN were measured by ELISPOT. Each symbol represents an individual mouse for a total of n = 8 VH147 and n = 7 tg neg mice. The mean number of ASCs for each is represented by a histogram. More anti-GPI ASCs are detected in the VH147 spleen compared with VH147 LN (P = 0.0002) or tg neg spleen or LN (P = 0.0003). (C) Spontaneous ASCs from purified follicular or MZ VH147 tg splenic B cells were measured by ELISPOT. Each symbol represents a pool of three mice with the mean number of ASCs represented by a histogram. Anti-GPI ASCs are found only in the MZ population (P = 0.0286). This experiment was repeated three times for a total of four pools of mice.
Anti-GPI B cells in the LN require T cell help to be recruited into the autoimmune response
In addition to the GPI-specific B cells in the MZ, the recirculating follicular anti-GPI B cells are also a potentially dangerous population of nontolerized autoreactive B cells with full functional capacity. However, unlike the anti-GPI B cells in the MZ, follicular and LN anti-GPI B cells were not activated nor did they secrete autoantibody in nonautoimmune mice. This leaves open the question of whether these ignorant anti-GPI B cells can play a role in the autoimmune response in arthritic mice. To address whether recirculating follicular anti-GPI B cells could be recruited into the autoimmune response by autoreactive T cells in vivo, VH147 tg mice were given cognate T cell help by breeding them to KRN (anti-GPI TCR) tg mice on a (C57BL/6xNOD) F1 background. All of the VH147/KRN tg mice developed arthritis between 4–5 wk of age (Fig. 7 A). This was slightly delayed compared with their KRN littermates without the VH147 tg, most likely because of the IgM transgene skewing the B cells away from the pathogenic anti-GPI IgG1 Ig. Consistent with this, VH147 tg mice also developed slightly lower serum titers of anti-GPI IgG1 (non-tg Ig) and total anti-GPI Ig titers (Fig. 7 B and not depicted). Importantly, however, VH147/KRN tg mice did develop arthritis with the same severity as their VH147 negative littermates (Fig. 7 A), and their serum anti-GPI Ig titers increased as disease progressed, unlike the constant low level of anti-GPI Ig detected in nonautoimmune VH147 tg mice (Fig. 7 B). Therefore, these mice allowed us to determine whether the anti-GPI B cells in the recirculating follicular pool could be engaged in the autoimmune response.
Figure 7.

Anti-GPI LN cells are recruited into the autoimmune response by in vivo T cell help. VH147 tg mice were bred to KRN tg mice on a (C57BL/6.K x NOD)F1 background and followed from 3–10 wk for the development of arthritis (A) and serum anti-GPI autoantibodies (B). VH147/KRN, KRN, and control mice (VH147 and tg neg) are shown. Symbols represent the mean ± SEM. n = a minimum of 8 mice per genotype. (C) Spontaneous anti-GPI IgM (top) or total Ig (bottom) ASCs from the spleen or LN were measured by ELISPOT. Prearthritic mice were 3 wk old, arthritic mice were 5–8 wk old, and control nonarthritic mice were 6–8 wk old. Each symbol represents an individual mouse for a minimum of 5 prearthritic, 10 arthritic, and 5 nonarthritic control mice of each genotype.
To measure the anti-GPI B cell response in VH147/KRN tg mice, the number of anti-GPI ASCs was assayed in the spleens and LNs of prearthritic and arthritic mice (Fig. 7, C and D). To distinguish between tg and non-tg Ig, both IgM (tg only; Fig. 7 C) and total Ig (tg + endogenous Ig; Fig. 7 D) anti-GPI ASCs were counted. Pre-arthritic VH147/KRN tg, like VH147 tg alone, mice contained IgM anti-GPI ASCs in the spleen (35.9 ± 9.6 ASCs/105 spleen cells), but not LN. Importantly, in arthritic VH147/KRN tg mice, IgM anti-GPI ASCs were now present in large numbers in the LNs (242.1 ± 64 ASCs/105 LN cells). In contrast, IgM anti-GPI ASCs were low to undetectable in prearthritic or arthritic KRN mice without the VH147 transgene, confirming that the ASCs were VH147 tg in origin. When total anti-GPI ASCs were compared between VH147/KRN and KRN tg alone arthritic mice, a similar large number of ASCs was found in the LN (VH147/KRN, 465.5 ± 116.2 ASCs/105 LN cells; KRN, 512.3 ± 106 ASCs/105 LN cells). This indicated that the VH147 tg anti-GPI ASCs were representative of the anti-GPI ASCs found in non-Ig tg arthritic mice with respect to both timing and location (Fig. 7 D) (33). Collectively, these data demonstrate that although anti-GPI B cells in the recirculating B cell pool of VH147 tg mice were ignorant in nonautoimmune mice, they could be recruited into an autoimmune response by T cell help in vivo.
DISCUSSION
In this study we described a new Ig tg model, VH147, and used it to follow the fate of an oligoclonal population of low affinity GPI-reactive B cells. These mice revealed two functionally distinct populations of anti-GPI B cells, one enriched in the splenic MZ and the other in the recirculating follicular pool. We demonstrated that, despite evidence of Ag encounter beginning in the BM and continued Ag binding in the periphery, none of the GPI-reactive B cells were rendered tolerant in nonautoimmune mice. However, only the anti-GPI B cells in the MZ were activated and spontaneously secreted autoantibody in vivo. We also showed, using a H+L tg mouse, that the selective activation of these MZ GPI-binders was caused by increased exposure to GPI autoantigen in the MZ and not to differences in the relative affinity of these B cells compared with those in the follicular pool. Finally, we demonstrated that the recirculating follicular anti-GPI B cells remained functionally competent and could be activated to secrete autoantibodies when given GPI-specific T cell help in vitro and in vivo. This study shows that low affinity anti-GPI B cells can contribute to autoantibody production in both T cell–dependent and –independent ways and suggests that they have the potential to induce an arthritogenic autoimmune response.
Our low affinity VH147 tg model allowed us to make the important and surprising finding that B cells were not tolerant to the ubiquitously expressed and circulating autoantigen GPI, despite evidence for binding to it in vivo. Stable and functional GPI exists as a homodimer (34) and therefore should be of sufficient valency to cross-link the BCR, as opposed to the monomeric Ags previously described to induce tolerance in B cells (35). However, the nontolerant B cells comprised two distinct populations. The GPI-specific B cells in the MZ were activated because of their higher exposure to the circulating autoantigen. In contrast, those in the follicle/LN were ignorant but could become activated when provided with T cell help. Ignorant self-reactive B cells with low affinity Ig receptor transgenes have been described previously (17, 19). In these models, it was proposed that the low affinity interaction was not strong enough to either activate or tolerize the B cells. This appears to be the case for the low level of Ag encountered by anti-GPI B cells in the recirculating follicular B cell pool. Strikingly, however, low affinity VH147 tg anti-GPI B cells in the MZ did show evidence of a productive Ag encounter. They had GPI bound to their Ig receptor and, in response, had both up-regulated activation markers and differentiated into ASCs. The concept of differential ignorance/tolerance to autoantigens has also been described before in the case of peritoneal B-1 B cells compared with their splenic B-2 counterparts and has been attributed to either increased susceptibility of B-2 B cells to tolerance induction (36) or lack of Ag exposure in the peritoneum (37, 38). What is unique about our system is that we demonstrated both ignorant and activated anti-GPI B cell populations, with compartmentalization into the MZ allowing these particular GPI-reactive B cells more access to their autoantigen. Because MZ B cells can be activated by lower levels of stimulation (22), the anti-GPI B cells in the MZ had a dual advantage over their counterparts in the follicle. They required less stimulation to become activated and, because of their anatomical location, encountered enough Ag to exceed that threshold.
Our findings differ considerably from a recently published high affinity anti-GPI Ig knock-in model in which the anti-GPI B cells were tolerized, either by developmental arrest or through receptor editing (39). These features of tolerance have been described previously and are well-characterized fates for high affinity autoreactive B cells (1–5, 9–13). A striking difference between the two models was that we found low affinity VH147 tg anti-GPI B cells were enriched in the splenic MZ, whereas they found no MZ enrichment of their anti-GPI B cells. This is consistent with the concept that there is an affinity threshold required for MZ B cell development (21, 22, 24, 25). Finally, we demonstrated that our low affinity GPI-reactive B cells remained functionally competent to proliferate and secrete autoantibody in response to in vitro and in vivo stimuli, whereas their high affinity anti-GPI B cells were poorly responsive in vitro (39). Together, this reinforces the importance of studying B cells with low affinity for self Ags.
The precise role of either the activated MZ or ignorant follicular/LN anti-GPI B cells in the pathogenic autoantibody response has not been established. Arthritis in K/BxN mice is dependent on high affinity IgG1 anti-GPI Igs (7). Not surprisingly then, the low affinity IgM autoantibodies secreted by the MZ B cells described here did not by themselves induce disease. In VH147 tg mice, the activated MZ and ignorant follicular/LN autoreactive B cells exist concurrently, raising the possibility that they could act coordinately to initiate the autoimmune response. Interestingly, MZ B cells have been shown to initiate immune responses by transporting IgM Ag-immune complexes into the follicle where they deposit them on follicular dendritic cells via receptors such as Cr1 and Cr2 (40). These experiments suggest that MZ B cells are in position to be the first responders to foreign Ags circulating in the blood and may be the catalyst for driving follicular B cell responses to these Ags. Likewise, MZ B cells would also be in close contact with autoantigens circulating in the blood and, if activated, could trigger autoimmune responses. Indeed, we demonstrated that more anti-GPI B cells in the MZ than in the recirculating follicular compartment have bound GPI. This raises the interesting possibility that these low affinity anti-GPI MZ B cells could augment the activation of pathogenic follicular B cells through the formation and transport of IgM/GPI immune complexes. Alternatively, it is also possible that the low affinity anti-GPI IgM could play an inhibitory role, acting instead to clear GPI from the circulation via a complement-mediated process (41, 42). Future experiments will be required to distinguish between these possibilities.
In K/BxN mice, the autoimmune response involves GPI-reactive KRN T cells as well as anti-GPI B cells (7). We demonstrated that the LN/follicular anti-GPI B cells were able to be stimulated to produce anti-GPI autoantibodies by KRN T cells both in vitro and in vivo. The potential for T cell help to rescue/activate autoreactive B cells has been controversial. Activation of autoreactive B cells has been demonstrated in autoimmune mice; however, because of the lack of reagents to identify Ag-specific T cells, the role of cognate T cell help could only be inferred (43–45). Other studies used surrogate forms of cognate T cell help and demonstrated that anergy in autoreactive B cells was able to be prevented or even reversed in some systems, whereas in others anergic B cells were not able to be rescued (46–49). These outcomes were attributed to differences in the valency or avidity of the self-Ag studied and/or the timing and quality of T cell help. Our model is unique in that we are able to provide our self-reactive B cells with help from T cells recognizing the same well-defined self-Ag, and all of this could be done during the development of autoimmunity. Therefore, we were able to show directly that GPI-specific B cells were stimulated to produce autoantibody by GPI-specific T cell help during the development of arthritis in vivo. Although the anti-GPI B cells in our study had a low affinity for self-Ag and therefore did not directly induce disease, we contend that these nontolerant B cells are representative of autoreactive B cells that escape tolerance induction and, under the appropriate conditions, could initiate autoimmune responses. There is precedence in another autoantigen response suggesting that high affinity autoreactive B cells can be derived from low affinity precursors (50). We propose then, that low affinity anti-GPI B cells can contribute to the autoantibody response by two distinct mechanisms depending on their compartmentalization. MZ anti-GPI B cells are spontaneously activated, whereas those in the follicle/LN require T cell help.
MATERIALS AND METHODS
Mice.
KRN TCR tg C57BL/6 and K/BxN mice have been described previously (6). KRN and Rag-1−/− mice were crossed onto an H-2k background (B6.K; The Jackson Laboratory). HELμMT were obtained from Herbert W. Virgin (Washington University, St. Louis, MO) (51). Cr2-deficient mice, originally obtained from Hector Molina (Washington University, St. Louis, MO) (28), were bred to VH147 tg mice and maintained in our colony. To obtain arthritic mice, VH147 tg B6.K mice were crossed to KRN B6.K mice and then subsequently crossed to NOD to generate VH147/KRN BxN mice. Tg mice were generated in the Washington University Department of Pathology transgenic core facility. Age-matched tg neg B6.K or tg neg (B6.K × NOD) F1 mice were used as controls in all experiments. All mice were bred and housed under specific pathogen-free conditions in the animal facility at the Washington University Medical Center. Experiments were performed in accordance with the National Institutes of Health and the Association for Assessment and Accreditation of Laboratory Animal Care guidelines.
Hybridoma generation.
Unmanipulated spleen and LN cells from individual arthritic K/BxN mice (6 wk old) or pooled VH147 B6.K mice (5 mice per fusion aged 6–8 wk each) were fused to the Ig− myeloma, SP2/0, as described previously (52). Cells were immediately plated at limiting dilution, and wells bearing single colonies were expanded for analysis. Hybridomas were screened by ELISA for binding to GPI-his protein and for Ig isotype. The KRN5-147 hybridoma bound specifically to GPI and not to the other cytoplasmic proteins, enolase, GAPDH, or hexokinase (unpublished data).
Flow cytometry.
BM, spleen, peritoneal cavity, LN, and peripheral blood cells (106 each) were stained with commercially available antibodies (see Supplemental materials and methods, available at http://www.jem.org/cgi/content/full/jem.20060701/DC1). mGPI-his protein was conjugated to biotin using N-hydroxysuccinimidobiotin (Sigma-Aldrich) in our laboratory. For GPI receptor occupancy studies, cells were stained with anti-B220–APC, anti-CD21/35–PE (BD Biosciences), and an FITC-conjugated pool of anti-GPI mAbs generated in our laboratory (KRN5-19, -28, -46, and -50.1) that bind to epitopes distinct from the KRN5-147 Ig, either with or without prior incubation with GPI-his.
All samples were analyzed on a flow cytometer (FACSCalibur; BD Biosciences) with CellQuest software (BD Biosciences). In some experiments the vital dye 7-amino-actinomysin D (Sigma-Aldrich) was added to exclude dead cells. Gating on live lymphocytes was based on forward and side scatter, and 50,000–100,000 events were collected for each sample.
BrdU labeling.
Mice were labeled with 0.8 mg/ml BrdU (Sigma-Aldrich) in their drinking water for 15 d. The BrdU water was changed daily. Cells with an increased turnover rate as well as cells that were actively proliferating in the spleen and LN were labeled. To distinguish between proliferation and turnover, mice were pulsed with 1 mg/ml BrdU i.p. for 2 h to assess the in vivo proliferation status of the B cells. Only a small percentage of the GPI-binding B cells in the spleen and LN incorporated BrdU during the 2-h pulse (0.8% VH147 vs. 1.2% tg neg; n = 3), indicating that they are not actively proliferating in vivo. Therefore, any BrdU+ cells detected during the 15-d labeling were caused by replacement from labeled BM precursors.
BrdU staining was performed as previously described (12). In brief, spleen and BM cells from mice were isolated and surface stained to identify GPI-binding MZ or follicular B cells as described in Flow cytometry. The cells were then fixed and permeabilized with 1% paraformaldehyde containing 0.1% Tween 20. The DNA was denatured using 10 μM HCl and 100 U/ml DNaseI. The incorporated BrdU was then detected using an anti-BrdU–FITC antibody (PRB-1) from eBioscience.
Anti-GPI ELISA.
Serum samples, plated at an initial dilution of 1:50 and diluted serially 1:2 (nonarthritic mice) or 1:10 (arthritic mice), or undiluted culture supernatants were plated in plates (Immulon II; Fisher Scientific) coated with 5 μg/ml GPI-his. Donkey anti–mouse total Ig-horseradish peroxidase (HRP; Jackson ImmunoResearch Laboratories) and goat anti–mouse IgM, IgG1, IgG2a, IgG2b, and IgG3-HRP (Southern Biotechnology Associates, Inc.) were used as secondary antibodies. Serum antibody was detected using a substrate (ABTS; Roche Applied Science). The serum titer was defined as the reciprocal of the last dilution which gave an OD >3× background.
Anti-GPI affinity ELISA.
Igs were tested for their relative affinity for GPI by titration of binding in an ELISA assay as described previously (53). Hybridoma IgM from tissue culture supernatant were purified using ImmunoPure immobilized mannan-binding protein according to manufacturer's instructions, except that the supernatant was concentrated before dialysis into binding buffer (Pierce Chemical Co.). 200 μg/ml purified hybridoma Igs were then tested in triplicate for binding to GPI-his, plated at an initial dilution of 10 μg/ml, and diluted serially 1:2 on Immulon II plates coated with 10 μg/ml neutravidin. Goat anti–mouse IgM-HRP was used as a secondary antibody and detection was done as described for the anti-GPI ELISA.
Anti-GPI ELISPOT assay.
Spleen or LN cells were plated at 4 × 105 cells/well and diluted serially 1:4 in mixed cellulose ester membrane plates (Multiscreen HA; Millipore) coated with 5 μg/ml GPI-his. The cells were incubated on the Ag-coated plates at 37°C for 4 h. The Ig secreted by the plated cells was detected by alkaline phosphatase–conjugated goat anti–mouse total Ig or IgM secondary antibodies (Southern Biotechnology Associates) and visualized using NBT/BCIP substrate (Sigma-Aldrich).
Purification of MZ versus follicular B cells.
Pooled spleens from VH147 tg mice (n = 3 per sample) were enriched for follicular or MZ B cells using a separation system (MACS; Miltenyi Biotech) in a two-step process. First, the cells were stained with anti-CD21–FITC and anti-CD23–PE, followed by anti-PE paramagnetic beads (Miltenyi Biotech). The CD23+ (PE+) follicular B cells were collected as the positive fraction. The CD23− fraction was then bound to anti-FITC paramagnetic beads (Miltenyi Biotech). The CD23−CD21+ MZ B cells were then collected as the positive fraction. Cell purity for follicular B cells was >90% with less than 1% contaminating MZ B cells. MZ B cells were enriched to 65% with 1–2% contaminating follicular B cells.
In vitro stimulation.
B cells from tg neg or VH147 tg B6.K or (B6.K × NOD)F1 spleen and LN and KRN T cells were purified using the MACS separation system with paramagnetic anti-CD43 or anti-CD4 beads, respectively. Cell purity was >95%. Cells were labeled with 5 μM CFSE (Invitrogen). 106 B cells were cultured in a 1-ml total volume in either media alone (Iscoves-DME, 10% FCS, 5 × 10−5 M 2-mercaptoethanol), 50 μg/ml LPS (Escherichia coli strain 0111:B4; Sigma-Aldrich), 10 μg/ml anti-CD40 (clone 1C10; R&D Systems) + 100 U/ml IL-4 (grown as supernatant from IL-4–transfected P815 cells), or 0.5 × 106 KRN T cells. To determine if the VH147 tg anti-GPI B cells could respond to cognate T cell help, VH147 tg (B6.K × NOD) F1 mice were generated to obtain B cells that express the I-Ag7 MHC class II molecule for presentation of GPI peptide to KRN tg T cells (6). VH147 tg anti-GPI B cells have an identical surface phenotype in B6.K and (B6.K × NOD) F1 mice and therefore were used interchangeably (unpublished data). Proliferation was measured on day 3 by flow cytometry as a decrease in CFSE intensity relative to unstimulated cells. Anti-GPI Ig secretion in tissue culture supernatants was determined on days 3 and 7 by anti-GPI ELISA.
Mixed BM chimeras.
Lethally irradiated (1,000 Rad) B6.K mice were reconstituted with 20 × 106 total BM cells containing either 95% VH147 + 5% tg neg, 100% VH147 tg, or 100% tg neg cells. The resulting mixed chimeras contained ∼20% VH147 tg B cells. Chimeras were allowed to reconstitute for 6 wk and were then labeled with BrdU for 10 d before analysis.
Cloning and sequencing of VH147 L chains.
GPI-binding B cells from VH147 tg spleen and LN, pooled from 15 mice each, were sorted using a high speed flow cytometer (MoFlo; DakoCytomation). RNA from the sorted cell populations was isolated using the RNeasy kit (QIAGEN) and stored at −70°C before analysis. cDNAs were generated using a Cκ primer. L chain sequences were then amplified using the Cκ 3′ primer with the degenerate 5′ primer, L3′ (52). The resulting PCR products were then cloned using a cloning kit for sequencing (TOPO TA; Invitrogen) and sequenced using a terminator mix (Big Dye; Applied Biosystems) and the M3 reverse primer from the vector sequence. L chain designation was determined by comparison to published sequences in the GenBank database using the Ig BLAST (available at http://www.ncbi.nlm.nih.gov/projects/igblast) and IMGT/V-Quest (http://imgt.cines.fr) programs. Alignment of L chain sequences using the same Vκ gene showed several nucleotide differences within the V region (most yielding silent mutations) and usage of different Jκ genes, demonstrating that they were derived from independent clones (unpublished data).
Arthritis incidence.
The two rear ankles were measured starting at the age of 3 wk. Measurement of ankle thickness was made above the footpad, axially across the ankle joint using a metric pocket thickness gauge (Fowler; Ralmikes Tool-A-Rama). Ankle thickness was rounded off to the nearest 0.05 mm.
Statistical analysis.
Statistical significance was determined using an unpaired Student's t test or the Mann-Whitney nonparametric test.
Online supplemental material.
Fig. S1 shows the allelic exclusion of endogenous Ig by the VH147 transgene. Fig. S2 shows immunofluorescence staining demonstrating expanded MZs in VH147 tg mice. Fig. S3 shows phenotypic marker staining on control non-GPI binding B cells in VH147 tg mice. Supplemental materials and methods include the cloning of the H and L chain genes for generating the tg Ig, generation of the recombinant GPI-his protein, antibodies used for flow cytometry, and the immunofluorescence staining protocol. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20060701/DC1.
Supplemental Material
Acknowledgments
We thank D. Nemazee for the IgM expression vector; M. White for pronuclei injections; W. Eades and J. Hughes for cell sorting; D. Kreamalmeyer for managing the mouse colony; S. Horvath for technical assistance; D. Donermeyer for Biacore analysis; S. Nayak and K. Matsui for help with the tg constructs; J. Smith for administrative assistance; and F. Martin, S. Kang, L. Norian, J. Erikson, C. Lindsley, and W. Swat for discussions and critical readings of the manuscript.
This work was supported by National Institutes of Health grant AI031238. The Siteman Cancer Center is supported in part by National Cancer Institute Cancer Center Support grant P30 CA91842.
The authors have no conflicting financial interests.
Abbreviations used: Ag, antigen; ASC, antibody-secreting cell; Cr, complement receptor; HELμMT, hen egg lysozyme Ig tg mice on an IgM-deficient background; GPI, glucose- 6-phosphate-isomerase; MZ, marginal zone; tg, transgenic; tg neg, transgene negative.
References
- 1.Nemazee, D.A., and K. Burki. 1989. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature. 337:562–566. [DOI] [PubMed] [Google Scholar]
- 2.Hartley, S.B., J. Crosbie, R. Brink, A.B. Kantor, A. Basten, and C.C. Goodnow. 1991. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature. 353:765–769. [DOI] [PubMed] [Google Scholar]
- 3.Goodnow, C.C., J. Crosbie, S. Adelstein, T.B. Lavoie, S.J. Smith-Gill, R.A. Brink, H. Pritchard-Briscoe, J.S. Wotherspoon, R.H. Loblay, K. Raphael, et al. 1988. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 334:676–682. [DOI] [PubMed] [Google Scholar]
- 4.Tiegs, S.L., D.M. Russell, and D. Nemazee. 1993. Receptor editing in self-reactive bone marrow B cells. J. Exp. Med. 177:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gay, D., T.L. Saunders, S. Camper, and M. Weigert. 1993. Receptor editing: an approach by autoreactive B cells to escape tolerance. J. Exp. Med. 177:999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kouskoff, V., A.-S. Korganow, V. Duchatelle, C. Degott, C. Benoist, and D. Mathis. 1996. Organ-specific disease provoked by systemic autoimmunity. Cell. 87:811–822. [DOI] [PubMed] [Google Scholar]
- 7.Korganow, A.-S., H. Ji, S. Mangialaio, V. Duchatelle, R. Pelanda, T. Martin, C. Degott, H. Kikutani, K. Rajewsky, J.-L. Pasquali, et al. 1999. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 10:451–461. [DOI] [PubMed] [Google Scholar]
- 8.Matsumoto, I., A. Staub, C. Benoist, and D. Mathis. 1999. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 286:1732–1735. [DOI] [PubMed] [Google Scholar]
- 9.Erikson, J., M.Z. Radic, S.A. Camper, R.R. Hardy, C. Carmack, and M. Weigert. 1991. Expression of anti-DNA immunoglobulin transgenes in non-autoimmune mice. Nature. 349:331–334. [DOI] [PubMed] [Google Scholar]
- 10.Chen, C., Z. Nagy, M.Z. Radic, R.R. Hardy, D. Huszar, S.A. Camper, and M. Weigert. 1995. The site and stage of anti-DNA B-cell deletion. Nature. 373:252–255. [DOI] [PubMed] [Google Scholar]
- 11.Wang, H., and M.J. Shlomchik. 1997. High affinity rheumatoid factor transgenic B cells are eliminated in normal mice. J. Immunol. 159:1125–1134. [PubMed] [Google Scholar]
- 12.Mandik-Nayak, L., A. Bui, H. Noorchashm, A. Eaton, and J. Erikson. 1997. Regulation of anti–double-stranded DNA B cells in nonautoimmune mice: localization to the T/B interface of the splenic follicle. J. Exp. Med. 186:1257–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santulli-Marotto, S., M.W. Retter, R. Gee, M.J. Mamula, and S.H. Clarke. 1998. Autoreactive B cell regulation: peripheral induction of developmental arrest by lupus-associated autoantigens. Immunity. 8:209–219. [DOI] [PubMed] [Google Scholar]
- 14.Manser, T., and M.L. Gefter. 1986. The molecular evolution of the immune response: idiotope-specific suppression indicates that B cells express germ-line-encoded V genes prior to antigenic stimulation. Eur. J. Immunol. 16:1439–1444. [DOI] [PubMed] [Google Scholar]
- 15.Coutinho, A., M.D. Kazatchkine, and S. Avrameas. 1995. Natural autoantibodies. Curr. Opin. Immunol. 7:812–818. [DOI] [PubMed] [Google Scholar]
- 16.Koenig-Marrony, S., P. Soulas, S. Julien, A.M. Knapp, J.C. Garaud, T. Martin, and J.L. Pasquali. 2001. Natural autoreactive B cells in transgenic mice reproduce an apparent paradox to the clonal tolerance theory. J. Immunol. 166:1463–1470. [DOI] [PubMed] [Google Scholar]
- 17.Hannum, L.G., D. Ni, A.M. Haberman, M.G. Weigert, and M.J. Shlomchik. 1996. A disease-related rheumatoid factor autoantibody is not tolerized in a normal mouse: implications for the origins of autoantibodies in autoimmune disease. J. Exp. Med. 184:1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borrero, M., and S.H. Clarke. 2002. Low-affinity anti-Smith antigen B cells are regulated by anergy as opposed to developmental arrest or differentiation to B-1. J. Immunol. 168:13–21. [DOI] [PubMed] [Google Scholar]
- 19.Rojas, M., C. Hulbert, and J.W. Thomas. 2001. Anergy and not clonal ignorance determines the fate of B cells that recognize a physiological autoantigen. J. Immunol. 166:3194–3200. [DOI] [PubMed] [Google Scholar]
- 20.Martin, F., and J.F. Kearney. 2002. Marginal-zone B cells. Nat. Rev. Immunol. 2:323–335. [DOI] [PubMed] [Google Scholar]
- 21.Pillai, S., A. Cariappa, and S.T. Moran. 2005. Marginal zone B cells. Annu. Rev. Immunol. 23:161–196. [DOI] [PubMed] [Google Scholar]
- 22.Oliver, A.M., F. Martin, and J.F. Kearney. 1999. IgMhighCD21high lymphocytes enriched in the splenic marginal zone generate effector cells more rapidly than the bulk of follicular B cells. J. Immunol. 162:7198–7207. [PubMed] [Google Scholar]
- 23.Attanavanich, K., and J.F. Kearney. 2004. Marginal zone, but not follicular B cells, are potent activators of naive CD4 T cells. J. Immunol. 172:803–811. [DOI] [PubMed] [Google Scholar]
- 24.Kanayama, N., M. Cascalho, and H. Ohmori. 2005. Analysis of marginal zone B cell development in the mouse with limited B cell diversity: role of the antigen receptor signals in the recruitment of B cells to the marginal zone. J. Immunol. 174:1438–1445. [DOI] [PubMed] [Google Scholar]
- 25.Wen, L., J. Brill-Dashoff, S.A. Shinton, M. Asano, R.R. Hardy, and K. Hayakawa. 2005. Evidence of marginal-zone B cell-positive selection in spleen. Immunity. 23:297–308. [DOI] [PubMed] [Google Scholar]
- 26.Hao, Z., and K. Rajewsky. 2001. Homeostasis of peripheral B cells in the absence of B cell influx from the bone marrow. J. Exp. Med. 194:1151–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srivastava, B., W.J. Quinn III, K. Hazard, J. Erikson, and D. Allman. 2005. Characterization of marginal zone B cell precursors. J. Exp. Med. 202:1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molina, H., V.M. Holers, B. Li, Y. Fung, S. Mariathasan, J. Goellner, J. Strauss-Schoenberger, R.W. Karr, and D.D. Chaplin. 1996. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc. Natl. Acad. Sci. USA. 93:3357–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loder, F., B. Mutschler, R.J. Ray, C.J. Paige, P. Sideras, R. Torres, M.C. Lamers, and R. Carsetti. 1999. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J. Exp. Med. 190:75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goodnow, C.C., J. Crosbie, H. Jorgensen, R.A. Brink, and A. Basten. 1989. Induction of self-tolerance in mature peripheral B lymphocytes. Nature. 342:385–391. [DOI] [PubMed] [Google Scholar]
- 31.Fulcher, D.A., and A. Basten. 1994. Reduced life span of anergic self- reactive B cells in a double-transgenic model. J. Exp. Med. 179:125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cyster, J.G., and C.C. Goodnow. 1995. Antigen-induced exclusion from follicles and anergy are separate and complementary processes that influence peripheral B cell fate. Immunity. 3:691–701. [DOI] [PubMed] [Google Scholar]
- 33.Mandik-Nayak, L., B.T. Wipke, F.F. Shih, E.R. Unanue, and P.M. Allen. 2002. Despite ubiquitous autoantigen expression, arthritogenic autoantibody response initiates in the local lymph node. Proc. Natl. Acad. Sci. USA. 99:14368–14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Read, J., J. Pearce, X. Li, H. Muirhead, J. Chirgwin, and C. Davies. 2001. The crystal structure of human phosphoglucose isomerase at 1.6 A resolution: implications for catalytic mechanism, cytokine activity, and haemolytic anaemia. J. Mol. Biol. 309:447–463. [DOI] [PubMed] [Google Scholar]
- 35.Eynon, E.E., and D.C. Parker. 1992. Small B cells as antigen-presenting cells in the induction of tolerance to soluble protein antigens. J. Exp. Med. 175:131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ait-Azzouzene, D., L. Verkoczy, B. Duong, P. Skog, A.L. Gavin, and D. Nemazee. 2006. Split tolerance in peripheral B cell subsets in mice expressing a low level of Igkappa-reactive ligand. J. Immunol. 176:939–948. [DOI] [PubMed] [Google Scholar]
- 37.Qian, Y., C. Santiago, M. Borrero, T.F. Tedder, and S.H. Clarke. 2001. Lupus-specific antiribonucleoprotein B cell tolerance in nonautoimmune mice is maintained by differentiation to B-1 and governed by B cell receptor signaling thresholds. J. Immunol. 166:2412–2419. [DOI] [PubMed] [Google Scholar]
- 38.Murakami, M., and T. Honjo. 1996. Anti-red blood cell autoantibody transgenic mice: murine model of autoimmune hemolytic anemia. Semin. Immunol. 8:3–9. [DOI] [PubMed] [Google Scholar]
- 39.Huang, H., J.F. Kearney, M.J. Grusby, C. Benoist, and D. Mathis. 2006. Induction of tolerance in arthritogenic B cells with receptors of differing affinity for self-antigen. Proc. Natl. Acad. Sci. USA. 103:3734–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferguson, A.R., M.E. Youd, and R.B. Corley. 2004. Marginal zone B cells transport and deposit IgM-containing immune complexes onto follicular dendritic cells. Int. Immunol. 16:1411–1422. [DOI] [PubMed] [Google Scholar]
- 41.Walport, M.J., and P.J. Lachmann. 1988. Erythrocyte complement receptor type 1, immune complexes, and the rheumatic diseases. Arthritis Rheum. 31:153–158. [DOI] [PubMed] [Google Scholar]
- 42.Liszewski, M.K., and J.P. Atkinson. 1991. The role of complement in autoimmunity. Immunol. Ser. 54:13–37. [PubMed] [Google Scholar]
- 43.Wang, H., and M.J. Shlomchik. 1999. Autoantigen-specific B cell activation in Fas-deficient rheumatoid factor immunoglobulin transgenic mice. J. Exp. Med. 190:639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mandik-Nayak, L., S. Seo, C. Sokol, K.M. Potts, A. Bui, and J. Erikson. 1999. MRL-lpr/lpr mice exhibit a defect in maintaining developmental arrest and follicular exclusion of anti–double-stranded DNA B cells. J. Exp. Med. 189:1799–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santulli-Marotto, S., Y. Qian, S. Ferguson, and S.H. Clarke. 2001. Anti-Sm B cell differentiation in Ig transgenic MRL/Mp-lpr/lpr mice: altered differentiation and an accelerated response. J. Immunol. 166:5292–5299. [DOI] [PubMed] [Google Scholar]
- 46.Rathmell, J.C., M.P. Cooke, W.Y. Ho, J. Grein, S.E. Townsend, M.M. Davis, and C.C. Goodnow. 1995. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature. 376:181–184. [DOI] [PubMed] [Google Scholar]
- 47.Fulcher, D.A., D.A. Lyons, S.L. Korn, M.C. Cook, C. Koleda, C. Parish, B. Fazekas de St.Groth, and A. Basten. 1996. The fate of self-reactive B cells depends primarily on the degree of antigen receptor engagement and availability of T cell help. J. Exp. Med. 183:2313–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cook, M.C., A. Basten, and B. Fazekas de St. Groth. 1998. Rescue of self-reactive B cells by provision of T cell help in vivo. Eur. J. Immunol. 28:2549–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seo, S.J., M.L. Fields, J.L. Buckler, A.J. Reed, L. Mandik-Nayak, S.A. Nish, R.J. Noelle, L.A. Turka, F.D. Finkelman, A.J. Caton, and J. Erikson. 2002. The impact of T helper and T regulatory cells on the regulation of anti-double-stranded DNA B cells. Immunity. 16:535–546. [DOI] [PubMed] [Google Scholar]
- 50.Li, H., Y. Jiang, H. Cao, M. Radic, E.L. Prak, and M. Weigert. 2003. Regulation of anti-phosphatidylserine antibodies. Immunity. 18:185–192. [DOI] [PubMed] [Google Scholar]
- 51.McClellan, J.S., S.A. Tibbetts, S. Gangappa, K.A. Brett, and H.W. Virgin IV. 2004. Critical role of CD4 T cells in an antibody- independent mechanism of vaccination against gammaherpesvirus latency. J. Virol. 78:6836–6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roark, J.H., C.L. Kuntz, K.A. Nguyen, A.J. Caton, and J. Erikson. 1995. Breakdown of B cell tolerance in a mouse model of systemic lupus erythematosus. J. Exp. Med. 181:1157–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.William, J., C. Euler, and M.J. Shlomchik. 2005. Short-lived plasmablasts dominate the early spontaneous rheumatoid factor response: differentiation pathways, hypermutating cell types, and affinity maturation outside the germinal center. J. Immunol. 174:6879–6887. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}