

Abstract
The SN2 displacements of chloride ion from CH3Cl, C2H5Cl, and C2H4Cl2 by acetate and hydroxide ions have been investigated, using ab initio molecular orbital theory at the HF/6–31+G(d), MP2/6–31+G(d), and MP4/6–31+G(d) levels of theory. The central barriers (calculated from the initial ion–molecule complex) of the reactions, the differences of the overall reaction energies, and the geometries of the transition states are compared. Essential stereochemical changes before and after the displacement reactions are described for selected cases. The gas phase reactions of hydroxide with CH3Cl, C2H5Cl, and C2H4Cl2 have no overall barrier, but there is a small overall barrier for the reactions of acetate with CH3Cl, C2H5Cl, and C2H4Cl2. A self-consistent reaction field solvation model was used to examine the SN2 reactions between methyl chloride and hydroxide ion and between 1,2-dichloroethane and acetate in solution. As expected, the reactions in polar solvent have a large barrier. However, the transition state structures determined by ab initio calculations change only slightly in the presence of a highly polar solvent as compared with the gas phase. We also calibrated the PM3 method for future study of an enzymatic SN2 displacement of halogen.
Haloalkanes are widely used as herbicides, pesticides, refrigerants, or solvents. The haloalkane dehalogenase enzyme from Xanthobacter autothropicus GJ10 catalyzes the dehalogenation of n-RX compounds, where R is a C(1) to C(4) chain and X = Cl, Br, and I. The highest reactivity was observed with 1,2-dichloroethane (DCE) (1), and the dehalogenation proceeds via the formation of the corresponding alcohol (ROH). This observation has stimulated much interest because it offers an elegant way to decontaminate water and soil that have been spoiled by man-made haloalkanes (2, 3). The generally accepted mechanism for the haloalkane dehalogenase enzyme is a two-step process (4–7). First, the carboxylate of Asp-124 performs a nucleophilic attack on the halosubstituted carbon atom of the substrate, displacing the halogen ion and forming an alkyl-enzyme ester intermediate which is subsequently hydrolyzed by a water (4). In this hydrolysis step, the imidazole of His-289 serves as a general base catalyst (7). Ultimately, we wish to understand the catalytic mechanism of the Asp-124–CO2− SN2 displacement on alkyl chlorides in the enzymatic reaction. We begin with the present theoretical investigation of the reactions of CH3CO2− with a number of alkyl chlorides.
Experimental energetic data concerning halogenated compounds are scarce. Empirical rules have been devised to estimate the free energy of formation of chlorinated aliphatic compounds (8). Although this empirical approach could provide reasonable estimates for reaction energy, it is unable to provide useful information regarding the reaction barrier. Knowledge of the nonenzymatic dehalogenation reactions are critical for the understanding of the catalytic efficiency of haloalkane dehalogenase, and theoretical studies using molecular orbital theory have provided valuable information concerning the mechanism and reaction profiles of SN2 reactions (9–16). Thus, we used ab initio molecular orbital theory to investigate the nonenzymatic SN2 reactions of alkyl chlorides with acetate.
COMPUTATIONAL METHODS
All calculations were carried out with Gaussian 94 program (17) using the PM3 Hamiltonian (18) for the semiempirical calculations and the Hartree–Fock (HF) method using the 6–31+G(d) basis set for all ab initio geometry optimizations. Minima and transition states were characterized by vibrational frequency calculations. To account for electron correlation, additional energy calculations were performed using Møller–Plesset (MP) perturbation theory [at the MP2/6–31+G(d) and MP4/6–31+G(d) levels of theory]. The frozen core approximation was applied at both correlation levels. All ab initio energies mentioned in the paper refer to the MP2/6–31+G(d) level of theory, unless otherwise indicated. The energies are corrected by the HF zero point vibrational energies. Wiberg bond indices and natural atomic charges were calculated at the HF/6–31+G(d) level by using the natural bond orbital (NBO) analysis (19) option as incorporated in the Gaussian 94 program. For the calculations in solution, the self-consistent isodensity polarized continuum model (SCI-PCM) were used (20). The dielectric constant of water (78.3) was used.
RESULTS AND DISCUSSION
Chart 1 shows the reactions investigated in the present study.
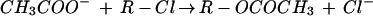 |
 |
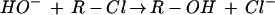 |
 |
 |
The optimized geometries for the ion/molecular complexes (IMCs) and transition states (TSs) are given in Fig. 1 for the acetate (Reactions 1–3). Fig. 2 shows the optimized geometries of the TSs of the hydroxide reactions (Reactions 4–6). The derived relative energies (in kcal/mol) are listed in Table 1.
Figure 1.

HF/6–31+G(d) calculated geometries for the IMCs and TSs for the acetate Reactions 1–3 (Chart 1). Selected C—O and C—Cl bond lengths are included. Bonds which are not completely formed are drawn in bold dashed lines.
Figure 2.

HF/6–31+G(d) calculated geometries for the TSs for the hydroxide Reactions 4–6 (Chart 1). Selected C—O and C—Cl bond lengths are included. Bonds that are not completely formed are drawn in bold dashed lines.
Table 1.
Calculated relative energies (kcal/mol) using the basis set 6-31+G(d)
| Reaction | ΔH1
|
ΔH‡
|
ΔH2
|
ΔHR
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HF | MP2/HF | MP4/HF | HF | MP2/HF | MP4/HF | HF | MP2/HF | MP4/HF | HF | MP2/HF | MP4/HF | |
| 1 | −11.5 | −11.5 | 11.8 | 15.5 | −25.3 | −14.5 | −25.0 | −10.6 | ||||
| 2 | −12.1 | −13.0 | 14.1 | 19.7 | −28.0 | −17.9 | −25.9 | −11.2 | ||||
| 3 | −19.5 | −20.8 | 19.7 | 23.2 | −27.9 | −14.6 | −27.7 | −12.1 | ||||
| 4 | −15.3 | −14.2 | −14.5 | 2.8 | 4.6 | 3.0 | −56.6 | −38.8 | −37.0 | −69.1 | −48.4 | −48.5 |
| 5 | −17.1 | −16.8 | −17.2 | 4.7 | 8.4 | 6.1 | −57.9 | −40.6 | −38.2 | −70.3 | −49.0 | −49.2 |
| 6 | −26.9 | −26.7 | −27.1 | 9.7 | 12.3 | 9.8 | −56.3 | −36.4 | −33.8 | −73.5 | −50.8 | −51.0 |
The energy designations are given in Fig. 3.
Reaction 4 has been investigated previously by Evanseck et al. (14) at a similar level. To simplify the comparison, we will use our own results for comparison with the data of Reactions 1–3, 5, and 6 (Chart 1).
Acetate as the Nucleophile.
As shown in Fig. 1, the C⋅⋅⋅O distance in the IMC1 of Reaction 1 is 298.7 pm (bond index, 0.01), whereas the corresponding calculated distances for IMC2 and IMC3 are about 320 pm (with a similar bond index). In the optimized structure of IMC3, the acetate ion is positioned symmetrically over the DCE (Fig. 1). Therefore, the distances of each of the acetate oxygen atoms to a carbon atom of DCE are virtually the same.
In comparison to the IMCs of the acetate Reactions 1–3, the C⋅⋅⋅O distances in TSs TS1–TS3 decrease to 208.3, 214.6, and 210.9 pm (with bond indices of 0.23, 0.21, and 0.23). In these TSs, the C—Cl distance is increased to 230.2, 240.8, and 235.1 pm with the corresponding bond indices of 0.46, 0.38, and 0.44. The pentacoordinated carbon atom is nearly planar, and the NBO analyses characterize it as almost sp2 hybridized.
In the acetate Reactions 1–3 (Chart 1), the calculated natural charge of the pentacoordinated carbon atom in the TSs is −0.20, +0.06, and −0.01. In the reaction with methyl chloride (Reaction 1), the carbon atom carries a higher negative charge than in the ethyl cases (Reactions 2 and 3). Furthermore, the difference in the charge at the chlorine atom between the haloalkane reactant and the TS is 0.59, 0.63, and 0.62.
As shown in Table 1, the calculated complexation energies for the formation of IMCs change only slightly when electron correlation effect was included. However, electron correlation seems to have a larger effect on the calculated barriers, which is in agreement with previous theoretical studies on similar reactions (15). The calculated energy differences (Table 1 and Fig. 3) show that the complexation energies for Reactions 1 and 2 are very similar, but it is 20.8 kcal/mol for Reaction 3 at the MP2/6–31+G(d) level of theory. The larger complexation energy for Reaction 3 is probably due to the larger dipole moment of the gauche form of DCE. The TSs are higher in energy than the sum of the energies of the reactants, indicating that there is a small barrier for the overall reactions.
Figure 3.

Calculated reaction profiles for the acetate reactions at MP2//HF/6–31+G(d) level of theory.
The perturbation theory based analyses of the nonbonded interactions within the NBO analyses reveal that, in the IMCs, a lone pair of the attacking acetate oxygen atom overlaps with the σ* orbital of the C—Cl bond. In the TSs for the C⋅⋅⋅O bond formation, the attacking orbital of the oxygen has high p character (about 80%), and the carbon atom is sp2 hybridized. The estimated energy for this interaction is, of course, much larger in the TSs than in the IMCs because the carbon σ* orbital in the IMC can accept more electron density than the carbon p orbital in the TS. This “donor/acceptor” type of interaction (nO → σC—Cl∗) is by far the most significant part of the nonbonding interactions.
Hydroxide as the Nucleophile.
The distance in the IMCs between the carbon atom of the haloalkane and the oxygen atom of the approaching hydroxide (C⋅⋅⋅O) is 263.4, 283.8, and 283.7 pm for Reactions 4–6, respectively. The bond indices for these C⋅⋅⋅O bonds in IMC4–IMC6 are 0.02, 0.01, and 0.01, respectively. The bond indices are increased in the TSs (TS4–TS6) to 0.16, 0.15, and 0.20 with C⋅⋅⋅O distances of 227.2, 232.0, and 222.8 pm (Fig. 2), whereas the bond indices of the elongated C—Cl bonds are 0.66, 0.57, and 0.57. In the TSs, the pentacoordinated carbon atom is almost planar—i.e., almost sp2 hybridized—as revealed by the NBO analyses.
The calculated charges show a significant difference between Reaction 4 and Reactions 5 and 6. The differences in the charges on the pentacoordinated carbon in the TSs for the HO− Reactions 4, 5, and 6 (−0.31, −0.04, and −0.08 for TS4, TS5, and TS6, respectively) reflect the charge differences of the carbons in the alkyl halide substrates. In the TSs, the chlorine atoms have increased their negative charges relative to the haloalkane reactants by 0.43, 0.49, and 0.51 for TS4, TS5, and TS6, respectively.
For Reaction 4, we found a product-side IMC where the
chloride is opposite to the hydroxyl (Scheme
I). However, for all other
reactions, the intrinsic reaction coordinate calculations (21) resulted
in a carbon chlorine distance which increased to ≈320 pm, and then
the chloride ion started to associate with either methylene or
hydroxide hydrogen atoms.
![]()
The calculated energy differences for Reactions 4–6 (Table 1) show an increasing trend in the complexation energy on the formation of the IMC (ΔH1) and the activation energy (ΔH‡). The potential energy profile for DCE is significantly different from the potential energy profile of chloromethane and chloroethane, which have similar relative energies for ΔH1 and ΔH‡. The overall reaction energy, ΔHR, is about −50 kcal/mol for Reactions 4–6. This is in good agreement with the experimentally estimated value of ΔH° for Reaction 4 of −47.5 kcal/mol (13). All TSs are lower in energy than the sum of the corresponding reactants energies.
The relative energies at MP2 and MP4 level reveal that there is a large influence of electron correlation on the energy differences, especially for ΔH2. However, only the activation energies ΔH‡ are influenced by including third and fourth order MP correlation energy corrections [MP4(SDTQ)].
Comparison Between the Reactions of CH3CO2− and HO−.
When comparing the C⋅⋅⋅O and C—Cl distances for the IMCs (IMC1–IMC3 vs. IMC4–IMC6), it is conspicuous that the C⋅⋅⋅O distances in the acetate reactions are longer than in the corresponding hydroxide structures. In contrast, the corresponding distances in the TSs of the acetate (TS1–TS3) are always shorter than the ones in the hydroxide TSs (TS4–TS6), in accord with the Hammond postulate (22). This might be explained by the difference in nucleophilicity of the two ions and the partial charge on the corresponding oxygen atoms.
Comparing the MP2//HF energy differences (Table 1), the acetate reactions (ΔHR ≅ −11 kcal/mol) are much less exothermic than the reactions with hydroxide (ΔHR ≅ −50 kcal/mol). This is probably due to the charged reactant species compared with the charged product species—i.e., the chloride is preferred over the hydroxide—whereas the delocalized charge of acetate is preferred over chloride and hydroxide. This difference in the properties of hydroxide and acetate ions is also reflected in the difference in the gas phase acidity of water and acetic acid (23).
The relative energies given in Table 1 provide information about the influence of the presence of a methyl and a chloromethyl group on the α-carbon atom. The substrate change from chloromethane to chloroethane increases the complexation energy ΔH1 by 1.5 (2.6) kcal/mol for the acetate (hydroxide) reaction. A similar trend has been noted in calculations for the reaction of fluoride ion with different alkyl chlorides (11). The effect on ΔH1 is significant in the reactions with dichloroethane. The chloromethyl group at the α-carbon atom increases ΔH1 by 9.5 (12.5) kcal/mol for the acetate (hydroxide) reaction (Reactions 3 and 6 vs. Reactions 1 and 4). A similar effect can be seen in the relative energies of the intrinsic reaction barrier ΔH‡. In contrast, the overall reaction energies ΔHR are not significantly different. Most likely, the ΔHRs are comparable because the changes in ΔH1, ΔH‡, and ΔH2 compensate each other.
Stereochemical Considerations.
Considering DCE as a reactant, the hydroxide ion will unlikely attack while the DCE is in its favored conformation, having C2h symmetry. In this case, repulsive Cl⋅⋅⋅O interactions might prevent the formation of the IMC and the TS. Therefore, DCE has to be in a C2-like (gauche) conformation to undergo HO− attack. The energy difference between the C2h-symmetrical (trans) conformation and the C2-symmetrical (gauche) conformation of DCE is 1.5 kcal/mol at MP4//HF/6–31+G(d) level of theory (24). The experimentally determined gas phase energy difference is in the 0.9–1.3 kcal/mol range (25). Because the gauche conformer has a larger dipole moment, the energy difference between the two conformations is expected to decrease in solvent. Indeed, in liquid DCE, the energy difference is only about 0.3 kcal/mol (25). Note, for the estimation of all relative energies of Reactions 3 and 6, the gauche conformation of DCE has been considered as the reference.
In the case of chloroethane reactions (Reactions 2 and 5; Chart 1), it is noteworthy that the methyl group must undergo a rotation because chloroethane as well as the products ethyl acetate and ethanol prefer to have a trans conformation (Cs symmetry). This is a rotation of 60° if chloroethane is not substituted in the 2-position. If there is a substituent, this would be a rotation of 180° or the C—C bond would have to rotate partly before the reaction takes place (see the DCE example above). The intrinsic reaction coordinate calculations reveal that the entire rotation in Reaction 5 takes place after the SN2 reaction is virtually finished. Fig. 4 shows the calculated minimum energy pathway of Reaction 5 (solid line) and the corresponding dihedral angle (dotted line) which is the degree of freedom for the rotation of the methyl group. The SN2 reaction itself is finished at reaction coordinate unit 9 (vertical dashed line in Fig. 4), and the reaction has almost reached a stationary point on the potential energy surface. Up to this point, there is almost no change in the dihedral angle of the methyl group. Subsequently, the energy drops slightly again, and the dihedral angle undergoes a rotation of about 60°.
Figure 4.

Plot of the energy and the H—C—C—Cl dihedral angle vs. position along the reaction coordinate at HF/6–31+G(d) level of theory for Reaction 5. The dihedral angle does not change until the reaction has almost reached a stationary point. (Insets) Shown are the HF/6–31+G(d) calculated structures for the reaction of chloroethane with hydroxide (Reaction 5) at reaction coordinate unit 9.0 and unit 15.4.
SCI-PCM Solvation Studies.
It is well known that solvation has a large effect on the rate constants for the SN2 reaction (26). To examine the solvation effect on the reaction profile, we examined Reactions 3 and 4 using a self-consistent reaction field model. The geometries of the reactants and products were optimized with the SCI-PCM at the HF/6–31+G(d) level of theory; TSs were located and characterized by calculating the harmonic vibrational frequencies. As shown above, electron correlation effects are very important for the energetic description of these reactions, and further SCI-PCM energy calculations were done on the HF/6–31+G(d) optimized geometries at the MP2/6–31+G(d) level. Following the previous approaches (20, 27), the free energy surfaces at the MP2/6–31+G(d) level were also calculated (see Fig. 5), based on the HF/6–31+G(d) harmonic frequencies using standard techniques.
Figure 5.

The calculated potential and free energy (in parenthesis) profiles for Reactions 3 (a) and 4 (b).
The calculated potential barrier and free energy barrier for Reaction 4 (between methyl chloride and hydroxide ion) using the SCI-PCM solvation model at the MP2/6–31+G(d) level are 9.9 kcal/mol and 18.0 kcal/mol, respectively. The previously reported experimental barrier for this reaction in aqueous solution is about 24.6 kcal/mol (28), which is about 6.6 kcal/mol larger than the value we calculated. In view of the inability of the SCI-PCM algorithm to model explicit hydrogen bonding between the substrate and the solvent, this calculated value agrees quite well with the experimental value for the reaction between HO− and CH3Cl in aqueous solution (28). The SCI-PCM calculated C⋅⋅⋅Cl and C⋅⋅⋅O distances in the TS are 212.8 and 225.6 pm, which differ from corresponding distances in the gas phase TS by −6.7 pm (−0.067 Å) and 2.8 pm (0.028 Å), respectively.
As in the gas phase, the reaction between DCE and acetate as calculated by the SCI-PCM model is less exothermic than the reaction between methyl chloride and hydroxide ion. The calculated potential and free energy barriers for Reaction 3 are 21.0 and 32.0 kcal/mol, respectively. Reaction 3 seems to have a larger computed barrier than Reaction 4. Because there are no experimental data concerning the barrier of this reaction in solution, it is difficult to assess the calculated values. However, assuming the error in the calculated data for the reaction between acetate and DCE is similar to the reaction between HO− and CH3Cl, the free energy barrier for the former reaction is probably about 38 kcal/mol. Apparently, according to our calculations, Reaction 3 is extremely slow in aqueous solution. The calculated reaction energy and reaction free energy in the presence of reaction field are about −12.0 and −5.7 kcal/mol, respectively. According to the present calculations, Reaction 3 is much less exothermic than Reaction 4. The optimized TS for Reaction 3 in the presence of a continuum solvent model, is very similar to the gas phase geometry. The C⋅⋅⋅Cl distance is about 1 pm (0.01 Å) shorter in the presence of the reaction field, and the C⋅⋅⋅O distance is about 4.5 pm (0.045 Å) longer in the presence of the reaction field than the distance in the gas phase TS.
Calibration of the PM3 Semiempirical Method for Future Study of an Enyzmatic SN2 Displacement of Halogen.
We have chosen
the semiempirical PM3 (18) method to investigate the structure of the
TS formed in the reaction of an enzyme
-CH2CO2− side chain with DCE in the active
site of haloalkane dehalogenase. To do so requires a comparison of
appropriate gas phase ab initio derived results to results
from PM3 calculations. Therefore, we calculated the gas phase
stationary points for the reaction of
CH3CO2− with DCE (Reaction 3)
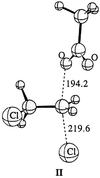
with the PM3 Hamiltonian. The relative energies are ΔH1 =
−18.5, ΔH‡ = 30.5, ΔH2 = −20.7, and
ΔHR = −8.8 kcal/mol. This is in qualitative agreement
with the ab initio results (Table 1). The geometry of the TS
(TS3PM3) is shown below (Scheme
II).
 Most important to our future
plans is a comparison of the percentage of bond breaking and forming in
the TSs with respect to the ground state C—Cl bond of the DCE and the
C—O bond of the esters and the data of the different TSs are
summarized in Table 2. In general, they are
very similar, with the PM3 TS being tighter than the HF/6–31+G(d)
and HF-SCRF/6–31+G(d) TSs.
Most important to our future
plans is a comparison of the percentage of bond breaking and forming in
the TSs with respect to the ground state C—Cl bond of the DCE and the
C—O bond of the esters and the data of the different TSs are
summarized in Table 2. In general, they are
very similar, with the PM3 TS being tighter than the HF/6–31+G(d)
and HF-SCRF/6–31+G(d) TSs.
Table 2.
Calculated ratio of bond breaking and making for different transition states with DCE substrate
| HF/6-31+G(d), TS3 | HF-SCRF/6-31+G(d), TS378.3 | PM3, TS3 | |
|---|---|---|---|
| r(C···Cl)/r(C—Cl) | 1.32 | 1.30 | 1.24 |
| r(C—O)/r(C···O) | 0.69 | 0.66 | 0.74 |
CONCLUSIONS
The calculated ab initio reaction profiles of the gas phase SN2 displacement reactions of alkyl chlorides with acetate and hydroxide ions have been compared. The reactions with HO− as the nucleophile are highly exothermic (≈50 kcal/mol) and have a central barrier that is lower in energy than the sum of the energies of the reactants. In contrast, with the CH3CO2− ion as the nucleophile, the reactions are much less exothermic (≈11 kcal/mol), and the TSs have a higher energy than the reactants. Electron correlation is shown to be important for the energetical description of the reactions.
The C⋅⋅⋅O bond in the hydroxide/molecule complexes is shorter in comparison to the acetate complexes. In accord with the Hammond postulate (22), the TS C⋅⋅⋅O distance in the hydroxide reactions is longer than in the acetate reactions. Conformational changes, which have to occur along with the displacement reactions, have been determined to take place before or after the actual substitution. DCE as a reactant has to change its conformation from trans (C2h symmetry) to gauche (C2 symmetry) prior the reaction to avoid repulsive interactions.
The TSs associated with carboxylate SN2 displacement of Cl− from DCE and hydroxide SN2 displacement of Cl− from chloromethane remain about the same when computed in the gas phase and surrounded by a dielectric continuum with a dielectric constant of 78.3. Reactions 3 and 4 have large free energy barriers in the presence of a polar solvent. The calculated barrier for Reaction 4 is about 18.0 kcal/mol, which is in reasonable agreement with the experimental value of 24.6 kcal/mol (28).
Acknowledgments
We acknowledge the National Center for Supercomputing Applications (Urbana, IL) for the allocation of computing time. We would also like to thank the Regionales Rechenzentrum in Cologne, Germany, for support and computing resources. This work was supported by the National Institutes of Health.
ABBREVIATIONS
- HF
Hartree–Fock
- MP
Møller–Plesset
- NBO
natural bond orbital
- SCI-PCM
self-consistent isodensity polarized continuum model
- IMC
ion/molecular complex
- TS
transition state
- DCE
1,2-dichloroethane
References
- 1.Keuning S, Janssen D B, Witholt B. J Bacteriol. 1985;163:635–639. doi: 10.1128/jb.163.2.635-639.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fetzner S, Lingens F. Microbiol Rev. 1994;58:641–685. doi: 10.1128/mr.58.4.641-685.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leisinger T. Curr Opin Biotechnol. 1996;7:295–300. doi: 10.1016/s0958-1669(96)80033-4. [DOI] [PubMed] [Google Scholar]
- 4.Verschueren K H G, Franken S M, Rozeboom H J, Kalk K H, Dijkstra B W. J Mol Biol. 1993;232:856–872. doi: 10.1006/jmbi.1993.1436. [DOI] [PubMed] [Google Scholar]
- 5.Verschueren K H G, Seljée F, Rozeboom H J, Kalk K H, Dijkstra B W. Nature (London) 1993;363:693–698. doi: 10.1038/363693a0. [DOI] [PubMed] [Google Scholar]
- 6.Kennes C, Pries F, Krooshof G H, Bokma E, Kingma J, Janssen D B. Eur J Biochem. 1995;228:403–407. [PubMed] [Google Scholar]
- 7.Pries F, Kingma J, Krooshof G H, Jeronimus-Stratingh C M, Bruins A P, Janssen D B. J Biol Chem. 1995;18:10405–10411. doi: 10.1074/jbc.270.18.10405. [DOI] [PubMed] [Google Scholar]
- 8.Dolfing J, Janssen D B. Biodegradation. 1994;5:21–28. [Google Scholar]
- 9.Shaik S S, Schlegel H B, Wolfe S. Theoretical Aspects of Physical Organic Chemistry: The SN2 Mechanism. New York: Wiley; 1992. [Google Scholar]
- 10.Harder S, Streitwieser A, Petty J T, Schleyer P v R. J Am Chem Soc. 1995;117:3253–3259. [Google Scholar]
- 11.Gronert S. J Am Chem Soc. 1993;115:652–659. [Google Scholar]
- 12.Hu W-P, Truhlar D G. J Am Chem Soc. 1996;118:860–869. [Google Scholar]
- 13.Ohta K, Morokuma K. J Phys Chem. 1985;89:5845–5849. [Google Scholar]
- 14.Evanseck J D, Blake J F, Jorgensen W L. J Am Chem Soc. 1987;109:2349–2353. [Google Scholar]
- 15.Glukhovtsev M N, Bach R D, Pross A, Radom L. Chem Phys Lett. 1996;260:558–564. [Google Scholar]
- 16.Truong T N, Stefanovich E V. J Phys Chem. 1995;99:14700–14706. [Google Scholar]
- 17.Frisch M J, Trucks G W, Schlegel H B, Gill P M W, Johnson B G, et al. Gaussian 94. Pittsburgh; 1995. [Google Scholar]
- 18.Stewart J J P. J Comput Chem. 1989;10:209–220. [Google Scholar]
- 19.Reed A E, Curtiss L A, Weinhold F. Chem Rev. 1988;88:899–926. [Google Scholar]
- 20.Wiberg K B, Keith T A, Frisch M J, Murcko M. J Phys Chem. 1995;99:9072–9079. [Google Scholar]
- 21.Gonzales C, Schlegel H B. J Phys Chem. 1990;94:5523–5527. [Google Scholar]
- 22.Hammond G S. J Am Chem Soc. 1955;77:334–338. [Google Scholar]
- 23.Lias, S. G., Bartmess, J. E., Liebman, J. F., Holmes, J. L., Levin, R. D. & Mallard, W. G. (1988) J. Phys. Chem. Ref. Data 17 (Suppl. 1).
- 24.Wong M W, Frisch M J, Wiberg K B. J Am Chem Soc. 1991;113:4776–4782. [Google Scholar]
- 25.Eliel E L, Wilen S H, Mander L N. Stereochemistry of Organic Compounds. New York: Wiley; 1994. pp. 607–608. [Google Scholar]
- 26.Parker A J. Chem Soc Q Rev. 1962;16:163–187. [Google Scholar]
- 27.Lim D, Jorgensen W L. J Phys Chem. 1996;100:17490–17500. [Google Scholar]
- 28.Albery W J, Kreevoy M M. Adv Phys Org Chem. 1978;16:87–157. [Google Scholar]