

Abstract
A combination of experimental and modeling approaches was used to study cellular–molecular mechanisms underlying the expression of short-term potentiation (STP) and long-term potentiation (LTP) of glutamatergic synaptic transmission in the hippocampal slice. Electrophysiological recordings from dentate granule cells revealed that high-frequency stimulation of perforant path afferents induced a robust STP and LTP of both (±)-α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) and N-methyl-d-aspartic acid (NMDA) receptor-mediated synaptic responses. However, the decay time constant for STP of the AMPA receptor-mediated excitatory postsynaptic potential was approximately 6 min, whereas the decay time constant for STP of the NMDA receptor-mediated excitatory postsynaptic potential was only 1 min. In addition, focal application of agonists during the expression of STP revealed that the magnitude of conductance change elicited by NMDA application was significantly enhanced, whereas the magnitude of conductance change elicited by application of AMPA remained constant. These findings are most consistent with a postsynaptic mechanism of STP and LTP. Different putative mechanisms were evaluated formally using a computational model that included diffusion of glutamate within the synaptic cleft, different kinetic properties of AMPA and NMDA receptor/channels, and geometric relations between presynaptic release sites and postsynaptic receptor/channels. Simulation results revealed that the only hypothesis consistent with experimental data is that STP and LTP reflect a relocation of AMPA receptor/channels in the postsynaptic membrane such that they become more closely “aligned” with presynaptic release sites. The same mechanism cannot account for STP or LTP of NMDA receptor-mediated responses; instead, potentiation of the NMDA receptor subtype is most consistent with an increase in receptor sensitivity or number.
Keywords: [long-term potentiation, dentate gyrus, ±-α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor, N-methyl-d-aspartic acid receptor, synaptic model]
Long-term potentiation (LTP) is a widely studied form of use-dependent synaptic plasticity expressed robustly by glutamatergic synapses of the hippocampus. The initial stage of LTP expression, typically identified as short-term potentiation (STP), is characterized by a rapid decay in the magnitude of potentiation to an asymptotic, steady-state level. Although there is a convergence of evidence concerning the cellular/molecular mechanisms mediating the induction of N-methyl-d-aspartic acid (NMDA) receptor-dependent STP and LTP (1), there remains substantial debate as to whether the expression of potentiation reflects change in presynaptic release mechanisms or postsynaptic receptor-channel function. Because of the synaptic coexistence of (±)-α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) and NMDA glutamatergic receptor subtypes (2), substantial differences in the magnitude of LTP expressed by AMPA and NMDA receptors would favor a mechanism that is postsynaptic in origin. Several studies have reported a more substantial induction of AMPA receptor-mediated LTP compared with NMDA receptor-mediated LTP (3–7), though in the presence of low concentrations of extracellular magnesium (8) or upon depolarization of the postsynaptic neuron (9), procedures that relieve blockade of the NMDA receptor/channel, equivalent magnitude increases in AMPA and NMDA receptor-mediated LTP consistently are observed. The possibility of an exclusively AMPA receptor-mediated postsynaptic expression of LTP also has been weakened by persistent difficulties in detecting an increased responsivity of AMPA receptors to exogenously applied glutamate or receptor agonist (10–13). In the studies reported here, we have investigated the potential differential expression of STP by AMPA and NMDA receptors and found that the decay time course of STP is markedly different for AMPA and NMDA receptor-mediated EPSPs. Furthermore, during both STP and LTP, we found evidence of a differential responsivity of AMPA and NMDA receptors to focal application of their respective agonists. With these results strongly supportive of a postsynaptic expression mechanism of STP and LTP, a computational model incorporating parameters for the synaptic space, the kinetic properties of AMPA and NMDA receptor/channels, and the relative locations of the two receptor/channel subtypes was used to investigate several more specific hypotheses. Results suggest a unique expression mechanism not previously proposed, namely, receptor/channel relocalization in the postsynaptic membrane. Our findings demonstrate that this hypothesis is sufficient to explain both STP and LTP of the AMPA receptor/channel, though not the NMDA receptor/channel, and moreover, can explain experimental observations that cannot be accounted for by previously hypothesized mechanisms.
METHODS
Standard techniques (14) were used to prepare transverse hippocampal slices from male New Zealand White rabbits and to evoke and record extracellular EPSPs and whole cell excitatory postsynaptic currents (EPSC) from dentate gyrus. Perfusing medium included reduced Mg2+ (0.1 mM) and picrotoxin (50 μM) to enhance NMDA receptor-gated conductance (15, 16). 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX) (10 μM, Tocris Neuramin, Bristol, U.K.), an AMPA receptor antagonist (17), was added as indicated. In some experiments, the AMPA receptor agonist, AMPA (500 μM), or the NMDA receptor agonist, NMDA (500 μM), was microejected into the middle 1/3 dendritic region of the granule cell (single pulse, 5 psi, 100 msec in duration, pH 7.3) via a multibarrel pipette. Whole cell EPSCs were recorded from dentate granule cell soma. The pipettes were filled with 120 mM cesium gluconate, 5 mM KCl, 2 mM MgSO4, 10 mM Hepes, 0.1 mM CaCl2, 1 mM 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate, and 3 mM MgATP. The membrane potential of the recorded cells (n = 20) was held at −80 mV except during high-frequency stimulations at which the membrane potential was reduced to between −40 and −20 mV.
The mathematical model of a glutamatergic synapse includes the representations of a synaptic cleft as a discretized two-dimensional array where the neurotransmitter release sites are located at the uppermost row and the kinetic models of AMPA and NMDA receptor channels. The diffusion of neurotransmitter (glutamate) in the cleft is described by the following equation:
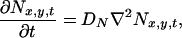 |
1 |
where Nx,y,t denotes the concentration of neurotransmitter molecules at the point (x, y) at time t and DN = 7.6 × 10−6 cm2/s (18). Eq. 1 was solved numerically with appropriate boundary conditions (19).
The EPSCs were computed from kinetic models of AMPA and NMDA receptor/channels. The kinetic model of the AMPA receptor/channel was adopted from Ambros-Ingerson and Lynch (20), while that of the NMDA receptor/channel followed the model developed by Lester and Jahr (21). The AMPA model includes one glutamate binding site whereas the NMDA model assumes two binding sites.
RESULTS
Differential Expression of STP by AMPA and NMDA Receptors.
The first experiments examined the possibility of a differential expression of STP by AMPA and NMDA receptors. Magnitude of the NMDA response was enhanced by reducing the extracellular concentration of magnesium ([Mg2+]o) to 0.1 mM (Fig. 1A), and NMDA receptor-mediated EPSPs were pharmacologically isolated by blocking AMPA receptors using the specific antagonist CNQX (10 μM). In reduced [Mg2+]o, a negative slope conductance is still clearly present in the I–V curve (Fig. 1B), and thus the receptor/channel still behaves in a physiological manner characteristic of higher concentrations of [Mg2+]o (15). Under these conditions, high-frequency stimulation induced both NMDA STP and LTP (Fig. 1C). The decay time courses were fitted with a single exponential curve, and the averaged STP decay time constant for NMDA STP was found to be 1.2 ± 0.20 min (mean ± SEM, n = 8).
Figure 1.

NMDA STP decays faster than AMPA STP. (A) Before (solid traces) and after (dotted traces) d-2-amino-5-phosphonovaleric acid (50 μM) at 0.1 mM Mg2+ (Upper) and 1.0 mM Mg2+ (Lower). (B) I–V curve of NMDA receptor-mediated EPSC at 0.1 mM Mg2+ (in presence of CNQX). (C) STP and LTP expressed by NMDA-mediated EPSPs (in presence of CNQX). (D) STP and LTP expressed by AMPA-mediated EPSPs (in absence of CNQX).
In the absence of CNQX and in the presence of 1.0 mM [Mg2+]o, EPSPs recorded in response to 0.1 Hz synaptic activation were almost exclusively AMPA receptor-mediated (Fig. 1A). High-frequency stimulation induced both STP and LTP of the AMPA response (Fig. 1D, ○), though the decay time constant of STP was found to be substantially longer than for NMDA receptor-mediated EPSPs (6.39 ± 0.86 min, n = 12). The difference in STP decay time constants for AMPA and NMDA responses was not due to the differences in [Mg2+]o. When AMPA STP was examined in 0.1 mM [Mg2+]o, the averaged decay time constant was found to be 6.30 ± 0.86 min (n = 11) (Fig. 1D, •), virtually identical to that observed in the presence of 1.0 mM [Mg2+]o.
An increase in the response of the AMPA receptor-mediated EPSP, with much less change in the response of the NMDA receptor-mediated EPSP is classically interpreted as indicating a postsynaptic locus of expression. However, it has been argued that the expression of STP still might be due to increased presynaptic transmitter release, even if NMDA and AMPA receptor responses show different changes because they have different kinetic properties. To test the hypothesis, we conducted a series of simulations by systematically varying the quantal content within reported physiological range. Our model, which takes into account the different kinetics of the NMDA and AMPA receptor/channels, predicts that the decay time course of STP of the NMDA component is longer than that of the AMPA component, contrary to our experimental finding. This is an additional evidence supporting that STP results from modifications in the postsynaptic mechanisms.
Differential Responsivity to Agonist by AMPA and NMDA Receptors During STP and LTP Expression.
The above results strongly suggest that the expression mechanism for STP is postsynaptic in origin, and as a consequence, we examined whether or not receptor responsivity was increased during STP. A multibarrel pipette was placed in the dendritic region of the recorded granule cell and membrane conductance changes elicited by focal application of AMPA or NMDA were measured before and after induction of LTP (Fig. 2). Conductance change was not altered in response to AMPA (−0.7 ± 3.5%; P > 0.20, n = 8); however, membrane conductance change elicited by NMDA was found to be enhanced (24.1 ± 7.2%; P < 0.02, n = 7) after LTP was induced. These results demonstrate that during LTP, NMDA receptor-mediated EPSCs are increased both in response to focal application of NMDA and perforant path stimulation. In contrast, AMPA receptor-mediated synaptic responses are increased, but AMPA receptor-mediated responses to focal application of agonist are unchanged.
Figure 2.

(A) Membrane conductance in response to microapplication of NMDA, but not AMPA, is increased during STP. (Upper) The upper two are responses induced by pressure ejection of NMDA at the moments indicated by downward arrowheads, (500 μM, 100 msec in duration); the lower two are responses induced by AMPA (500 μM, 100 msec in duration); before (Left) and after (Right) induction of LTP. (B) STP was expressed by whole cell EPSCs in both experimental groups. (Upper) The upper two EPSCs are obtained from the same hippocampal slice as the one shown in A for focal application of NMDA; the lower two are from that for focal application of AMPA before (Dotted) and during (Solid) STP.
Effect of Relative Position of Presynaptic Release Site and Postsynaptic Receptors.
Factors that could affect synaptic efficacy include changes in (i) mechanisms of presynaptic glutamate release, (ii) number of postsynaptic receptors, (iii) kinetic properties of postsynaptic receptors, and (iv) the geometry of the synaptic junction. Because alteration of presynaptic release mechanisms would be expected to affect AMPA and NMDA receptor-mediated responses equivalently, the observed differences in expression of STP and LTP by the two receptor subtypes strongly suggest that the mechanisms are postsynaptic in origin. The fact that potentiation of the AMPA response is not observed during focal application of agonist further suggests that the mechanism of AMPA STP and LTP involves changes other than an increase in receptor responsivity or number. Because the contribution of changes in synaptic geometry is more difficult to evaluate, we conducted a series of computer simulations to investigate the possible consequences of changes in synaptic morphology.
Initial simulation studies revealed that the extreme narrowness of the synaptic cleft (20 nm) leads to a highly localized distribution of glutamate molecules along the postsynaptic membrane (Fig. 3A). As a consequence, we tested the hypothesis that the relative positions of the presynaptic release sites and postsynaptic receptor/channels will be a significant factor in determining the magnitude and time course of the EPSCs. Specifically, we simulated AMPA and NMDA receptor-mediated EPSCs for different lateral displacements of release sites and receptor/channels. Results showed that the AMPA receptor-mediated EPSC is 42% larger and the NMDA receptor-mediated EPSC is 17% larger in magnitude when the release sites and receptors are in complete alignment (a zero lateral displacement) compared with the EPSCs simulated for a 40-nm displacement (Fig. 3 B and C). Furthermore, when release sites and receptors are in complete alignment, AMPA receptor-mediated EPSCs are characterized by a faster rate of onset and decay, in agreement with experimental findings (Figs. 2 and 3). The differential sensitivity of AMPA and NMDA receptors to the degree of “alignment” with presynaptic release sites can be explained by their different affinities to glutamate (30 μM for AMPA receptor/channel and 0.9 μM for NMDA receptor/channel in our model; also see refs. 20–22). Simulation results showed that due to NMDA receptors’ higher affinity, more than 98% of NMDA receptors are bound when a 40-nm displacement is assumed. As a consequence, changes in the postsynaptic location of the NMDA receptor have little effect on the probability of binding with glutamate in changing the magnitude of the EPSC. In contrast, because only 23% of the AMPA receptors are liganded when a 40-nm displacement is assumed, lateral movement that increases alignment with the presynaptic release site will result in a higher proportion of bound receptors.
Figure 3.

The effect of redistribution of receptors. (A) Simulation of neurotransmitter release from a vesicle into the synaptic cleft. Because the cleft is extremely narrow, the concentration of glutamate at the postsynaptic membrane is highly localized. (B) Simulated AMPA response (Upper) and experimental whole cell EPSCs (Lower). They exhibit a similar profile (note the normalized results). In simulation, the AMPA-mediated current is 43% greater when the receptor is perfectly aligned with the release site than when they are 40 nm away (Left). The two traces are normalized to show that the time course of the larger response (solid trace) is faster (Right). (C) A smaller change is seen in the NMDA-mediated EPSC (Left) with virtually no change in its time course (Right).
When glutamate is delivered to the synapse by means of perfusion, such as during focal application of agonists, its distribution would be more uniform and the influence of receptor relocation should be reduced for both AMPA and NMDA receptor-mediated EPSCs. This condition was simulated by assuming that glutamate diffuses into the synaptic cleft from its perimeter: virtually no difference was seen in either AMPA or NMDA receptor-mediated EPSCs. To test the plausibility that changes in receptor/channel kinetics account for STP and LTP of AMPA receptor-mediated responses, opening and closing rate were increased in magnitude as proposed by Ambros-Ingerson and Lynch (20). Simulation results showed that the AMPA receptor-mediated response to focal application of agonist increases by 31%. These results strongly support the hypothesis that potentiation of AMPA receptor-mediated EPSCs reflect a relocation of the receptor/channel in the postsynaptic membrane so as to become more aligned with presynaptic release sites. The same mechanism cannot account for STP or LTP of the NMDA receptor-mediated EPSP.
DISCUSSION
Different Expression Mechanisms Underlie Potentiation of AMPA and NMDA Receptors.
The major findings of the present study are that STP of the NMDA receptor exhibits a much shorter decay time course than that for the AMPA receptor. In addition, during the expression of both STP and LTP, only the postsynaptic response to microejection of NMDA is enhanced; the response to microejection of AMPA is unchanged. Thus, there are different mechanisms of expression for STP and LTP of AMPA and NMDA receptor/channels. Assuming colocalization of the two glutamatergic receptor subtypes, these data also strongly argue that, for both AMPA and NMDA receptors, the expression of STP and LTP involve mechanisms that are postsynaptic in origin. Only the extremely rapid decay rate for NMDA STP suggests the possibility of a presynaptic mechanism, i.e., that STP of the NMDA receptor/channel reflects primarily PTP. This hypothesis has been dealt with elsewhere (23). Results of our simulation studies showed that either sensitization of the NMDA receptor or an increase in number of receptors is sufficient to account for the increased postsynaptic responsivity to agonist, and thus, the LTP expressed by the NMDA receptor. The failure of others to detect a similar change in NMDA receptor responsivity with focal application of glutamate (10–13) is most likely due to the use of high concentrations of Mg2+ in the perfusing media, suppressing NMDA channel opening.
Enhancement of synaptically evoked AMPA responses in the absence of increased responsivity to agonist further suggests that the expression mechanisms of AMPA STP and LTP involve changes other than an increase in number or sensitivity of the receptor. Because receptors and other membrane proteins can diffuse laterally in the plane of the cytoplasmic membrane (24, 25), the effects of changes in position of postsynaptic AMPA and NMDA receptors were investigated using computer simulations of a glutamatergic synapse. Results revealed that the relative positions of the presynaptic release site and the postsynaptic receptor can have a profound effect on the magnitude and time course of the EPSCs. Detailed studies found that properties of AMPA receptor-mediated STP and LTP were most consistent with the hypothesis that, in response to high-frequency stimulation, the receptor/channel becomes relocalized in the postsynaptic membrane so as to be more closely aligned with the presynaptic release site.
Postsynaptic AMPA Receptor Realignment Model.
Thus, we propose the following model for the mechanism of expression of NMDA receptor-dependent STP and LTP of AMPA receptor-mediated synaptic responses. NMDA receptor activation is known to be critical for glutamatergic synapse stabilization during development (26), and thus, we assume that a close alignment of NMDA receptors and presynaptic release sites emerges from the process of synapse formation. For synapses in the unpotentiated state, we further assume that AMPA receptors have a wider spatial distribution, with many beyond the range of effective concentration of released neurotransmitter. Ca2+ influx through the NMDA receptor/channel activates several calcium-dependent processes, among them, the calcium-dependent protease calpain (27). Calpain-mediated degradation of cytoskeletal proteins has been shown to play an important role in the redistribution of cell surface receptors (28), and we postulate that it allows the lateral diffusion of AMPA receptors in synaptic membranes, and their migration toward the region of recently activated NMDA receptor/channels. This initial clustering of AMPA receptors in membrane locations more closely aligned with presynaptic release sites is the basis for STP.
As the mechanisms that promote receptor aggregation decay with time, AMPA receptors diffuse away from membrane locations proximal to the initially activated NMDA receptors (and thus to locations more displaced from the release site), and there is a corresponding time-dependent decay in the magnitude of the potentiated AMPA receptor-mediated response. Activation of other calcium-dependent processes and second messenger systems, including those related to adhesion molecules (29, 30), have been shown to be required for the stabilization of STP into LTP. If AMPA receptors are “anchored” in their new position before relaxation of STP is complete, then the newly stabilized locations relative to the release sites leads to LTP. Without activation of the anchoring mechanisms, however, recently aggregated receptors will diffuse back to their original positions (or equivalently distant positions), with the resulting synaptic potentiation expressed only as STP. Thus, the magnitude of LTP is determined by the decay time course of the clustering process and the onset time course of the anchoring process.
The proposed model is consistent with previous conceptualizations of two parallel and independent processes for STP and LTP (31–33), as well as numerous studies indicating that inhibition of second messenger pathways effectively blocks stabilization of LTP while having minimal effect on the magnitude of STP. In particular, the proposed model is consistent with our recent observation that STP can be induced repetitively in the presence of saturating levels of LTP (23). Saturation of LTP is presumed to represent the maximum density of receptors that can be anchored in the synaptic region. If the processes underlying clustering (increased receptor mobility and lateral displacement) are independent of the mechanisms underlying anchoring, then neither the magnitude of STP nor its repetitive induction should be altered by saturation of LTP.
A variant of this model is that new AMPA receptors are inserted in close proximity to NMDA receptors, and thus, at membrane positions in closer alignment with presynaptic release sites. To account for the differential effects of exogenous AMPA and NMDA found in the present studies, however, it also must be postulated that the total number of AMPA receptors remains constant, i.e., that some receptors distant from the NMDA receptors are internalized. Recent studies have shown that the C-terminal domain of various AMPA receptor subunits is truncated after NMDA receptor stimulation and the resulting activation of the calcium-dependent protease calpain (34, 35). Furthermore, several sources of evidence indicate the existence of a cytoplasmic pool of AMPA receptors that could be inserted in postsynaptic membranes in a calcium-dependent manner (36). Though a change in channel kinetic properties, such as an increase in the opening and closing rates (20), has been shown to reproduce the EPSP waveforms characteristic of LTP expression, such a mechanism cannot explain why no increase is observed with focal application of AMPA or glutamate in the present or in the majority of past studies (10–13, 37).
Predictions of the Postsynaptic AMPA Realignment Model.
There are a number of results that can be predicted or explained by our model. First, increased responses to microejection of AMPA may occur during later phases of LTP. As shown in Fig. 4, receptor aggregation may cause an overall receptor redistribution on the membrane surface. This redistribution may serve as a signal for more receptors to be supplied to the membrane. As a result, the absolute number of AMPA receptors could gradually increase over time. Davies et al. (38) observed such a progressive increase in AMPA responsivity, which started about 30 min after the induction of LTP with the peak magnitude appearing approximately 90 min later.
Figure 4.

Schematic representation of our STP-LTP model. Rest, patches of presynaptic and postsynaptic membrane in a nonpotentiated state. HFS, presynaptic LTP-inducing pulses arrive that cause NMDA receptor/channel activation and Ca2+ influx, which, in turn, trigger the receptor attraction mechanism. STP, as a result, AMPA receptors are attracted toward the activated NMDA receptor/channel, which is aligned previously with the presynaptic release zone. LTP, depending on the effectiveness of the anchoring mechanism, all or part AMPA receptors will diffuse again. An electrophysiological observer then will interpret it as only STP or STP plus LTP correspondingly. Note that due to diffusion, AMPA receptors used to be farther located may enter this domain and can be used for further potentiation of synaptic strength.
Second, if AMPA receptors are poorly aligned relative to presynaptic site, the synapse can be functionally silent. Our simulation results showed that the amplitude of EPSP produced by an AMPA receptor/channel displaced by 100 nm from the presynaptic release site is 32% of that generated by a receptor/channel aligned with the release site, whereas a receptor/channel displaced by 200 nm will generate an EPSP only 6% in amplitude, making it almost undetectable. This prediction is consistent with reports that LTP arises from recruitment of inactive synapses or latent receptors (39–43). More recently, Liao et al. (44) and Isaac et al. (45) have confirmed that a high proportion of synapses in hippocampal area CA1 exhibits NMDA receptor-mediated transmission only, making these synapses effectively nonfunctional at normal resting potentials. These “silent” synapses acquire AMPA receptor-mediated response characteristics after LTP induction.
Third, our results show that the dynamics of neurotransmitter distribution in the synapse varies as a function of distance from the release site, namely, the greater the distance from the release site, the slower the variation in concentration of neurotransmitter. Because the model predicts that synaptic potentiation reflects the migration of AMPA receptors to positions closer to the release site, the rise and decay times of AMPA EPSCs should be more rapid during the expression of LTP, as found in the present and previous studies (46). More generally, the location of AMPA receptor/channels in the postsynaptic membrane should range from optimal alignment with the presynaptic release site to beyond the range of effective concentration of released neurotransmitter, i.e., functionally “silent.” Within this spectrum, the rise-fall time and peak amplitude of the AMPA EPSC waveform should correlate positively with proximity to the presynaptic release site.
Finally, the model predicts a nonuniform distribution of AMPA and NMDA receptors in the synapse. It is expected that NMDA receptors are at the most focal point opposite a given release site, with an “annulus” of more peripherally located AMPA receptors. The degree of nonuniformity should be more pronounced for potentiated synapses.
Acknowledgments
We thank M. Yeckel for his generous advice and technical help. This work was supported by the Office of Naval Research, National Institute of Mental Health, National Center for Research Resources, the Human Frontiers Science Organization, and a National Research Service Award fellowship to J.-S.L.
ABBREVIATIONS
- AMPA
(±)-α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid
- NMDA
N-methyl-d-aspartic acid
- LTP
long-term potentiation
- STP
short-term potentiation
- EPSP
excitatory postsynaptic potential
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- [Mg2+]o
extracellular concentration of magnesium
- EPSC
excitatory postsynaptic current
References
- 1.Bliss T V P, Collingridge G L. Nature (London) 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 2.Bekkers J M, Stevens C F. Nature (London) 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- 3.Kauer J A, Malenka R C, Nicoll R A. Neuron. 1988;1:911–917. doi: 10.1016/0896-6273(88)90148-1. [DOI] [PubMed] [Google Scholar]
- 4.Muller D, Joly M, Lynch G. Science. 1988;242:1694–1697. doi: 10.1126/science.2904701. [DOI] [PubMed] [Google Scholar]
- 5.Muller D, Lynch G. Proc Natl Acad Sci USA. 1988;85:9346–9350. doi: 10.1073/pnas.85.23.9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perkel D J, Nicoll R A. J Physiol. 1993;471:481–500. doi: 10.1113/jphysiol.1993.sp019911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asztely F, Wigström H, Gustafsson B. Eur J Neurosci. 1992;4:681–690. doi: 10.1111/j.1460-9568.1992.tb00177.x. [DOI] [PubMed] [Google Scholar]
- 8.Xie X, Berger T W, Barrionuevo G. Soc Neurosci Abstr. 1992;18:628.5. [Google Scholar]
- 9.O’Connor J J, Rowan M J, Anwyl R. J Neurosci. 1995;15:2013–2020. doi: 10.1523/JNEUROSCI.15-03-02013.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lynch G, Gribkoff V, Deadwyler S A. Nature (London) 1976;263:151–153. doi: 10.1038/263151a0. [DOI] [PubMed] [Google Scholar]
- 11.Murali M P, Sastry B R. Eur J Pharmacol. 1985;114:335–341. doi: 10.1016/0014-2999(85)90378-4. [DOI] [PubMed] [Google Scholar]
- 12.Taube J S, Schwartzkroin P A. J Neurosci. 1988;8:1632–1644. doi: 10.1523/JNEUROSCI.08-05-01632.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malgaroli A, Tsien R W. Nature (London) 1992;357:134–139. doi: 10.1038/357134a0. [DOI] [PubMed] [Google Scholar]
- 14.Xie X, Berger T W, Barrionuevo G. J Neurophysiol. 1992;67:1009–1013. doi: 10.1152/jn.1992.67.4.1009. [DOI] [PubMed] [Google Scholar]
- 15.Nowak L, Bregestovski P, Ascher P. Nature (London) 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 16.Mayer M L, Westbrook G L. J Physiol. 1987;394:501–527. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drejer J, Honore T. Neurosci Lett. 1988;87:104–108. doi: 10.1016/0304-3940(88)90153-x. [DOI] [PubMed] [Google Scholar]
- 18.Longsworth L G. J Am Chem Soc. 1953;75:5705–5709. [Google Scholar]
- 19.Carnahan B, Luther H A, Wilkes J O. Applied Numerical Methods. New York: Wiley; 1969. [Google Scholar]
- 20.Ambros-Ingerson J, Lynch G. Proc Natl Acad Sci USA. 1993;90:7903–7907. doi: 10.1073/pnas.90.16.7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lester R A J, Jahr C E. J Neurosci. 1992;12:635–643. doi: 10.1523/JNEUROSCI.12-02-00635.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patneau D K, Mayer M L. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie X, Barrionuevo G, Berger T W. Learn Mem. 1996;3:115–123. doi: 10.1101/lm.3.2-3.115. [DOI] [PubMed] [Google Scholar]
- 24.Froehner S C. Annu Rev Neurosci. 1993;16:347–368. doi: 10.1146/annurev.ne.16.030193.002023. [DOI] [PubMed] [Google Scholar]
- 25.Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson J D. Molecular Biology of the Cell. 3rd Ed. New York: Garland; 1994. pp. 475–506. [Google Scholar]
- 26.Debski E A, Cline H T, Constantine-Paton M. J Neurobiol. 1990;21:18–32. doi: 10.1002/neu.480210103. [DOI] [PubMed] [Google Scholar]
- 27.Vanderklish P, Saido T C, Gall C, Lynch G. Mol Brain Res. 1995;32:25–35. doi: 10.1016/0169-328x(95)00057-y. [DOI] [PubMed] [Google Scholar]
- 28.Croall D E, Demartino G N. Physiol Rev. 1991;71:813–847. doi: 10.1152/physrev.1991.71.3.813. [DOI] [PubMed] [Google Scholar]
- 29.Luthi A, Laurent J P, Figurov A, Muller D, Schachner M. Nature (London) 1994;372:777–779. doi: 10.1038/372777a0. [DOI] [PubMed] [Google Scholar]
- 30.Xiao P, Bahr B A, Staubli U, Vanderklish P, Lynch G. Neuroreport. 1991;2:461–464. doi: 10.1097/00001756-199108000-00013. [DOI] [PubMed] [Google Scholar]
- 31.Gustafsson B, Asztely F, Hanse E, Wigström H. Eur J Neurosci. 1989;1:382–394. doi: 10.1111/j.1460-9568.1989.tb00803.x. [DOI] [PubMed] [Google Scholar]
- 32.Nicoll R A, Malenka R C. Nature (London) 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- 33.Arai A, Larson J, Lynch G. Brain Res. 1990;511:353–357. doi: 10.1016/0006-8993(90)90184-d. [DOI] [PubMed] [Google Scholar]
- 34.Bi X, Chang V, Tocco G, Baudry M. Brain Res. 1996;726:98–108. doi: 10.1016/0006-8993(95)01360-1. [DOI] [PubMed] [Google Scholar]
- 35.Bi X, Tocco G, Baudry M. Neuroreport. 1994;6:61–64. doi: 10.1097/00001756-199412300-00017. [DOI] [PubMed] [Google Scholar]
- 36.Standley S, Bi X, Baudry M. In: Long-Term Potentiation 3. Baudry M, Davis J L, editors. Cambridge, MA: MIT Press; 1996. pp. 17–40. [Google Scholar]
- 37.Poolos N P, Kocsis J D. Brain Res. 1990;524:342–346. doi: 10.1016/0006-8993(90)90714-m. [DOI] [PubMed] [Google Scholar]
- 38.Davies S N, Lester R A J, Reymann K G, Collingridge G L. Nature (London) 1989;338:500–503. doi: 10.1038/338500a0. [DOI] [PubMed] [Google Scholar]
- 39.Edwards F. Nature (London) 1991;350:271–272. doi: 10.1038/350271a0. [DOI] [PubMed] [Google Scholar]
- 40.Liao D, Jones A, Malinow R. Neuron. 1992;9:1089–1097. doi: 10.1016/0896-6273(92)90068-o. [DOI] [PubMed] [Google Scholar]
- 41.Manabe T, Renner P, Nicoll R A. Nature (London) 1992;355:50–55. doi: 10.1038/355050a0. [DOI] [PubMed] [Google Scholar]
- 42.Kullmann D M. Neuron. 1994;12:1111–1120. doi: 10.1016/0896-6273(94)90318-2. [DOI] [PubMed] [Google Scholar]
- 43.Andrew R D, Macvicar B A. Neurosci. 1994;62:371–383. doi: 10.1016/0306-4522(94)90372-7. [DOI] [PubMed] [Google Scholar]
- 44.Liao D, Hessler N A, Malinow R. Nature (London) 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- 45.Isaac J T R, Nicoll R A, Malenka R C. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 46.Ambros-Ingerson J, Xiao P, Larson J, Lynch G. Brain Res. 1993;620:237–244. doi: 10.1016/0006-8993(93)90161-f. [DOI] [PubMed] [Google Scholar]