

Abstract
The deep sea hydrothermal tube worm Riftia pachyptila possesses a multihemoglobin system with three different extracellular hemoglobins (Hbs; V1, V2, and C1): two dissolved in the vascular blood, V1 and V2, and one in the coelomic fluid, C1. V1 consists of four heme-containing chains and four linker chains. The globin chains making up V2 and C1 are, with one exception, common to V1. Remarkably these Hbs are able to bind oxygen and sulfide simultaneously and reversibly at two different sites. Two of the globin chains found in these three Riftia Hbs possess one free Cys residue and for at least one of the globins, the b-Cys75 is conserved among vestimentifera (Lamellibrachia sp.) and pogonophora (Oligobrachia mashikoi). By selectively blocking the free Cys with N-ethylmaleimide and using electrospray ionization mass spectrometry experiments, we show that these Cys residues are involved in sulfide binding by Riftia Hbs. Moreover, we also demonstrate that the larger V1 Hb can form persulfide groups on its linker chains, a mechanism that can account for the higher sulfide-binding potential of this Hb.
Riftia pachyptila (1), the giant tube worm, is one of the most studied species living at deep sea hydrothermal vents (2). The nutritional needs of this mouthless and gutless organism are satisfied by chemolitoautotrophic sulfide-oxidizing bacteria located intracellularly in a specialized organ, the trophosome (3–5). Riftia supplies these symbionts with oxygen and sulfide via extracellular hemoglobins (Hbs), two dissolved in the vascular blood (V1 ≈ 3,500 kDa and V2 ≈ 400 kDa) and one in the coelomic fluid (C1 ≈ 400 kDa). Remarkably, Riftia Hbs simultaneously and reversibly bind oxygen and sulfide at two different sites (6, 7). Furthermore, sulfide binding does not affect the simultaneous binding of oxygen, appearing to occur at a site removed from the heme (7–9). These Hbs also protect animal tissues from sulfide toxicity by binding sulfide with a higher affinity than cytochrome c oxidase, the site responsible for toxic effects (10, 11).
Despite these singular properties, the quaternary structure of V1 Hb is similar to those of some annelid Hbs (i.e., hexagonal bilayer structure). In a previous paper (12), we proposed models for Riftia Hbs based on mass spectrometry measurements. In these models, V1 Hb has 180 polypeptide chains consisting of 144 globin chains made up of two monomers (b and c) and one dimer (D1 = d + e) and 36 linker chains of four different types, L1–L4 (12). The smaller Hbs, V2 and C1, have only 24 globin-like chains, either monomeric (a–c) or dimeric (D1 or D2 = e + f). The number of polypeptide chains for each hemoglobin was estimated from the measured masses of each hemoglobin and polypeptide chain and by assuming that the V1 molecule possesses a D6 point group of symmetry. The estimates of the number of globin chains for the molecules V1, V2, and C1 are 165 ± 36, 26 ± 6, and 23 ± 2, respectively, and 35 ± 5 for V1 linker chains (12). As is the case for other vestimentifera (Lamellibrachia sp.) (13) and pogonophora (Oligobrachia mashikoi) (14), there are free Cys residues on the globin chains b and e (12). The number of Cys, and whether they are in disulfide bridges or free, is especially important in Riftia because Cys residues have been proposed as the sulfide-binding sites in vestimentiferan (7, 13, 15) and pogonophoran (14) Hbs. However, this hypothesis has not been directly addressed previously.
Fourteen years after the discovery of the ability of Riftia Hbs to bind sulfide (6), we report here the identity of the sulfide-binding sites. The results presented were obtained from sulfide-binding experiments on purified Riftia Hbs after pretreatment of Hbs with a specific alkylant reagent, N-ethylmaleimide (NEM) or other treatments. In addition, we used electrospray ionization mass spectrometry (ESI-MS) to monitor the formation of an S-sulfohemoglobin component after exposure of V1 Hb to sulfide, testing an earlier suggestion (16). Furthermore, we have tested for disulfide-exchange processes as proposed for Riftia V1 Hb (7, 16). This latter mechanism was tested for by following changes in A335, indicative of the production of persulfide groups (i.e., R—SSH) resulting from the cleavage of disulfide bridges by sulfide, according to the reaction (17): R—SS—R + H2S ⇄ R—SSH + R—SH.
MATERIALS AND METHODS
Animal Collection.
R. pachyptila were collected by the French submersible Nautile during the HOT’96 expedition in the eastern Pacific at 13°N (12°46′N–103°56′W and 12°50′N–103°57′W) at an average depth of 2,600 m. Worms were pulled from the rocks by the remote control arm of the submersible and brought to the surface in a thermally insulated container. On board ship the worms were bled, and the samples were stored in liquid nitrogen (18).
Purification Technique.
Gel filtration was carried out with a 1 × 30 cm Superose 6-C (Pharmacia LKB Biotechnology) by using a low-pressure fast protein liquid chromatography system (Pharmacia). Riftia saline buffer (50 mM BTP/400 mM NaCl/3 mM KCl/32 mM MgSO4/11 mM CaCl2 at pH 7.0) (18, 19) was used as the eluent. Running conditions and sample treatments were similar to those used previously (18). After purification, the heme and protein concentrations of Riftia V1, V2, and C1 Hbs, as well as Lumbricus terrestris Hb, were measured (20, 21).
Sulfide-Binding Analysis.
The measurement of sulfide binding was carried out by equilibrium dialysis of purified Hb samples against Riftia saline at pH 7.5 and 5°C (6, 16). Total sulfide (ΣH2S, the sum of the different species) was analyzed by gas chromatography (6, 8). Similar experiments were performed on purified Riftia Hbs after modification of their free sulfydryl groups by incubation in 3 mM NEM, for 30 min at room temperature before dialysis, a procedure effective for the b-Cys93 residue of human Hb (22–24). Sulfide binding was standardized to heme for convenience, as sulfide is not bound to the heme (7).
Mass Spectrometry.
After V1 purification, one sample was equilibrated with 2.0 mM Na2S at 4°C for 24 h. This and a no-sulfide control sample were then dialyzed against deoxygenated distilled water at 4°C and lyophilized. One milligram of each of these samples was dissolved in distilled water (concentration, 10 μg/μl). Aliquots (10 μl) of these solutions were diluted with 20 μl of 1% formic acid, 20 μl of distilled water, and 20 μl of acetonitrile to make the working solutions for the analysis. ESI-MS analysis was performed on a VG Quattro II (Micromass, Altrincham, U.K.) scanning over the m/z range 700–2,500 at 15 sec per scan, and 24 scans were summed over approximately 6 min from three 10-μl aliquots of the working solutions to produce the final data. The flow rate into the electrospray source was 5 μl/min. The sampling cone voltage was ramped from 40 V at m/z 700 to 100 V at m/z 2,500 during each scan. The electrospray source temperature was set to 80°C. Mass scale calibration used the multiply-charged series from horse heart myoglobin (Mr = 16,951.5; Sigma product M-1882). Molecular masses are based on the following atomic weights of the elements: C, 12.011; H, 1.00794; N, 14.00674; O, 15.9994; S, 32.066 (25).
Spectrophotometry.
The formation of persulfide (—R—SSH) was monitored by following the A335 (17) on a Shimadzu UV 160 U spectrophotometer at room temperature (approximately 20°C). This temperature roughly corresponds to this species’ natural habitat temperature (2). Persulfide formation is normally described as occurring at alkaline pH (17) outside the physiological range of Riftia. To determine whether this reaction might take place at physiological pH values near 7.5 (8, 9), we tested for the appearance of persulfide groups in deoxygenated Riftia saline buffer, by incubating 5 mM cystamine, cystine, cysteamine, or cysteine solutions (all reagents from Sigma) with 2 mM Na2S. Riftia Hbs are quite stable and there was no evidence of denaturation of the Hb during the long experiments. The saline buffer contains Ca2+ and Mg2+ among other ions that maintain the quaternary structure of these hemoglobins (18, 26). The A335 was recorded until a maximum was reached. A similar experiment was performed on 3 mg of purified Riftia V1 Hb. An identical experiment on L. terrestris Hb served as a control because its quaternary structure is closely related to that of Riftia V1 Hb. Anaerobic conditions were maintained by closing each spectrophotometer cuvette with a rubber septum and flushing nitrogen gas through the space above the solution before each experiment.
Another way to confirm the relation between A335 and the formation of persulfides was to measure the decrease in A335 on addition of cyanide, represented by the reaction (27):
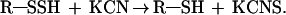 |
When the absorbance at 335 was almost maximal, we added 100 ml of 0.01 M KCN dissolved in 0.01 M NaOH to the 3 ml of Hb solution in the cuvette. Further confirmation was obtained by adding ferric nitrate reagent [2 g of Fe(NO3)⋅9H2O in 20 ml of 65% nitric acid] (28) after treatment with KCN. The presence of thiocyanate was indicated by the development of a red color.
We have also tested the effects of denaturation (with 3 M guanidine hydrochloride at pH 7.5) and pH (from 5.5 to 9.5) on the persulfide group formation for V1 Hb. The buffers were made in saline solution containing 50 mM citric acid phosphate (pH 5.5), 50 mM Tris⋅HCl (pH 6.5, 7.5, and 8.5), or 50 mM glycine (pH 9.5).
RESULTS AND DISCUSSION
Fig. 1 summarizes the sulfide-binding experiments performed on purified Riftia Hbs. Recently, we found that V1 Hb could bind 2.87 mol of ΣH2S per mol of heme (ratio ≈ 3.0). V2 and C1 possessed a lower capacity for sulfide and could bind only 0.63 and 0.49 mol of ΣH2S per mol of heme (ratio ≈ 0.50), respectively (16). In an earlier paper, we tested the effect of NEM on whole blood [i.e., a mixture of V1, V2, and C1 (16)] because NEM reacts specifically with free Cys residues as shown for the b-Cys93 residue of human Hb (22–24). We found that NEM inhibited approximately two-thirds of the sulfide-binding capacity of this mixed fluid, and we suggested that the one-third unaltered by NEM was caused by the V1 linker chains (16). In the present experiments, we used purified Riftia Hbs with the extracellular Hb from L. terrestris as a control, because all of its globin chains are devoid of free Cys residues but possess a quaternary structure similar to Riftia V1 Hb (i.e., the association of globin and linker chains, ref. 29). We confirmed the sulfide-binding capacity data described above for each of the three Hbs and also showed the inability of L. terrestris Hb to bind sulfide, apparently caused by the absence of free Cys. Indeed, after blocking the free thiol residues on Riftia Hbs, we observed a total inhibition of sulfide binding for both V2 and C1 Hbs, whereas for V1 Hb a 75% decrease was observed (i.e., 0.70 instead of 2.85 mol of HS− per mol of heme; range, 0.51–0.89 mol of HS− per mol of heme; n = 3). These experiments demonstrate that the free Cys present on the Riftia Hbs are involved in sulfide binding. This mechanism alone accounts for the sulfide-binding capacity of V2 and C1 Hbs but, for V1 Hb, a second mechanism must exist. Because Riftia Hbs are built by different associations of almost the same globin chains (12), this second mechanism probably involves the linker chains that are present in V1 but not in V2 and C1. The involvement of the disulfide bridges present on linker chains in sulfide binding was previously hypothesized for Lamellibrachia sp. Hb (13, 15) and for Riftia Hb (7). In addition, by using our published stoichiometry data and proposed Hb models (12, 16), we showed that the binding of 2.85 mol HS− per mol of heme for V1 could account for the saturation of 516 sulfide-binding sites or 516 Cys residues including 84 free. If we consider the lower value measured for V1 (i.e., 0.51 HS− per mol of heme), after NEM pretreatment (Fig. 1), this value saturates 92 sites, which is actually very close to the number of free Cys present on V1 globin chains (16).
Figure 1.

Modification of the free sulfydryl groups using NEM as a thiol blocking reagent. ○, Without NEM pretreatment; •, with NEM. Error bars correspond to the maximum and minimum binding recorded for each Hb (n = 3).
To demonstrate the formation of S-sulfohemoglobin
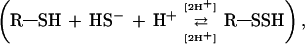 |
we used ESI-MS (Fig. 2). The protons above and below the arrows correspond either to an elimination or to an addition needed to satisfy the equilibrium. The mass measured for the “native” b chain, i.e., without chemical modification of the protein (16,135.29 ± 0.33 Da; Fig. 2A) agreed well with that previously measured by using the same protocol [16,133.5 ± 0.7 Da without the mass of the heme group, i.e., 616.5 Da) (12)]. After incubation with sulfide (Fig. 2B), two species were observed, the first one was the native b globin (16,136.01 ± 1.70 Da, expected mass as on Fig. 2A), whereas the second one had a mass of 16,166.50 ± 0.85 Da with a shift of Δm = +30.49 ± 2.55 mass units. This indicates the addition of a single HS− moiety to the b chain [Mr of HS− component = 33.07 Da (25)], confirming the presence of one reactive free Cys residue on this chain (12). Further, this adduct is not present when native V1 was carbamidomethylated, blocking all free Cys present, using the same conditions as for carboxymethylation (30) followed by incubation with sulfide (data not shown). This adduct appears very unstable because it can be observed for only a short time after preparation, perhaps because of the acidic conditions (i.e., around pH 4.0) for mass spectrometry analysis. Indeed, sulfide release can be observed in vitro from Riftia Hbs when the pH is decreased from 7.5 to 5.8 (8), in agreement with the reaction proposed above. During ESI-MS experiments, V1 linker chains were not observed in either set of experiments (i.e., with or without sulfide), probably because of either ionization difficulties or signal suppressions induced by the hydrophobicity of these chains. This observation has already been reported for studies under reducing conditions (12) but not in the “native” state used in this report.
Figure 2.

ESI-MS spectra of chain b from V1 Hb in “native” condition (A) and after incubation with sulfide (B).
To further investigate the existence of a second sulfide-binding mechanism on V1, we followed the A335 in a variety of experiments (17). Cystamine and cystine, which possess disulfide bridges, were able to form persulfide groups in contrast to cysteamine and cysteine (Fig. 3). These results show that disulfide exchange or S-thiolation processes can occur under the conditions used in this study, even if the pH is close to neutrality. Similar experiments were performed on purified Riftia Hbs (i.e., V1, V2, and C1) and on L. terrestris Hb (Fig. 4). The correlation of the A335 with persulfide groups produced by reacting Hbs with sulfide is based on the similarity of the absorption elicited by sulfide when added to cystamine and cystine (Fig. 3) (17, 31). Fig. 4 shows that only V1 is able to form persulfide groups at a significant level, in contrast to V2, C1, and L. terrestris Hbs, which cannot. This set of experiments also demonstrates that the A335 is related to persulfide group formation and there is no contribution of the V1 Hb itself at this wavelength because V2, C1, and Lumbricus Hb don’t show significant A335.
Figure 3.

Change of A335 for compounds with or without disulfide bridges. -SS-: •, cystamine; □, cystine. -SH: ▴, cysteamine; ▵, cysteine.
Figure 4.

Change of A335 for R. pachyptila Hbs. •, V1; ▴, V2; ▵, C1; ◊, L. terrestris Hbs.
Another confirmation of the presence of persulfide groups came from the use of potassium cyanide. As shown in Fig. 5, the addition of cyanide to V1 Hb, when the A335 was almost maximal, readily abolished the absorbance in accord with the formulation R—SSH + KCN → R—SH + KCNS (27), confirming the presence of persulfide groups. Surprisingly, the rate of formation of persulfide groups on Hb V1 appears to be very slow in comparison to cystamine and cystine. This could be caused by low reactivity of V1 disulfide groups or to a steric effect between the native Hb and the disulfide bonds, which restricts the access of sulfide to them. Our finding that 3 M guanidine hydrochloride greatly accelerates persulfide-group formation (Fig. 6) supports the latter hypothesis. This slow process probably reflects a compromise between sulfide binding and maintenance of the quaternary structure of this molecule, because linker chains play an important role in the specific architecture of V1, characterized by an hexagonal-bilayer structure (32). In addition, such lability of disulfide bridges could explain the relative instability of V1 Hb in solution (18, 33), compared with other annelid Hbs. Furthermore, it seems unlikely that all disulfide bridges involved in linker chains are occupied by sulfide in vivo, in contrast to in vitro experiments, because the molecule must maintain its quaternary structure.
Figure 5.

Decomposition by potassium cyanide of persulfide groups on R. pachyptila V1 Hb.
Figure 6.

Change of A335 for R. pachyptila V1 Hb. Curves recorded without denaturant (•) and with denaturant (▵).
To determine the number of linker disulfide bridges involved with sulfide on V1 Hb, we performed a similar experiment with the same conditions, using BSA, which possesses 17 disulfide bridges (34). BSA is also able to bind sulfide at approximately 19 mmol of ΣH2S per mg of protein (F.Z. and J.J.C., unpublished observation). To date, persulfide-group formations on BSA have been reported only under alkaline conditions (34). The maximum A335 recorded for BSA and V1 at the same protein concentrations and conditions was 0.093 and 1.212, respectively. If we consider that BSA absorbance accounts for 17 persulfide groups according to the reaction R—SS—R + H2S ⇄ R—SSH + R—SH (17), we can calculate that only 221 Cys residues were involved in disulfide-exchange processes in Riftia V1. However, stoichiometric data showed that 432 Cys residues were apparently involved in this mechanism for V1 Hb (16). These results suggest that both Cys making disulfide bridges are involved in the formation of persulfide groups (i.e., 34 Cys residues instead of 17 for BSA) and suggest the following reaction:
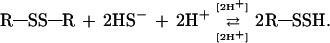 |
Hence, we estimate that 443 Cys residues are involved in this mechanism, a value very close to the number of Cys in linker chains (432) proposed in a V1 Hb model (12, 16).
Although alkaline pH has been described as a prerequisite for the interaction of sulfide with protein disulfide bonds (17, 31, 34), this condition is not required for Riftia V1 Hb or BSA. Thus proteins can form persulfide groups at conditions close to neutral pH. We also found that the formation of persulfide groups on V1 is strongly pH-dependent and that the maximum A335 occurred at pH 7.5 (Fig. 7), with absorbance falling off at pH values above and below this, in agreement with the pH dependency of sulfide binding by whole blood (8). These authors suggested that this result may reflect the availability of HS− in solution, because the dissociation constant for H2S is 6.6 (35).
Figure 7.

Effect of pH on persulfide formation (change in A335) in R. pachyptila V1 Hb in various buffers. ○, pH 5.5; •, pH 6.5; ▵, pH 7.5; ▴, pH 8.5; □, pH 9.5.
Using our data and those published previously (7, 12, 16), we can estimate the sulfide-binding capacity of Riftia Hbs and body fluids (Table 1). Hence, maximum sulfide concentrations near 10 mM and 1 mM could be bound by the Hbs contained in the vascular blood and the coelomic fluid, respectively. These values agree well with experimental values (9).
Table 1.
Estimation of maximum sulfide-binding capacity by R. pachyptila Hbs
| V1 | V2 | C1 | Ref. | |
|---|---|---|---|---|
| Proportion, (range) | 62.17 | 37.83 | 100 | 7 |
| (53.27–71.07) | (46.73–28.93) | |||
| Heme, mM (range) | 2.17 | 1.32 | 1.9 | 7 |
| (1.86–2.48) | (1.63–1.01) | |||
| Heme, no. | 144 | 24 | 24 | 12 |
| Free Cys, no. | 84 | 14 | 14 | 18 |
| Cys on linkers, no. | 432 | 0 | 0 | 18 and this study |
| No. Cys/no. heme | 3.5 | 0.58 | 0.58 | This study |
| Sulfide saturation, mM (range) | 7.59 | 0.76 | 1.10 | This study |
| (6.51–8.68) | (0.94–0.58) |
In conclusion, two sulfide-binding mechanisms have been demonstrated for Riftia Hbs (i.e., S-sulfohemoglobin and persulfide group formation). In addition, V1 Hb can make persulfide groups near neutral pH. Furthermore, because the optimum pH to load sulfide is 7.5, the unloading of sulfide to the trophosome could occur in response to a lower pH in the trophosome vasculature due to the elimination of protons as end products of sulfide oxidation by the symbionts. Recently, free Cys groups, which were previously unknown in annelid globins, have been found in the extracellular Hbs of Arenicola marina (36) and Alvinella pompejana (37), species that live in sulfide-rich environments. Hence, the presence of both free Cys residues and disulfide groups on Hb appears to be an important adaptation for animals living in sulfidic environments, enabling them to bind this substance either for detoxification and/or transport.
Acknowledgments
We thank the Conseil Régional d’Alsace, the Centre National de la Recherche Scientifique (Unité de Recherche Associée 31), ULP, and Bioavenir (Rhône Poulenc Santé) for financial support of the mass spectrometry study (to E.L. and A.V.D.). We thank the members of the Nautile group, the captains and crews of the N/O Nadir and R/V New Horizon, and the chief scientist of the HOT’96 research cruise, F. Gaill, who allowed us to conduct this work. We are very grateful to S. N. Vinogradov for supplying L. terrestris Hb. We also thank S. Goffredi and P. Girguis for their critical reviews of this article and the two anonymous referees who have helped us to highlight some unclear passages of the last version of this manuscript. This research was supported by research grants for the Ministère des Affaires Étrangères (“Lavoisier” program) and the Conseil Régional de Bretagne grants (to F.Z.), U.S. National Science Foundation (OCE 9632861) to J.J.C. and Centre National de la Recherche Scientifique (UPR 9042), Institut National des Sciences de l’Univers, IFREMER (URM 7), MENESR (ACC-SV3) (to F.H.L. and A.T.).
ABBREVIATIONS
- Hb
hemoglobin
- NEM
N-ethylmaleimide
- ESI-MS
electrospray ionization mass spectrometry
References
- 1.Jones M L. Science. 1981;213:333–336. doi: 10.1126/science.213.4505.333. [DOI] [PubMed] [Google Scholar]
- 2.Childress J J, Fisher C R. Oceanogr Mar Biol Annu Rev. 1992;30:337–410. [Google Scholar]
- 3.Cavanaugh C M, Gardiner S L, Jones M L, Jannasch H W, Waterbury J B. Science. 1981;213:340–342. doi: 10.1126/science.213.4505.340. [DOI] [PubMed] [Google Scholar]
- 4.Felbeck H, Childress J J, Somero G N. Nature (London) 1981;293:291–293. [Google Scholar]
- 5.Felbeck H. Science. 1981;213:336–338. doi: 10.1126/science.213.4505.336. [DOI] [PubMed] [Google Scholar]
- 6.Arp A J, Childress J J. Science. 1983;219:295–297. doi: 10.1126/science.219.4582.295. [DOI] [PubMed] [Google Scholar]
- 7.Arp A J, Childress J J, Vetter R D. J Exp Biol. 1987;128:139–158. [Google Scholar]
- 8.Childress J J, Arp A J, Fisher C R J. Mar Biol. 1984;83:109–124. [Google Scholar]
- 9.Childress J J, Fisher C, Favuzzi J, Kochevar R, Sanders N K, Alayse A. Biol Bull. 1991;180:135–153. doi: 10.2307/1542437. [DOI] [PubMed] [Google Scholar]
- 10.Powell M A, Somero G N. Science. 1983;219:297–299. doi: 10.1126/science.219.4582.297. [DOI] [PubMed] [Google Scholar]
- 11.Powell M A, Somero G N. Biol Bull. 1986;171:274–290. [Google Scholar]
- 12.Zal F, Lallier F H, Green B N, Vinogradov S N, Toulmond A. J Biol Chem. 1996;271:8875–8881. doi: 10.1074/jbc.271.15.8875. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki T, Takagi T, Ohta S. Biochem J. 1990;266:221–225. doi: 10.1042/bj2660221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuasa H J, Green B N, Takagi T. Biochim Biophys Acta. 1996;1296:235–244. doi: 10.1016/0167-4838(96)00081-7. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki T, Takagi T, Ohta S. J Biol Chem. 1990;265:1551–1555. [PubMed] [Google Scholar]
- 16.Zal F, Suzuki T, Kawasaki Y, Childress J J, Lallier F H, Toulmond A. Proteins Struct Funct Genet. 1997;29:562–574. [PubMed] [Google Scholar]
- 17.Rao G S, Gorin G. Org Chem. 1959;24:749–753. [Google Scholar]
- 18.Zal F, Lallier F H, Wall J S, Vinogradov S N, Toulmond A. J Biol Chem. 1996;271:8869–8874. doi: 10.1074/jbc.271.15.8869. [DOI] [PubMed] [Google Scholar]
- 19.Fisher C R, Childress J J, Sanders N K. Symbiosis. 1988;5:229–246. [Google Scholar]
- 20.Drabkins D L, Austin J H. J Biol Chem. 1935;112:51–65. [Google Scholar]
- 21.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 22.Winterbourn C C, Carrell R W. Biochem J. 1977;165:141–148. doi: 10.1042/bj1650141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Potuznik S, Gelvan D, Burda P, Saltman P. Biochim Biophys Acta. 1993;1164:289–298. doi: 10.1016/0167-4838(93)90261-o. [DOI] [PubMed] [Google Scholar]
- 24.Eguchi L A, Saltman P. Inorg Chem. 1987;26:3669–3672. [Google Scholar]
- 25.IUPAC. J Phys Chem Ref Data. 1993;22:1571–1584. [Google Scholar]
- 26.Lamy J N, Green B N, Toulmond A, Wall J S, Weber R E, Vinogradov S N. Chem Rev. 1996;96:3113–3124. doi: 10.1021/cr9600058. [DOI] [PubMed] [Google Scholar]
- 27.Cavallini D, De Marco C, Mondovi B, Mori R. Enzimologia. 1960;22:161. [PubMed] [Google Scholar]
- 28.Sörbo B. Biochim Biophys Acta. 1958;27:324–329. doi: 10.1016/0006-3002(58)90339-1. [DOI] [PubMed] [Google Scholar]
- 29.Martin P D, Kuchumov A R, Green B N, Oliver R W A, Braswell E H, Wall J S, Vinogradov S N. J Mol Biol. 1996;255:154–169. doi: 10.1006/jmbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 30.Crestfield A M, Moore S, Stein W H. J Biol Chem. 1963;239:622–627. [PubMed] [Google Scholar]
- 31.Villarejo M, Westley J. J Biol Chem. 1963;238:4016–4020. [PubMed] [Google Scholar]
- 32.Vinogradov S N, Kapp O H, Ohtsuki M. In: Electron Microscopy of Proteins. Harris J, editor. Vol. 3. New York: Academic; 1982. pp. 135–163. [Google Scholar]
- 33.Terwilliger R C, Terwilliger N B, Schabtach E. Comp Biochem Physiol. 1980;65B:531–535. [Google Scholar]
- 34.Cavallini D, Frederici G, Barboni E. Eur J Biochem. 1970;14:169–174. doi: 10.1111/j.1432-1033.1970.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 35.Millero F J, Plese T, Fernandez M. Limnol Oceanogr. 1988;33:269–274. [Google Scholar]
- 36.Zal F, Green B N, Lallier F H, Vinogradov S N, Toulmond A. Eur J Biochem. 1997;243:85–92. doi: 10.1111/j.1432-1033.1997.85_1a.x. [DOI] [PubMed] [Google Scholar]
- 37.Zal F, Green B N, Lallier F H, Toulmond A. Biochemistry. 1997;36:11777–11786. doi: 10.1021/bi9712899. [DOI] [PubMed] [Google Scholar]