

Abstract
Aspects of protein disulfide isomerase (PDI) function have been studied in yeast in vivo. PDI contains two thioredoxin-like domains, a and a′, each of which contains an active-site CXXC motif. The relative importance of the two domains was analyzed by rendering each one inactive by mutation to SGAS. Such mutations had no significant effect on growth. The domains however, were not equivalent since the rate of folding of carboxypeptidase Y (CPY) in vivo was reduced by inactivation of the a domain but not the a′ domain. To investigate the relevance of PDI redox potential, the G and H positions of each CGHC active site were randomly mutagenized. The resulting mutant PDIs were ranked by their growth phenotype on medium containing increasing concentrations of DTT. The rate of CPY folding in the mutants showed the same ranking as the DTT sensitivity, suggesting that the oxidative power of PDI is an important factor in folding in vivo. Mutants with a PDI that cannot perform oxidation reactions on its own (CGHS) had a strongly reduced growth rate. The growth rates, however, did not correlate with CPY folding, suggesting that the protein(s) required for optimal growth are dependent on PDI for oxidation. pdi1-deleted strains overexpressing the yeast PDI homologue EUG1 are viable. Exchanging the wild-type Eug1p C(L/I)HS active site sequences for C(L/I)HC increased the growth rate significantly, however, further highlighting the importance of the oxidizing function for optimal growth.
Formation of correct disulfide bonds is essential for proper folding of the majority of secretory proteins in eukaryotic cells. Refolding of proteins containing more than one disulfide bond in vitro is stimulated by the enzyme protein disulfide isomerase (PDI)1 (Givol et al., 1965). PDI of mammalian origin has a characteristic a-b-b′-a′-c domain structure (Edman et al., 1985). The a and a′ domains have sequence and structural homology to thioredoxin (Kemmink et al., 1996; Fig. 1 A). The b and b′ domains are characterized by mutual homology. The COOH-terminal c region is rich in acidic residues, and is thought to be involved in binding of calcium ions (Lebeche et al., 1994; Nigel Darby, EMBL, Heidelberg, personal communication).
Figure 1.

(A) Sequences of PDI and Eug1p from yeast and thioredoxin from E. coli surrounding the active site CXXC motifs (boxed). A section of the a and a′ domains of PDI and Eug1p is compared, and the location of the predicted secondary structure features is indicated by β2 (second β-sheet structure) and α2 (second α-helix structure). Residues that are found in a majority of the sequences are underlined. (B) Reaction cycle for oxidation of nascent polypeptide (ProteinSH/SH) catalyzed by PDI. The putative involvement of GSSG in the oxidation cycle is indicated. The upper SH group of PDI (PDI SH) indicates the NH2-proximal cysteine residue of the CXXC motif. This is the more reactive residue and will engage in the formation of mixed disulfides with the substrate, here shown only for the protein substrate although a similar reaction pathway exists for GSSG reduction. PDISH indicates the more COOH-proximal cysteine residue, which is involved in the formation of the internally oxidized form of PDI. In mutant forms of PDI lacking the COOH-proximal cysteine, oxidation of substrate protein can occur, albeit inefficiently, by reduction of a mixed disulfide between glutathione and the PDI NH2-proximal cysteine residue. (C) Isomerization reaction in which the NH2-proximal cysteine residue of the CXXC motif attacks a wrongly formed disulfide of a substrate protein. The formation of mixed intermediate I allows a free SH group of the substrate to form an alternative SS bond. Importantly, the more COOH-proximal cysteine residue does not participate in this reaction cycle.
Members of the thioredoxin family contain two catalytically active cysteine residues found in a CXXC motif. The identity of the two X residues varies among the family members, but catalysis is always mediated by reduction and oxidation of an internal disulfide bond between the cysteine residues of the active site (Martin, 1995). As the name would indicate, isomerization of disulfide bonds is an important feature of PDI. What is less widely acknowledged is that PDI may also play an important role in the oxidation of newly synthesized proteins. Detailed biochemical analysis has shown that the a and a′ domains of PDI are capable of catalyzing two kinds of disulfide reactions (Creighton et al., 1980; Darby and Creighton, 1995a ): (a) oxidation reactions in which the intramolecular disulfide bond of the CGHC motif is transferred to a pair of sulfhydryls in a substrate polypeptide (Fig. 1 B); and (b) isomerization reactions in which disulfides are rearranged through the formation of a mixed disulfide between the first cysteine residue of the CGHC motif and the substrate (Fig. 1 C).
In the eukaryotic cell, PDI is localized to the ER. Glutathione and glutathione disulfide (GSSG) are believed to function as the redox buffer since they are found at approximately equal millimolar concentrations in the ER, providing a much more oxidizing environment than the cytoplasm (Hwang et al., 1992).
In yeast, the structural gene for PDI, PDI1, is essential for viability (Farquhar et al., 1991; LaMantia et al., 1991; Günther et al., 1991; Scherens et al., 1991). When PDI is depleted from the cells, a secretory pathway marker protein accumulates in the ER, suggesting an impairment in folding (Günther et al., 1991; Tachibana and Stevens, 1992). Despite the potentially central role that PDI plays in the folding of newly synthesized secretory pathway proteins, little has been done to dissect the functional aspects of its role in folding in vivo. In the present work, we have analyzed the effects of various mutations in PDI1 on the folding of a yeast vacuolar protein, carboxypeptidase Y (CPY). CPY has a number of virtues that make it a very useful model for the study of in vivo folding and secretory transport. Changes in molecular mass that accompany its transport through different compartments of the secretory pathway can be followed in pulse-labeling experiments (Stevens et al., 1982). Upon translocation into the ER, the polypeptide receives four core N-glycosyl residues, resulting in the p1 form of proCPY, which has a molecular mass of 67 kD. The core glycosyl structures are further modified in the Golgi compartment to give the 69-kD p2 form of proCPY. This form is sorted to the vacuole, where it is processed to its mature form of 63 kD. The individual transport steps are easily followed in 35S pulse-labeling experiments, and the total sequence of events has a half time of ∼6 min. As in mammalian systems, proteins that do not fold into their correct three-dimensional structure are retained in the ER. Thus, ER retention is an indication of misfolding. In a more direct assay, the in vivo folding rate was monitored using a mutant form of CPY (CPY-3T) that contains a new glycosyl acceptor site that is inaccessible in the folded protein. Treatment of whole cells with DTT may block the folding of many disulfide bond–containing proteins by preventing oxidation of the cysteine residues of the newly synthesized proteins (Braakman et al., 1991). In yeast cells, DTT treatment results in accumulation of p1-proCPY (Simons et al., 1995) and, in the case of proCPY-3T, modification of the new glycosylation site (Holst et al., 1996). In strains expressing some mutant alleles of PDI1, we found this modification was present on CPY-3T at a level approaching that of DTT-treated cells. These observations are consistent with the view that not only is formation of the five disulfide bonds of the protein essential for correct folding, but also that mutations in PDI can affect the rate of folding in vivo even when no growth phenotype is obvious.
Materials and Methods
Strains
Saccharomyces cerevisiae strains W303-1BαΔpdi1 (MATα ade2-1 can1-100 ura3-1 leu2-3,112 trp1-1 his3-11,15 Δpdi1::HIS3) and M4143 (MATα ade2-1 can1-100 ura3-1 leu2-3,112 trp1-1 his3-11,15 Δpdi1::HIS3 Δprc1-17) containing either pCT37, pCT40 (Kurjan, 1985; Tachibana and Stevens, 1992), or pBH1464 was used for plasmid shuffling and characterization of the different pdi1 mutants. M4143 was constructed by gene replacement (Scherer and Davis, 1979) of PRC1 using plasmid pWI-17 (Holst et al., 1996).
Escherichia coli strains.
DH5α (Sambrook et al., 1989) was used for plasmid propagation, BMH71-18mutS (Kramer et al., 1984; Zell and Fritz, 1987), CJ236 (Kunkel et al., 1987), and JM109 (Yanisch-Perron et al., 1985) were used for site-directed mutagenesis. Yeast strain JWY33-2B (MATα ura3-52 leu2-3,112 trp1-1 sec18-1 Δpdi1::HIS3 Δprc1-17) was constructed by crossing M4143 with XCR101-12 (MAT a ura3-52 sec18-1 his3 leu2-3,112) using standard genetic procedures (Sherman, 1991).
Media and Materials
Yeast cells were grown in standard YPD and SC media (Sherman, 1991). E. coli was grown in LB, SOC, and 2× YT medium (Sambrook et al., 1989). Restriction enzymes, T4 DNA polymerase, T4 polynucleotide kinase, T4 DNA ligase, and Klenow polymerase were from Promega (Madison, WI). [35S]methionine was from DuPont NEN (Boston, MA). Zymolase 100-T was from Seikagaku Kogyo (Tokyo, Japan). Fixed Staphylococcus aureus cells were IgGsorb from The Enzyme Center (Malden, MA). Oligonucleotides were from DNA Technology (Aarhus, Denmark). Sequencing was performed using a Taq Dye Deoxy™ terminator cycle sequencing kit on an API 373A DNA Sequencer, both from Applied Biosystems (Foster City, CA).
Plasmid Construction and Mutagenesis
Subcloning and transformation of E. coli and yeast were carried out using standard procedures (Sambrook et al., 1989; Ito et al., 1983). The plasmids used in this study, the resulting PDI1 alleles, and the encoded enzymes are listed in Table I. pBH1464 was constructed by subcloning of a KpnI-SacI fragment from pCT38 (Tachibana and Stevens, 1992) into the same sites in pRS314 (Sikorski and Hieter, 1989). pBH1514 was constructed by subcloning a KpnI-XbaI fragment from pCT38 into the same sites in pSELECT (Lewis and Thompson, 1990). Site-directed mutagenesis was performed as described previously (Lewis and Thompson, 1990) with the modification described by Olesen and Kielland-Brandt (1993). For cloning purposes, a BamHI site was introduced by site-directed mutagenesis into the PDI1 promoter region, generating pBH1857 after reconstitution in an otherwise wild-type context. Pulse-labeling and growth experiments showed no change in phenotype, as compared to the wild-type PDI1 (data not shown). pBH1852, pBH1630, and pBH1692, containing serine residues instead of cysteine residues at the a site, the a′ site, and both the a and the a′ active sites, respectively, were generated by site-directed mutagenesis (Table I). pBH1680 was constructed by recloning of a KpnI-XbaI fragment from pBH1692 into pSELECT in the same sites. pBH1680 was used for site-directed mutagenesis to randomize the two middle amino acids in either of the two active sites (CGHC). Table I lists the mutants reconstituted into an otherwise wild-type PDI1 context, except for the already introduced alterations. Likewise, mutants with serine residues instead of cysteine residues in the most COOH-proximal cysteine in either of the two active sites or in both active sites, were generated by site-directed mutagenesis in pBH1680 followed by a similar reconstitution (Table I). EUG1 was mutated using conventional techniques (Kunkel et al., 1987; Herlitze and Koenen, 1990) and subcloned into pBH1865 to place the gene under PDI1 promoter control.
Table I.
Plasmids Used
| Name | PDI1 allele | Enzyme | ||
|---|---|---|---|---|
| pCT37 | GAL-PDI1 | CGHC-CGHC | ||
| pCT38 | PDI1 | CGHC-CGHC | ||
| pBH1464 | PDI1 | CGHC-CGHC | ||
| pBH1514 | PDI1-1514 | CGHC-CGHC | ||
| pBH1630 | pdi1-1630 | CGHC-SGHS | ||
| pBH1680 | pdi1-1680 | SGHS-SGHS | ||
| pBH1692 | pdi1-1692 | SGHS-SGHS | ||
| pBH1703 | pdi1-1703 | CGSC-SGHS | ||
| pBH1704 | pdi1-1704 | CWSC-SGHS | ||
| pBH1706 | pdi1-1706 | CRPC-SGHS | ||
| pBH1708 | pdi1-1708 | CGGC-SGHS | ||
| pBH1711 | pdi1-1711 | CRRC-SGHS | ||
| pBH1715 | pdi1-1715 | SGHS-CCPC | ||
| pBH1722 | pdi1-1722 | SGHS-CSGC | ||
| pBH1731 | pdi1-1731 | CSGC-SGHS | ||
| pBH1732 | pdi1-1732 | CLYC-SGHS | ||
| pBH1733 | pdi1-1733 | SGHS-CPKC | ||
| pBH1734 | pdi1-1734 | SGHS-CKAC | ||
| pBH1735 | pdi1-1735 | SGHS-CNCC | ||
| pBH1736 | pdi1-1736 | SGHS-CRSC | ||
| pBH1737 | pdi1-1737 | CGCC-SGHS | ||
| pBH1738 | pdi1-1738 | CWLC-SGHS | ||
| pBH1739 | pdi1-1739 | CKIC-SGHS | ||
| pBH1741 | pdi1-1741 | SGHS-CGTC | ||
| pBH1742 | pdi1-1742 | SGHS-CRNC | ||
| pBH1743 | pdi1-1743 | SGHS-CGEC | ||
| pBH1744 | pdi1-1744 | SGHS-CPSC | ||
| pBH1745 | pdi1-1745 | SGHS-CVDC | ||
| pBH1746 | pdi1-1746 | SGHS-CHQC | ||
| pBH1852 | pdi1-1852 | SGHS-CGHC | ||
| pBH1857 | PDI1-1857 | CGHC-CGHC | ||
| pBH1945 | pdi1-1945 | CGHS-SGHS | ||
| pBH1960 | pdi1-1960 | SGHS-CGHS | ||
| pBH1966 | pdi1-1966 | CGHS-CGHS | ||
| pCT58 | eug1-58 | CLHC-CIHC |
Characterization of pdi1 Mutants
Pulse labeling and immunoprecipitation were performed essentially as described by Winther et al. (1991). Yeast cells were grown in SC-trp-ura and starved for sulfur in SC-trp-ura without (NH4)2SO4. [35S]methionine was used for labeling instead of [35S]H2SO4. When indicated, DTT was added to give final concentrations of 5 mM. For pulse labeling, PRC1 alleles encoding wild-type CPY or CPY with an extra N-glycosylation site (CPY-3T; Holst et al., 1996) were reintroduced via plasmids pJW1433 (Ramos et al., 1994) and pBH994, respectively. The randomly mutagenized pdi1 mutants were screened for their ability to grow in the presence of different concentrations of DTT. The final concentration of DTT varied between 0 and 5 mM. Overnight cultures grown in the absence of DTT were diluted 104 times in water, aliquots of 10 μl were placed on freshly made SC plates buffered to pH 5 with 50 mM NaH2PO4, and the plates were subsequently incubated in a CO2 atmosphere to prevent oxidation of the DTT by O2. After 48 and 72 h at 30°C, the mutants were ranked according to the maximal DTT concentration at which they were able to grow. Several independent experiments showed variation in ranking of no more than two positions.
Results
Analysis of the Relative Importance of a and a′ Domains
To evaluate the relative importance of the two thioredoxin-like domains of PDI, mutant forms of the protein were constructed containing either of the two CGHC active site motifs converted to SGHS. Such mutations impair the ability of the mutated active site to engage in disulfide chemistry. Since PDI1 is an essential gene, we used a plasmid-shuffle procedure (Sikorski and Boeke, 1991) to introduce the mutant genes. Mutants were introduced on a plasmid containing a TRP1 selectable marker into a Δpdi1 strain that carried PDI1 on a plasmid with a URA3 marker. After transformation, the cells were forced to lose the plasmid-containing wild-type PDI1 by plating on medium containing 5-fluoro orotic acid, which selects cells that have lost URA3. Cells will be able to grow only if the newly introduced PDI mutant form can complement the chromosomal pdi1 deletion. Consistent with the theory that the essential activity of PDI is dependent on its ability to engage in disulfide chemistry, we found that a SGHS-SGHS2 mutant is not able to complement Δpdi1.
Cells producing the CGHC-SGHS or SGHS-CGHC mutant enzymes as their sole PDI forms showed no growth defect under normal conditions. To investigate protein folding in vivo in these strains, we monitored the rate of intracellular transport of CPY. Cells were pulse-labeled with 35S for 15 min, and were chased with nonradioactive sulfur for 0, 5, and 15 min before lysis and immunoprecipitation of CPY antigen. The precipitates were subjected to SDS-PAGE, and the gels were exposed to x-ray film or PhosphorImager screens. As described in the introduction, CPY undergoes a number of modifications in the secretory pathway that allow estimation of the rate of transport between the ER, Golgi, and vacuole. Fig. 2 A shows that while the rate of CPY maturation is essentially the same in wild-type and CGHC-SGHS mutants, the half time of CPY maturation is 15–20 min in the SGHS-CGHC mutant. Since only folded proteins are allowed to exit the ER, (Gething et al., 1986) this shows that the a domain plays a more important part in folding of CPY than the a′ domain.
Figure 2.

Inactivation of the PDI a domain results in reduced rate of CPY maturation and inability to recover from DTT treatment. (A) Cells were pulse-labeled with 35S-labeled amino acids for 15 min and chased with nonradioactive sulfur for the indicated periods of time. Intracellular CPY was immunoprecipitated, separated by 8% SDS-PAGE, and exposed to PhosphorImager screens. Mutants are identified according to the sequence of their CXXC motifs. (B) Cells were pulse labeled for 20 min as described above, but in the presence of 5 mM DTT. They were then washed with unlabeled DTT-free medium and chased in this medium for the indicated periods of time. The SGHS-CGHC mutant is not able to oxidize and mature p1-proCPY (lane 9), while the wild type (lanes 1–3) and the CGHC-SGHS mutant (lanes 4–6) show similar maturation rates. In accordance with earlier observations (Simons et al., 1995; Holst et al., 1996), the rate of maturation is considerably slower after treatment with DTT. A partial degradation of the CPY antigen, like the one seen in this experiment, is often found under folding defective conditions (Ramos and Winther, 1996).
Treatment of intact yeast or mammalian cells with DTT results in ER accumulation of newly synthesized, disulfide-containing secretory proteins in a reduced state (Braakman et al., 1991; Jämsä et al., 1994). To test the ability of the PDI mutants to oxidize proCPY after DTT treatment, we carried out the following experiment. Cells were treated with 5 mM DTT during labeling with 35S, after which they were washed and chased with nonlabeled, DTT-free medium. As seen in Fig. 2 B (lanes 1, 4, and 7), all strains accumulate p1-proCPY in the ER during a DTT pulse. We find that the rate of maturation is comparable in the wild-type cells and in those expressing the CGHC-SGHS form of PDI. In cells producing the SGHS-CGHC mutant, no mature CPY is found, even 60 min after DTT washout. The simplest explanation for this effect on CPY folding is that reoxidation of ER components is slower in this mutant than in the CGHC-SGHS mutant or the wild type.
Random Mutagenesis of the CGHC Sequence of the Thioredoxin Domains
The redox potential of thioredoxin and DsbA (an E. coli analogue of PDI; Bardwell et al., 1991) is strongly dependent on the identity of the two central amino acid residues of the CGHC motif (Grauschopf et al., 1995; Chivers et al., 1996). To investigate the importance of the redox potential for both growth and ability to fold CPY in vivo, a library of CχχC mutants (χ denotes a random amino acid residue) was constructed. The mutagenesis was carried out on a plasmid encoding the SGHS-SGHS mutant. Two independent libraries of mutants were made so that each library converted only one of the two active sites from SGHS to CχχC. Each pool of mutant plasmids was introduced into a Δpdi1 yeast strain and tested for complementation. Survival required the introduction of both the necessary cysteine residues as well as the mutagenesis of the intervening GH sequence. From the mutant pools, a collection of 10 and 12 clones, respectively, were isolated and sequenced (Table II). We have not analyzed the identity or frequency of noncomplementing mutants. All complementing mutants, along with wild-type PDI, the CGHC-SGHS, and the SGHS-CGHC mutants, were further characterized by sensitivity to DTT. This test was chosen since DTT is known to penetrate into the ER and interfere with the formation of disulfide bonds in nascent disulfide-containing proteins. We adjusted the pH in the plates to 5 and incubated them in a CO2 atmosphere to reduce the spontaneous oxidation of the DTT. The wild-type and the CGHC-SGHS mutant grow on 5 mM DTT. The SGHS-CGHC mutant, which showed increased sensitivity to DTT in the pulse-labeling experiment, also shows a weaker growth on plates containing DTT (Fig. 3). In Table II, the mutants are sorted according their DTT sensitivity. Since the DTT sensitivity is, in most cases, not an all-or-nothing relationship, many of the mutants are classified as having the same sensitivity threshold. Nevertheless, using media containing a spectrum of DTT concentrations, a clear order of the ranking of growth rates was observed within each class. No significant growth phenotype was found under aerobic or anaerobic conditions in the absence of DTT (Fig. 3). In the CχχC mutants of DsbA, an increased DTT sensitivity of the bacteria correlates with a more reducing redox potential (Grauschopf et al., 1995). By analogy, it is likely that a similar ranking of the yeast PDI mutants reflects a ranking of redox potential.
Table II.
DTT Sensitivity of PDI CχχC Mutants
| Enzyme (a domain mutants) | DTT sensitivity threshold | Enzyme (a′ domain mutants) | DTT sensitivity threshold | |||
|---|---|---|---|---|---|---|
| mM | mM | |||||
| CGHC-CGHC | 5 | CGHC-CGHC | 5 | |||
| CGHC-SGHS | 5 | SGHS-CGHC | 3 | |||
| CLYC-SGHS | 2 | SGHS-CCPC | 3 | |||
| CKIC-SGHS | 1 | SGHS-CPKC | 3 | |||
| CGSC-SGHS | 1 | SGHS-CRNC | 3 | |||
| CGCC-SGHS | 0.5 | SGHS-CVDC | 3 | |||
| CGGC-SGHS | 0.5 | SGHS-CHQC | 2 | |||
| CRPC-SGHS | 0.2 | SGHS-CGTC | 1 | |||
| CSGC-SGHS | 0.2 | SGHS-CKAC | 1 | |||
| CWSC-SGHS | 0.2 | SGHS-CGEC | 0.5 | |||
| CWLC-SGHS | 0.2 | SGHS-CNCC | 0.5 | |||
| CRRC-SGHS | 0.2 | SGHS-CRSC | 0.5 | |||
| SGHS-CSGC | 0.5 | |||||
| SGHS-CPSC | 0.5 |
Mutants were plated on selective media containing various concentrations of DTT, as described in Fig. 3, and were monitored for growth after 3 d. The sensitivity does not have an absolute threshold, but it gives a clear ranking (less than two positions) of the mutants as indicated above, even within the same sensitivity class.
Figure 3.
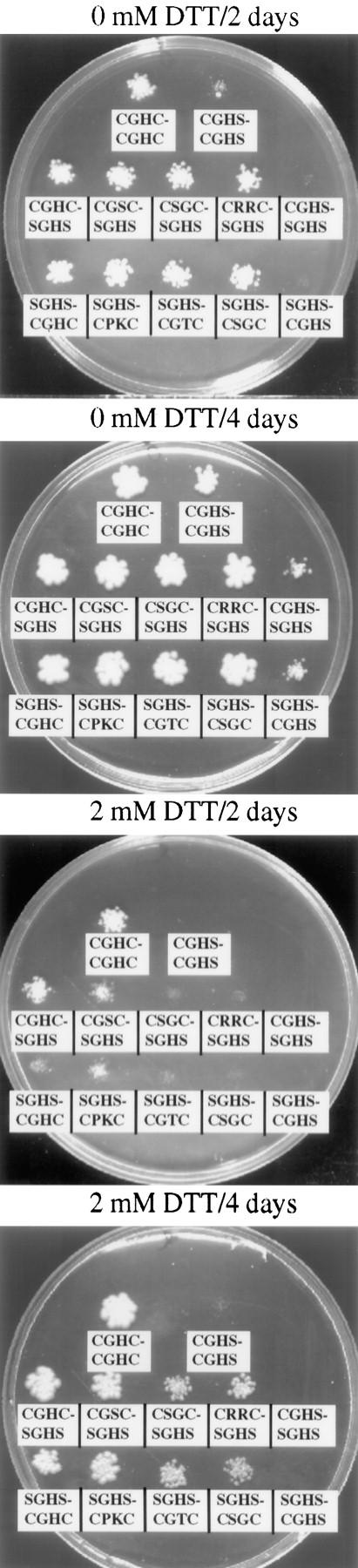
Sensitivity of CχχC mutants to DTT in the growth medium. Diluted suspensions of cells expressing selected PDI1 mutants were plated on media without DTT or with 2 mM DTT. Plates were incubated in a CO2 atmosphere at 30°C and photographed after 2 and 4 d. Mutants are identified by their active-site sequences.
All mutants were analyzed further by pulse-chase labeling and immunoprecipitation of CPY, as described above, and a correlation was found between rate of ER exit and in vivo sensitivity towards DTT. Labeled immunoprecipitates of CPY from representative mutants are shown in Fig. 4. The labeling shows that the rate of transport is reduced considerably for the mutants, as compared to the wild type. The CGSC-SGHS mutant, which is sensitive to 1 mM DTT (Table II), has a half time of maturation only slightly longer than the wild type (8–10 min). The CSGC-SGHS mutant has a half-time of maturation of ∼15 min and a DTT sensitivity threshold of 0.2 mM. In the most DTT-sensitive mutant (CRRC-SGHS), proCPY is matured with a half time of ∼30 min, as compared to 6 min for cells containing wild-type PDI.
Figure 4.

DTT-sensitive PDI mutants have a reduced rate of CPY maturation. Pulse labeling and immunoprecipitation was carried out as described in Fig. 2 A. Mutants are identified by their active-site CXXC motifs; the left panel represents the wild-type a domain.
Oxidation-deficient Mutants
The ability of enzymes in the thioredoxin family to cycle efficiently between an oxidized and reduced state depends on the presence of an intramolecular disulfide bond in the CXXC motif (Fig. 1 B). However, only the more NH2-proximal of the two cysteine residues plays an essential role in the isomerization reaction. This is because only this residue engages in the formation of mixed disulfides with the substrate polypeptide (Fig. 1 C; Wunderlich et al., 1995; Walker et al., 1996).
After considering the effect of random changes to the redox potential of PDI, we wished to analyze the most oxidation-deficient mutants that we could conceivably construct without the loss of isomerase activity. Efficient oxidation is possible only when both active-site cysteines are present (Fig. 1, B and C). Thus, mutants of the CGHS-type should not be able to perform the internal redox chemistry, but should still be able to function as isomerases. It has been shown previously that the CGHA mutant forms of PDI will complement a pdi1 deletion (LaMantia and Lennarz, 1993). Similarly, overexpressed EUG1, a PDI homologue with a CXXS motif in both thioredoxin domains, can support growth of a Δpdi1 strain (Tachibana and Stevens, 1992). We constructed the CGHS-CGHS, CGHS-SGHS, and SGHS-CGHS mutants and found, in accordance with previous results, that they did support growth. However, the three CGHS mutants all show reduced rates of growth. Furthermore, they are extremely sensitive to DTT on plates. While the slowest growth rate is seen for the CGHS-SGHS mutant (Fig. 5 A), pulse-chase experiments showed that the CPY folding was most severely affected in the SGHS-CGHS mutant (Fig. 5 B). This shows that there is no direct correlation between rate of growth and rate of intracellular CPY folding. This conclusion is supported further by the observation that the rate of CPY maturation in the CRRC-SGHS mutant is as slow as that of the CGHS-SGHS mutant, while the growth rate of the former is essentially the same as wild type. This also suggests the existence of specific PDI substrates that are essential for optimal cell growth and more dependent than CPY on PDI catalysis.
Figure 5.


PDI CGHS mutants show reduced rates of growth and CPY maturation. (A) Cells producing CGHS active-site mutant forms of PDI were streaked on YPD and photographed after 3 d. (B) The same mutants were subjected to pulse labeling and immunoprecipitation of CPY, as described in Fig. 2 A.
Reconstruction of a Thioredoxin-like Active Site in Eug1p
The EUG1 gene was originally isolated by its homology to PDI1 (Tachibana and Stevens, 1992). The Eug1p protein is uniquely different from PDI since it has CLHS-CIHS in place of the of CGHC-CGHC active site sequences. The expression of EUG1 under normal growth conditions is ∼5–10% of that of PDI1 (Tachibana and Stevens, 1992). We found that EUG1 complemented the growth defect of a pdi1 deletion when expressed under control of the PDI1 promoter (Fig. 6). It did so, however, only to an extent similar to that of the CGHS-CGHS mutant of PDI, i.e., the growth rate was considerably reduced compared to wild type. This suggested that the main reason that EUG1 does not fully complement Δpdi1 is the absence of the COOH-proximal active-site cysteines. To investigate this hypothesis, we attempted to convert Eug1p to a more PDI-like enzyme by changing the CLHS-CIHS sequences to genuine thioredoxin-type active sites CLHC-CIHC. Indeed, under control of the PDI1 promoter, this EUG1 mutant allowed a Δpdi1 strain to grow at an essentially wild-type rate (Fig. 6). The obvious interpretation of this result is that the mutant Eug1p gained the ability to form internal cystine bridges in the two active sites, allowing oxidative activity as well as disulfide isomerase activity.
Figure 6.

Mutants of Eug1p converting CXXS active site motifs to CXXC show improved growth. Shown are: yeast cells deleted for the chromosomal copies of PDI1 and EUG1 containing plasmids with PDI1 (PDI), wild-type EUG1 under control of the PDI1 promoter (Eug1p) or a mutant form of EUG1 containing a CIHC-CLHC active site sequences (Eug1p-CXXC). EUG1 expressed from the PDI1 promoter is able to complement a pdi1 deletion, albeit with a considerably reduced growth rate. The growth deficiency of this strain is essentially corrected by the introduction of CXXC active site motifs.
Assaying Folding In Vivo Using Glycosyl Modification of Buried Sites
To test the rate of folding in the PDI mutants more directly, we used a mutant form of CPY in which a new site for asparagine-linked glycosylation has been introduced at a buried position in the protein structure. The mutant protein, CPY-3T (Holst et al., 1996), is useful because the level of glycosylation at the new site is dependent on the folding rate of the protein. Thus, no glycosylation of this site takes place under normal growth conditions, while in the presence of DTT the mutant shows ∼40% modification of the buried acceptor site. Overglycosylation results in a shift in SDS-PAGE mobility approximately equal to the p2 modification, making precise evaluation of the level of overglycosylation difficult when the protein is not substantially retained in the ER. We have therefore expressed the mutants in a sec18 strain that is blocked in the ER exit at the nonpermissive temperature (Stevens et al., 1982). The only band immunoprecipitated from the PDI1 wild-type control strain corresponds to p1-proCPY (Fig. 7, lane 2), indicating that no modification is taking place at the buried site. The strains expressing various mutant PDIs show varying degrees of overglycosylation of CPY-3T. The level of overglycosylation is highest in the pdi1 mutants with the slowest rate of ER exit found for wild-type CPY. This confirms that the rate of CPY folding is reduced in these mutants.
Figure 7.

PDI1 mutants show glycosylation of a buried sequence for N-linked glycosylation in CPY-3T. Cells were 35S-labeled for 30 min without chase. Labeling was carried out at 37°C, which is restrictive for the temperature-sensitive sec18-1 mutation. This blocks ER exit and ensures that no p2proCPY is formed. The band marked p1* thus arises from glycosylation of the buried site. In cells expressing wild-type PDI1 or the CGHC-SGHS mutant, no p1* form is observed, while an increasing fraction of the newly synthesized CPY-3T is modified in lanes 4–7. Lane 1 shows wild-type CPY in the same strain at the permissive temperature.
Discussion
In this work, we have addressed some basic questions concerning the function of PDI in vivo. There is ample biochemical evidence that PDI activity is exerted through the action of two thioredoxin-like domains, a and a′, which have a very high level of mutual similarity. Using rat PDI expressed in yeast, Laboissiere et al. (1995) showed that the simultaneous mutation of the CGHC active-site sequences of these domains to SGHS generates an enzyme that is unable to support yeast growth. Our mutagenesis of yeast PDI does not support an earlier proposal that yeast PDI possesses an essential “chaperone” function (LaMantia and Lennarz, 1993). The combined evidence indicate that the ability of PDI to engage in disulfide chemistry is essential for the viability of yeast.
Considering the mutual sequence similarity and the in vitro evidence that both thioredoxin domains are able to engage in disulfide chemistry, we wished to investigate the relative importance of the two domains for folding of CPY. CPY is an exceedingly well-characterized model substrate in terms of biosynthesis and folding both in vivo and in vitro (Stevens et al., 1982; Winther et al., 1991; Ramos et al., 1994; Holst et al., 1996), and requires the presence of at least some of its five disulfide bonds for folding (Jämsä et al., 1994). We have used two methods for monitoring the folding rate in vivo. One relies on the presence of a quality control system in the ER (Ramos et al., 1994; Gething et al., 1986). This mechanism ensures that only correctly folded proteins are allowed to exit the ER and proceed through the secretory pathway. Our other assay for in vivo folding takes advantage of a mutant form of CPY that contains an N-linked glycosylation site at a buried position in the protein structure. This site becomes partially glycosylated if folding is compromised (Holst et al., 1996). In both assays for folding, we find that a mutant enzyme that contains only an active a site (the CGHC-SGHS mutant) is as efficient in folding CPY as the wild-type enzyme. The SGHS-CGHC mutant, on the other hand, shows a significantly slower CPY folding. This is the first in vivo evidence that the two thioredoxin domains are functionally different. This might reflect differences in substrate specificity between the domains, but as we shall see below, could also be explained by differences in redox potential.
The intramolecular disulfide bond between the two cysteine residues of thioredoxin-like proteins can be characterized with respect to its redox potential. Thioredoxins from various sources have fairly reducing redox potentials, while PDIs are generally characterized by a very oxidizing active-site cystine bond (Bardwell and Beckwith, 1993). This is true in spite of the observation that the three-dimensional structures of the various thioredoxin-like domains are virtually superimposable (Martin, 1995; Kemmink et al., 1996). In DsbA, an enzyme from E. coli with a function similar to that of PDI, the relevance of the two central residues in the CGHC motif has been investigated extensively (Grauschopf et al., 1995). Changing these residues had marked effects on the redox potential of the enzyme. DsbA mutants showed increased sensitivity to DTT for folding of a chimeric malF-β-galactosidase fusion protein in vivo. Importantly, the sensitivity was inversely related to the oxidative power, which was determined on the purified mutant enzymes in vitro. Thus, it appears that the wild-type E. coli version of PDI is able to overcome the reducing power of DTT in an in vivo folding reaction. In yeast and higher eukaryotes, DTT is able to penetrate into the cells and inhibit the folding of disulfide bonded proteins (Braakman et al., 1991). In particular, this has been shown for CPY in yeast (Jämsä et al., 1995; Simons et al., 1995). We find that wild-type cells are seriously inhibited for growth on plates containing 10 mM DTT at pH 5 under anaerobic conditions; however, cells will grow at 5 mM DTT. The ability of cells to grow on DTT-containing medium is strongly dependent on PDI function. We have shown that the CGHC-SGHS and SGHS-CGHC mutants have different growth rates on DTT plates. While the CGHC-SGHS mutant grew as well as the wild type on 5 mM DTT, the SGHS-CGHC mutant showed increased sensitivity and grew like the wild type only on plates containing 3 mM DTT or less. This is in accordance with the observation on human PDI that the a domain is more oxidizing than the a′ domain (Darby and Creighton, 1995b ). It is also interesting to compare these data with results from Walker et al. (1996), following the oxidative refolding of RNase using similar mutants of human PDI in vitro. In these experiments, the oxidative folding of RNase was faster in the presence of a PDI with a mutated a′ site than in the presence of a PDI with a mutated a site. Taken together, these results indicate that the initial oxidative formation of disulfide bonds may be a rather important aspect of PDI function. The difference in the rate of CPY folding by PDI mutants disrupted in the a and a′ domains might reflect differences in their ability to oxidize substrate. Mutagenesis of the cysteines engaged in disulfide bonds in CPY has shown that several of these are important for folding in vivo (Jakobsen, A., and J.R. Winther, unpublished observations). However, the actual rates of ER exit probably cannot be directly correlated to in vitro observations since characterization of CPY propeptide mutants has shown that rates of folding in vivo and in vitro can only be related qualitatively (Lunde, C., and J.R. Winther, unpublished observations).
To investigate the importance of the redox potential of PDI, we randomly mutagenized the two central amino acid residues in the CGHC motif in both the a and a′ domains. The mutants were initially selected for complementation of a chromosomal pdi1 deletion. Sequencing a number of randomly chosen mutants showed a clear overrepresentation of the helix-breaking residues Gly and Pro (Table II). This bias is not unreasonable considering the presence of the CGHC sequence at the beginning of helix 2 of the thioredoxin fold (Fig. 1 A). Apart from this trend, we do not find any obvious characteristics or similarities among these sequences. The mutants showed little or no growth phenotype under normally oxidizing conditions. The screening procedure, however, would probably implicitly select against mutant forms giving very slow growth phenotypes. Many of the mutants proved to be extremely DTT sensitive (Table II). A priori, it might not be obvious that DTT should inhibit growth of the cells because of its effect on disulfide bond formation. There could, in principle, be several reasons for the toxic effects of DTT. These results, however, indicate a direct link between PDI structure/function and growth rate on various concentrations of DTT, and they imply that toxicity is evoked specifically through inhibition of the folding of disulfide-bonded secretory proteins. In accordance with this hypothesis, invertase, a secretory protein without disulfide bonds, is secreted normally in the presence of DTT at concentrations that completely blocked proCPY ER exit (data not shown; Jämsä et al., 1994). This suggests that the secretory pathway as such is not perturbed by DTT treatment, but that the effect is specific to proteins containing disulfide bonds. By analogy to DsbA, we propose that our DTT-sensitive PDI mutants are altered in their redox potential (Fig. 4).
The mutants are affected in their rate of CPY folding in the ER, as demonstrated by the reduced rate of exit from the ER of CPY in the PDI mutant backgrounds. The reduced folding rate is also seen in a more direct way by the observation that CPY-3T (Holst et al., 1996) is glycosylated at a buried N-glycosylation site in the PDI mutants but not in the wild type. Thus, the overglycosylation of the CPY-3T mutant would strongly suggest that the PDI mutants are affected in early folding events associated with the introduction of disulfide bonds. The importance of the redox potential for “PDI function” has also been highlighted in studies by Chivers et al. (1996). Here, a signal peptide was introduced in front of E. coli thioredoxin to target it to the ER. In such a construct, thioredoxin is not able to complement a PDI deletion. However, CχχC mutants were selected by their ability to complement a pdi1 deletion, and it was found that only mutants with a more oxidizing redox potential could complement the yeast PDI1 deletion. This supports the notion that oxidation is an important, albeit not essential, function of PDI in terms of supporting yeast growth.
To examine extreme effects on deficiency in oxidative power, we constructed CGHS mutants that are unable to form the normal active site disulfide bonds. It is likely that such mutations would not abolish all PDI activity, since they are not lethal for yeast and CHGS mutants are still able to catalyze oxidation reactions by formation of a mixed disulfide with glutathione (Wunderlich et al., 1995; Walker et al., 1996). CPY folding in cells expressing the CGHS-SGHS and SGHS-CGHS mutants was not significantly slower than in the most DTT-sensitive CχχC mutant (e.g., CRRC-SGHS). Nascent proCPY might be oxidized even in the absence of the oxidative power of PDI by the endogenous GSH-GSSG redox buffer in the ER (Hwang at al., 1992) or by another other PDI homologue. MPD1, for example, is able to suppress the lethality of a pdi1 deletion when expressed from a high copy plasmid (Tachikawa et al., 1995) or under the control of the PDI1 promoter (Holst, B., and J.R. Winther, unpublished observations). Although this enzyme is not normally produced at levels that would sustain growth in the absence of PDI, it may provide a background of oxidizing power seen in the CGHS mutants.
Despite the similarity between the most extreme CχχC mutants and the CGHS mutants in the rate of CPY folding, there is a marked difference in the way they affect the growth rate. This suggests that other substrates, which are required for optimal growth rates, have a stronger requirement for the oxidative power of PDI. It is somewhat surprising that the CGHS-SGHS mutant grows more slowly than the SGHS-CGHS mutant, since it seems to contradict the conclusion that the a site is more important for CPY folding than the a′ site. However this may be because the a′ domain is more important for the growth-essential substrate(s) than the a domain.
The yeast PDI-like protein Eug1p does not contain the second cysteine residue of the CXXC motif, yet it is able to sustain viability when overexpressed. EUG1, under the control of the PDI1 promoter, complements a pdi1 deletion, and the growth rate of such cells is comparable to that of a CGHS-CGHS PDI mutant. In view of the current interest in the relevance of PDI activity for yeast survival, we investigated the effect of mutating the CLHS-CIHS active-site sequences of Eug1p “back” to thioredoxin-like structures: CLHC-CIHC. Presumably, wild-type Eug1p can isomerize disulfide bonds, but it can oxidize only by first forming a mixed disulfide with GSH. The altered Eug1p should be able to form an internal active-site disulfide bond, which would greatly facilitate its ability to act as an oxidant. The observation that these mutants, when expressed from the PDI1 promoter, complemented the pdi1 deletion to an almost wild-type growth rate indicates that genuine PDI activity can be achieved by the altered Eug1p. It also underscores the importance of the oxidative function of PDI. The implication that Eug1p is able to adapt a thioredoxin-type mechanism is interesting from both an enzymatic and an evolutionary point of view. Sequencing of the yeast genome has revealed an ancient duplication that is reflected the presence of two homologous copies of most genes. It is likely that EUG1 and PDI1 constitute such a pair since they belong to the same duplication “block” (http://acer.gen.tcd.ie/∼khwolfe/yeast/topmenu.html). Also, the apparent absence of Eug1p homologues of nonyeast origin suggests that Eug1p has evolved from PDI fairly recently. This may also explain why the enzyme mechanism can be changed by a single amino acid substitution. The fact that CPY folding occurs as slowly in the Eug1p mutant (not shown) as in a CRRC-SGHS PDI mutant is not surprising since it is probably not very oxidizing. In vitro characterization is in progress to compare biochemical properties of both wild-type and mutant forms of PDI and Eug1p.
Acknowledgments
We wish to thank Anette W. Bruun for excellent technical assistance, as well as Morten C. Kielland-Brandt, Mathilde Lerche, and Vibeke Westphal for critical reading of the manuscript. Nigel Darby is thanked for stimulating discussions and for providing data before publication.
Abbreviations used in this paper
- CPY
carboxypeptidase Y
- GSSG
glutathione disulfide
- PDI
protein disulfide isomerase
Footnotes
Address all correspondence to Dr. Jakob R. Winther, Department of Yeast Genetics, Carlsberg Laboratory, Gamle Carlsberg Vej 10, DK-2500 Copenhagen Valby, Denmark. Tel: +45 3327 5282. Fax: +45 3327 4765. E-mail: jrw@crc.dk
C. Tachibana was supported in part by the PSU Office of International Cooperative Programs Global Fund.
2. We have adopted a nomenclature for mutants in PDI in which the amino acid sequence of a and a′ active sites, respectively, are written out in single-letter abbreviation.
References
- Bardwell JC, Beckwith J. The bonds that tie: catalyzed disulfide bond formation [comment] Cell. 1993;74:769–771. doi: 10.1016/0092-8674(93)90455-y. [DOI] [PubMed] [Google Scholar]
- Bardwell JC, McGovern K, Beckwith J. Identification of a protein required for disulfide bond formation in vivo. Cell. 1991;67:581–589. doi: 10.1016/0092-8674(91)90532-4. [DOI] [PubMed] [Google Scholar]
- Braakman I, Hoover-Litty H, Wagner KR, Helenius A. Folding of influenza hemagglutinin in the endoplasmic reticulum. J Cell Biol. 1991;114:401–411. doi: 10.1083/jcb.114.3.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivers PT, Laboissiere MC, Raines RT. The CXXC motif: imperatives for the formation of native disulfide bonds in the cell. EMBO (Eur Mol Biol Orgin) J. 1996;15:2659–2667. [PMC free article] [PubMed] [Google Scholar]
- Creighton TE, Hillson DA, Freedman RB. Catalysis by protein-disulphide isomerase of the unfolding and refolding of proteins with disulfide bonds. J Mol Biol. 1980;142:43–62. doi: 10.1016/0022-2836(80)90205-3. [DOI] [PubMed] [Google Scholar]
- Darby NJ, Creighton TE. Characterization of the active site cysteine residues of the thioredoxin-like domains of protein disulfide isomerase. Biochemistry. 1995a;34:16770–16780. doi: 10.1021/bi00051a027. [DOI] [PubMed] [Google Scholar]
- Darby NJ, Creighton TE. Functional properties of the individual thioredoxin-like domains of protein disulfide isomerase. Biochemistry. 1995b;34:11725–11735. doi: 10.1021/bi00037a009. [DOI] [PubMed] [Google Scholar]
- Edman JC, Ellis L, Blacher RW, Roth RA, Rutter WJ. Sequence of protein disulphide isomerase and implications of its relationship to thioredoxin. Nature (Lond) 1985;317:267–270. doi: 10.1038/317267a0. [DOI] [PubMed] [Google Scholar]
- Farquhar R, Honey N, Murant SJ, Bossier P, Schultz L, Montgomery D, Ellis RW, Freedman RB, Tuite MF. Protein disulfide isomerase is essential for viability in Saccharomyces cerevisiae. . Gene (Amst) 1991;108:81–89. doi: 10.1016/0378-1119(91)90490-3. [DOI] [PubMed] [Google Scholar]
- Gething M-J, McCammon K, Sambrook J. Expression of wild-type and mutant forms of influenza hemagglutinin: the role of folding in intracellular transport. Cell. 1986;46:939–950. doi: 10.1016/0092-8674(86)90076-0. [DOI] [PubMed] [Google Scholar]
- Givol D, DeLorenzo F, Goldberger RF, Anfinsen CB. Disulfide interchange and the three-dimentional structure of proteins. Proc Natl Acad Sci USA. 1965;53:676–684. doi: 10.1073/pnas.53.3.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grauschopf U, Winther JR, Korber P, Zander T, Dallinger P, Bardwell JC. Why is DsbA such an oxidizing disulfide catalyst? . Cell. 1995;83:947–955. doi: 10.1016/0092-8674(95)90210-4. [DOI] [PubMed] [Google Scholar]
- Günther R, Bräuer C, Janetzky B, Förster HH, Ehbrecht IM, Lehle L, Küntzel H. The Saccharomyces cerevisiaeTRG1 gene is essential for growth and encodes a lumenal endoplasmic reticulum glycoprotein involved in the maturation of vacuolar carboxypeptidase. J Biol Chem. 1991;266:24557–24563. [PubMed] [Google Scholar]
- Herlitze S, Koenen M. A general and rapid mutagenesis method using polymerase chain reaction. Gene (Amst) 1990;91:143–147. doi: 10.1016/0378-1119(90)90177-s. [DOI] [PubMed] [Google Scholar]
- Holst B, Bruun AW, Kielland-Brandt MC, Winther JR. Competition between folding and glycosylation in the endoplasmic reticulum. EMBO (Eur Mol Biol Orgin) J. 1996;15:3538–3546. [PMC free article] [PubMed] [Google Scholar]
- Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science (Wash DC) 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jämsä E, Simonen M, Makarow M. Selective retention of secretory proteins in the yeast endoplasmic reticulum by treatment of cells with a reducing agent. Yeast. 1994;10:355–370. doi: 10.1002/yea.320100308. [DOI] [PubMed] [Google Scholar]
- Kemmink J, Darby NJ, Dijkstra K, Nilges M, Creighton TE. Structure determination of the N-terminal thioredoxin-like domain of protein disulfide isomerase using multidimensional heteronuclear 13C/15N NMR spectroscopy. Biochemistry. 1996;35:7684–7691. doi: 10.1021/bi960335m. [DOI] [PubMed] [Google Scholar]
- Kramer B, Kramer W, Fritz H-J. Different base/base mismatches are corrected with different efficiencies by the methyl-directed DNA mismatch-repair system in E. coli. . Cell. 1984;38:879–887. doi: 10.1016/0092-8674(84)90283-6. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–381. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- Kurjan J. Alpha-factor structural gene mutations in Saccharomyces cerevisiae: effects on alpha-factor production and mating. Mol Cell Biol. 1985;5:787–796. doi: 10.1128/mcb.5.4.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laboissiere MC, Sturley SL, Raines RT. The essential function of protein-disulfide isomerase is to unscramble non-native disulfide bonds. J Biol Chem. 1995;270:28006–28009. doi: 10.1074/jbc.270.47.28006. [DOI] [PubMed] [Google Scholar]
- LaMantia M, Miura T, Tachikawa H, Kaplan HA, Lennarz WJ, Mizunaga T. Glycosylation site binding protein and protein disulfide isomerase are identical and essential for cell viability in yeast. Proc Natl Acad Sci USA. 1991;88:4453–4457. doi: 10.1073/pnas.88.10.4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMantia ML, Lennarz WJ. The essential function of yeast protein disulfide isomerase does not reside in its isomerase activity [see comments] Cell. 1993;74:899–908. doi: 10.1016/0092-8674(93)90469-7. [DOI] [PubMed] [Google Scholar]
- Lebeche D, Lucero HA, Kaminer B. Calcium binding properties of rabbit liver protein disulfide isomerase. Biochem Biophys Res Commun. 1994;202:556–561. doi: 10.1006/bbrc.1994.1964. [DOI] [PubMed] [Google Scholar]
- Lewis MK, Thompson DV. Efficient site directed in vitro mutagenesis using ampicillin selection. Nucleic Acids Res. 1990;18:3439–3443. doi: 10.1093/nar/18.12.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JL. Thioredoxin—a fold for all reasons. Structure. 1995;3:245–250. doi: 10.1016/s0969-2126(01)00154-x. [DOI] [PubMed] [Google Scholar]
- Olesen K, Kielland-Brandt MC. Altering substrate preference of carboxypeptidase Y by a novel strategy of mutagenesis eliminating wild type background. Protein Engin. 1993;6:409–415. doi: 10.1093/protein/6.4.409. [DOI] [PubMed] [Google Scholar]
- Ramos C, Winther JR, Kielland-Brandt MC. Requirement of the propeptide for in vivo formation of active yeast carboxypeptidase Y. J Biol Chem. 1994;269:7006–7012. [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Scherens B, Dubois E, Messenguy F. Determination of the sequence of the yeast YCL313 gene localized on chromosome III. Homology with the protein disulfide isomerase (PDI gene product) of other organisms. Yeast. 1991;7:185–193. doi: 10.1002/yea.320070212. [DOI] [PubMed] [Google Scholar]
- Scherer S, Davis RW. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc Natl Acad Sci USA. 1979;76:4951–4955. doi: 10.1073/pnas.76.10.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Boeke JD. In vitro mutagenesis and plasmid shuffling: from cloned gene to mutant yeast. Methods Enzymol. 1991;194:302–318. doi: 10.1016/0076-6879(91)94023-6. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons JF, Ferro-Novick S, Rose MD, Helenius A. BiP/Kar2p serves as a molecular chaperone during carboxypeptidase Y folding in yeast. J Cell Biol. 1995;130:41–49. doi: 10.1083/jcb.130.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens TH, Esmon B, Schekman R. Early stages in the yeast secretory pathway are required for transport of carboxypeptidase Y to the vacuole. Cell. 1982;30:439–448. doi: 10.1016/0092-8674(82)90241-0. [DOI] [PubMed] [Google Scholar]
- Tachibana C, Stevens TH. The yeast EUG1gene encodes an endoplasmatic reticulum protein that is functionally related to protein disulfide isomerase. Mol Cell Biol. 1992;12:4601–4611. doi: 10.1128/mcb.12.10.4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachikawa H, Takeuchi Y, Funahashi W, Miura T, Gao XD, Fujimoto D, Mizunaga T, Onodera K. Isolation and characterization of a yeast gene, MPD1, the overexpression of which suppresses inviability caused by protein disulfide isomerase depletion. FEBS Lett. 1995;369:212–216. doi: 10.1016/0014-5793(95)00750-4. [DOI] [PubMed] [Google Scholar]
- Walker KW, Lyles MM, Gilbert HF. Catalysis of oxidative protein folding by mutants of protein disulfide isomerase with a single active-site cysteine. Biochemistry. 1996;35:1972–1980. doi: 10.1021/bi952157n. [DOI] [PubMed] [Google Scholar]
- Winther JR, Stevens TH, Kielland-Brandt MC. Yeast carboxypeptidase Y requires glycosylation for efficient intracellular transport, but not for vacuolar sorting, in vivo stability, or activity. Eur J Biochem. 1991;197:681–689. doi: 10.1111/j.1432-1033.1991.tb15959.x. [DOI] [PubMed] [Google Scholar]
- Wunderlich M, Otto A, Maskos K, Mucke M, Seckler R, Glockshuber R. Efficient catalysis of disulfide formation during protein folding with a single active-site cysteine. J Mol Biol. 1995;247:28–33. doi: 10.1006/jmbi.1995.0119. [DOI] [PubMed] [Google Scholar]
- Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene (Amst) 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- Zell R, Fritz H-J. DNA mismatch-repair in Escherichia colicounteracting the hydrolytic deamination of 5-methyl-cytosine residues. EMBO (Eur Mol Biol Orgin) J. 1987;6:1809–1815. doi: 10.1002/j.1460-2075.1987.tb02435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]