

Abstract
We have investigated the function of p55CDC, a mammalian protein related to Cdc20 and Hct1/Cdh1 in Saccharomyces cerevisiae, and Fizzy and Fizzy-related in Drosophila. Immunofluorescence studies and expression of a p55CDC-GFP chimera demonstrate that p55CDC is concentrated at the kinetochores in M phase cells from late prophase to telophase. Some p55CDC is also associated with the spindle microtubules and spindle poles, and some is diffuse in the cytoplasm. At anaphase, the concentration of p55CDC at the kinetochores gradually diminishes, and is gone by late telophase. In extracts prepared from M phase, but not from interphase HeLa cells, p55CDC coimmunoprecipitates with three important elements of the M phase checkpoint machinery: Cdc27, Cdc16, and Mad2. p55CDC is required for binding Mad2 with the Cdc27 and Cdc16. Thus, it is likely that p55CDC mediates the association of Mad2 with the cyclosome/anaphase-promoting complex. Microinjection of anti-p55CDC antibody into mitotic mammalian cells induces arrest or delay at metaphase, and impairs progression of late mitotic events. These studies suggest that mammalian p55CDC may be part of a regulatory and targeting complex for the anaphase-promoting complex.
Progression through mitosis requires regulated proteolysis. Anaphase onset and the transition from M to G1 phase are cued by protein destruction. A number of putative proteolysis targets have been identified, including Pds11 (precocious division of sister chromatids), Mcd1/Scc1 (mitotic chromosome determinant/sister chromatid cohesion), and Asc1 (anaphase spindle elongation) in S. cerevisiae (Cohen-Fix et al., 1996; Yamamoto et al., 1996; Guacci et al., 1997; Michaelis et al., 1997; Juang et al., 1997), Cut2 in Schizosaccharomyces pombe (Funabiki et al., 1996a ; Funabiki et al., 1996b ), and mitotic cyclins in a variety of species (Holloway et al., 1993; King et al., 1995; Sudakin et al., 1995). Proteolysis of target proteins occurs within the 26S proteosome. Substrates are first targeted for destruction by polyubiquitination through the sequential actions of three enzymes: a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin–ligase complex (E3). In M phase, an E3 complex known as the anaphase-promoting complex (APC) or cyclosome ubiquitinates, target proteins that contain a specific sequence motif termed the destruction box (Glotzer et al., 1991). Recent evidence indicates that the timing of substrate proteolysis in M phase occurs by regulating substrate ubiquitination by the APC. However, since different substrates (e.g., type A and B cyclins) appear to be proteolyzed at different times in mitosis (Sigrist et al., 1995), simply turning the APC on or off cannot serve as the only regulatory mechanism.
A potential pathway for regulating APC activity is through a mitotic checkpoint that monitors proper chromosome attachment to the spindle. This checkpoint, called the spindle checkpoint, guards the integrity of the cell division by monitoring interactions of the mitotic spindle and the kinetochores (reviewed in Rudner and Murray, 1996; Gorbsky, 1997; Nicklas, 1997; Hardwick, 1998). In the presence of one or more misaligned chromosomes, the checkpoint temporarily inhibits the onset of anaphase until all chromosomes achieve proper bipolar attachment to the spindle. Control of this checkpoint appears to be based on a mechanochemical system that senses the presence or absence of physical tension mediated by microtubule attachment at the kinetochores of chromosomes (Li and Nicklas, 1995). Tension also regulates expression of a kinetochore phosphoepitope detected by the 3F3/2 monoclonal antibody (Nicklas et al., 1995). Antibody microinjection experiments suggest that this phosphoepitope plays a functional role in maintaining the checkpoint signal until chromosome congression is complete (Campbell and Gorbsky, 1995). Additional elements of the spindle checkpoint were identified in budding yeast (reviewed in Rudner and Murray, 1996). Yeast mutants of MAD1 (mitosis arrest–deficient), MAD2, MAD3, BUB1 (budding uninhibited by benzimidazole), BUB2, and BUB3 genes exhibit defective checkpoint function, progressing through M phase when the spindle is disrupted with microtubule poisons. Human and Xenopus homologs of Mad2 and a murine homolog of Bub1 were identified, indicating that checkpoint components are conserved from yeast to mammals (Chen et al., 1996; Li and Benezra, 1996). Recently, Li et al. (1997) presented evidence that human Mad2 protein binds to the APC in mitotic extracts of HeLa cells. Moreover, depletion and add-back experiments in cycling Xenopus extracts reported in the same study suggest that Mad2 functions as an inhibitor of the APC.
Additional proteins appear to target substrates to the APC at appropriate times. Evidence from yeast and Drosophila suggest that substrate selection may be directed by a family of proteins related to Cdc20, first identified in S. cerevisiae (Hartwell and Smith, 1985). All the known members of the Cdc20 protein family: Cdc20 and Hct1/ Cdh1 of S. cerevisiae (Hartwell and Smith, 1985; Schwab et al., 1997; Visintin et al., 1997), Fizzy and Fizzy-related of Drosophila melanogaster (Dawson et al., 1993; Sigrist and Lehner, 1997), and Slp1 of S. pombe (Matsumoto, 1997), have previously been implicated in cell division regulation. Originally, Cdc20 was found to be required for APC- dependent degradation of Pds1 (Visintin et al., 1997), while the related protein Hct1 was used for targeting yeast mitotic cyclin Clb2 (Schwab et al., 1997). New evidence, however, suggests that Cdc20 may be required in M phase for degradation of both Pds1 and Clb2, while Hct1 may function during G1 (Lim et al., 1998). Recently, a study in S. pombe showed association of the Cdc20 homolog, Slp1, with Mad2 protein (Kim et al., 1998). Similarly, two hybrid and coimmunoprecipitation experiments indicate that Cdc20 interacts with Mad2 as well as with Mad3 in S. cerevisiae (Hwang et al., 1998). In Drosophila, mutation of the fizzy locus causes metaphase arrest in Drosophila embryos (Dawson et al., 1993) and prevents normal degradation of cyclins A and B during mitosis (Dawson et al., 1995; Sigrist et al., 1995), while the protein produced by the fizzy-related gene functions in the degradation of mitotic cyclins in G1 when cells have completed embryonic divisions (Sigrist and Lehner, 1997).
In mammals, p55CDC, a putative member of the Cdc20 protein family, was identified by Weinstein et al. (1994). The level of p55CDC protein fluctuates during the cell cycle. Synthesis is initiated at G1/S; protein levels peak in M phase, and the protein is abruptly degraded at the M/G1 transition (Weinstein, 1997). Degradation is blocked by proteosome inhibitors, suggesting that p55CDC is targeted by ubiquitin-mediated proteolysis. The p55CDC protein also exhibits cell cycle changes in phosphorylation, immunolocalization, and associations with other unknown proteins, including some with kinase activity (Weinstein et al., 1994; Weinstein, 1997).
In this study we investigate the function of the p55CDC protein in M phase in mammalian cells. We demonstrate that p55CDC is part of a complex containing both the Mad2 protein, a component of the spindle checkpoint signaling pathway, and the APC. Moreover, p55CDC appears to mediate the association between the Mad2 protein and the APC. By antibody microinjection experiments we present evidence that p55CDC is required for normal metaphase-to-anaphase transition and for late mitotic events. Our evidence is consistent with a role for p55CDC as a component of a cell cycle–regulated inhibitory complex of APC or in targeting the APC to mitotic substrates whose timely degradation is required for the metaphase-to-anaphase transition, for anaphase chromatid movement, and for exit from mitosis.
Materials and Methods
Cell Culture
Adherent and suspension cell cultures were maintained as previously described (Campbell and Gorbsky, 1995; Renzi et al., 1997). For microinjection experiments, cells were cultured on gridded coverslips sealed with silicone grease over holes cut in the bottoms of 60-mm plastic Petri dishes. The chambers were sterilized by inverting them on a 365-nm UV light box for 20 min before use.
Immunofluorescence
Antibodies were prepared to the COOH-terminal region of p55CDC as previously described (Weinstein et al., 1994). The antibody to Xenopus Mad2 protein (Chen et al., 1996) was provided by Drs. Rey-Huei Chen and Andrew Murray, and the antibody to human Cdc27 (Tugendreich et al., 1995) was provided by Drs. Stuart Tugendreich and Phil Hieter. Monoclonal 3F3/2 antibody, originally produced by Cyert et al. (1988), was used as previously reported (Gorbsky and Ricketts, 1993). Immunofluorescence was essentially performed as described earlier (Gorbsky and Ricketts, 1993; Campbell and Gorbsky, 1995). In brief, for p55CDC and 3F3/2 immunofluorescence, cells on coverslips were simultaneously fixed and extracted for 15 min in 0.2% Triton X-100 in PHEM (60 mM Pipes, 25 mM Hepes at pH 6.9, 10 mM EDTA, and 4 mM MgCl2) containing 2.0% formaldehyde and 100 nM of phosphatase inhibitor microcystin LR (Calbiochem Novabiochem, San Diego, CA). Cells on the coverslips were incubated in primary antibodies for 45 min at a dilution of 1:450 of anti-p55CDC (1.0 μg/ml) or 1:2,500 of 3F3/2 ascites. In some experiments, human scleroderma autoimmune serum (a gift of Dr. J.B. Rattner, University of Calgary) was used at a dilution of 1:500 to label kinetochores. Cy3-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) was used at 1:650 to visualize 3F3/2; fluorescein-conjugated goat anti– rabbit (Jackson ImmunoResearch Laboratories) at 1:450 to visualize anti-p55CDC, and fluorescein-conjugated goat anti–human (Jackson ImmunoResearch Laboratories) at 1:400 to visualize CREST antibodies. DNA was counterstained with DAPI (Sigma Chemical Co., St. Louis, MO) at 0.3 μg/ml in distilled water, or with 0.1 nM Yo-Pro (Molecular Probes, Eugene, OR).
Fluorescently labeled cells were analyzed with a Diaphot™ microscope (Nikon Inc., Melville, NY) equipped with a microchannel plate image intensifier and video rate CCD camera (DAGE-MTI, Michigan City, IN), and was digitized with Image 1 software (Universal Imaging Corp., West Chester, PA). Some images were obtained with a digitally cooled CCD camera (Photometrics Ltd., Tucson, AZ) using Metamorph software (Universal Imaging Corp.). Images were further processed using Metamorph, Corel Photopaint, and Corel Draw software and printed with an 8600 printer (Eastman Kodak Co., Rochester, NY).
Transfection with p55CDC-GFP
The full-length coding sequence for p55CDC was inserted between the HindIII and BamHI sites of pEGFP-N3 obtained from Clontech (Palo Alto, CA). LLC-PK1 cells were subcultured on 22-mm coverslips in 60-mm plastic Petri dishes, and were transfected by calcium phosphate coprecipitation. Control cultures were transfected with the plasmid pEGFP-N1 (Clontech) that coded for GFP without p55CDC. 8–24 h after transfection, the cells were fixed with 4% formaldehyde, counterstained with DAPI, and mounted with Prolong antifade kit (Molecular Probes).
Immunoprecipitation
HeLa cells were obtained from unsynchronized populations, or from cells arrested in M phase with colcemid as described (Renzi et al., 1997). Cells were washed with PBS and used immediately, or were frozen in liquid nitrogen and stored at −70°C. The cells were lysed in APC buffer (20 mM Tris-HCl, pH 7.7, 100 mM KCl, 50 mM sucrose, 0.1 mM CaCl2, 1 mM MgCl2, 0.5% Triton X-100) with 400 nM microcystin LR (Calbiochem Novabiochem) and 5 μg/ml of protease inhibitors (Leupeptin, Pefablock and Pepstatin A) for 5 min on ice with intermittent vortexing. The extracts were centrifuged at 15,000 g for 10 min at 4°C, and the supernatants were used for immunoprecipitation. Polyclonal anti-p55CDC antibody prepared against the full-length bacterially expressed p55CDC (Weinstein et al., 1994), polyclonal anti-Cdc27 prepared against a COOH-terminal fragment of Cdc27 protein (Tugendreich et al., 1995), and a monoclonal anti-Cdc27 antibody obtained commercially (Transduction Laboratories, Lexington, KY) were used for immunoprecipitation experiments. Antibodies were precoupled to either Protein A or Protein G Sepharose (Pierce Chemical Co., Rockford, IL) by incubating 10 μg of antibody with 200 μl of protein bead slurry in the APC buffer for ∼1 h at 4°C. 1,000 μl of the extract was mixed with 200 μl of the antibody/protein bead complex in the APC buffer, and was incubated at 4°C for 3 h. After incubation, the washed immunoprecipitate and the supernatant were collected for Western blotting analysis and immunoprecipitations. In some cases, supernatants from one immunoprecipitation were used a second time for another immunoprecipitation with a different antibody. After the second immunoprecipitations, the washed immunoprecipitate and the remaining supernatants were collected for immunoblotting analysis.
Immunoblotting
Whole cell lysates, cell extracts, immunoprecipitations, and supernatants remaining from immunoprecipitation experiments were prepared in sample loading buffer with β-mercaptoethanol. Electrophoresis was carried out on 5–20% SDS-polyacrylamide gradient gels. The proteins were transferred to an Immobilon-P membrane (Millipore Corp., Bedford, MA). The membranes were blocked with 5% BSA in TBST (10 mM Tris, 150 mM NaCl, 0.05% Tween 20) for 30 min, washed, and incubated either with rabbit anti-p55CDC (1 μg/ml in TBST), mouse monoclonal anti-CDC27 (125 ng/ml in TBST), rabbit anti-Cdc27 (1:6,000 in TBST), rabbit anti-Cdc16 (1:6,000 in TBST), or with rabbit anti-Mad2 (100 ng/ml in TBST) for 1.5 h. The washed membranes were incubated with HRP-conjugated AffiniPure donkey anti–rabbit IgG, AffiniPure goat anti–rabbit IgG, or goat anti–mouse IgG (Jackson ImmunoResearch Laboratories) 1:2,5000 in TBST for 1 h. The proteins were visualized using chemiluminescent Renaissance Kit (Dupont-NEN, Boston, MA). Some immunoblots were reprobed using anti-Cdc16 antibodies after stripping with 0.2 N NaOH.
Live Cell Experiments and Microinjection
Anti-p55CDC antibody concentration was determined with the BCA assay (Pierce Chemical Co.). Antibody was injected in either Tris-glycine (0.05 M Tris, 0.1 M glycine) or KCl-PO4 (0.1 M KCl, 1.7 mM NaCl, 8.1 mM Na2HPO4, 1.5 mM KH2PO4) buffer. Neither buffer showed any effect on normal mitotic progression when injected without antibody. For control microinjections, rabbit IgG (Sigma Chemical Co.) diluted to 8.0 mg/ml with either Tris-glycine or KCl-PO4 was used. Cells were cultured on marked glass coverslips, and microinjections were performed essentially as previously described (Campbell and Gorbsky, 1995). In brief, the medium in the culture chamber containing the coverslip was overlaid with mineral oil to prevent evaporation. The dish was placed on the prewarmed stage of a Diaphot™ microscope (Nikon Inc., Rockville, MD) and maintained at 36–37°C with a warm air curtain incubator (Sage, Boston, MA).
For live cell observations and microinjection, a dry 40× objective (Nikon, Inc.) was used with a long working distance condenser and video rate, charge-coupled device camera. Microinjections were performed with a micromanipulator (Narishige USA, Inc., Greenvale, NY) using freshly pulled glass microneedles. Cells were microinjected at different phases of division, ranging from late prophase to early anaphase. After microinjection, cells were monitored using planapochromat 60× (N.A. 1.4) or neofluar 100× (N.A. 1.3) objectives (Nikon, Inc.) with a short working distance condenser. Images were recorded in one of two ways. Images captured with a video rate CCD camera and 16-frame averages were digitized with Image 1 software after subtracting background. Images were captured alternatively with a digital cooled CCD camera using Metamorph software.
Results
Characterization of Antibodies
Western blotting analysis of cycling and mitotic HeLa cells and cycling PtK1 cells with antibody against the COOH-terminal region of p55CDC reveals a protein band at 55 kD (Fig. 1). Two additional faint protein bands were present in PtK1 cell at ∼40 and 70 kD.
Figure 1.

Expression of p55CDC in proliferating mammalian cells. The antibody recognizes a 55-kD band (arrow) in whole HeLa cell extracts from both cycling cultures (cycl) and after mitotic arrest (mitot). In whole cycling PtK1 cell extracts, the anti-p55CDC antibody recognizes primarily a 55-kD band, although slight reactivity is seen with two other bands at ∼40 and ∼70 kD.
p55CDC is Present Diffusely and Concentrates at the Kinetochores of M Phase Chromosomes
To examine cell cycle–dependent changes in the localization of p55CDC, we performed immunofluorescence studies in cultured HeLa and PtK1 cells. In interphase of HeLa and Ptk1 cells, faint diffuse labeling throughout the cell was observed (Fig. 2). In early prophase, diffuse labeling throughout the cell was evident with a concentration of label within the nucleus. Beginning with late prophase, double-dot labeling was observed at the primary constriction of all chromosomes. Colocalization with human autoimmune antibodies confirmed that double-dot labeling was at the kinetochores. p55CDC was present at the kinetochores of chromosomes from late prophase to telophase in both HeLa (Fig. 2 A) and PtK1 cells (Fig. 2 B). The p55CDC label at the kinetochores was most intense during prometaphase and metaphase. During anaphase, the kinetochore labeling gradually diminished, and ultimately disappeared by late telophase. In cells extracted with 1% CHAPS detergent before fixation, kinetochore labeling in mitotic cells was diminished.
Figure 2.

Immunofluorescent localization of p55CDC in HeLa and PtK1 cells. (A) HeLa cells. The left column shows DNA (DAPI staining), the middle column shows kinetochores (human autoimmune antibody), and the right column shows p55CDC labeling. At prophase, diffuse labeling is seen throughout the cells with a concentration at the kinetochores. The kinetochore localization is apparent from late prophase to telophase. During anaphase, kinetochore labeling diminishes and is lost by the end of telophase. The diffuse cytoplasmic labeling persists until the M-to-G1 transition. (B) PtK1 cells. The left column shows DNA (Yo-Pro staining) and kinetochores (human autoimmune antibody). The middle column demonstrates p55CDC staining at different stages of mitosis where intensity and contrast settings have been optimized for each individual panel. This technique permits visualization of the staining at anaphase and telophase. The right column shows p55CDC labeling in which intensity and contrast settings were kept constant for all stages, allowing comparison of the relative intensity of labeling at different stages of mitosis. The top set of panels, taken at low magnification, shows a field of cells with some in G2 (arrowheads) and one in prophase (arrow). p55CDC relocalizes from the diffuse cytoplasmic/nuclear pool to the kinetochores of chromosomes just before NEB. Kinetochores show different intensities of labeling during prometaphase. At prometaphase, the kinetochores near the spindle poles often possess brighter signals (arrows) compared with ones that are closer to the spindle equator (arrowhead). The box (3 μm on either side of the spindle equator) used to define metaphase is shown. During anaphase, the anti-p55CDC signal gradually diminishes at the kinetochores, and ultimately disappears by late telophase. Bars, 5 μm.
Similarly, in LLC-PK1 cells expressing p55CDC-GFP chimeric protein, the kinetochores were p55CDC-positive from late prophase to late anaphase (Fig. 3 A). In addition to the bright labeling at the kinetochores, p55CDC-GFP was present diffusely, and also concentrated at the spindle poles and along the kinetochore fiber microtubules throughout M phase (Fig. 3 A). Interphase centrosomes also contained concentrations of p55CDC-GFP. In prometaphase cells, the intensity of p55CDC-GFP at the kinetochores varied among chromosomes depending on their degree of congression to the metaphase plate. Chromosomes near one of the spindle poles showed brighter kinetochore labeling than did chromosomes at the spindle equator (Fig. 3 A). In many cases, one or a few chromosomes remained misaligned when all the others had moved to the metaphase plate. These lagging chromosomes always exhibited more intense p55CDC-GFP signals (Fig. 3 A). Similar differences in apparent concentration of p55CDC at kinetochores were seen with immunofluorescence experiments (Fig. 2), but were more evident with p55CDC-GFP. No apparent difference was observed in the intensity of the p55CDC-GFP signal between the leading kinetochore and the trailing kinetochore of individual prometaphase chromosomes (Fig 3 A). Control cells expressing the GFP alone showed only a diffuse fluorescence, and did not exhibit concentrations of fluorescence on mitotic organelles (Fig. 3 B).
Figure 3.
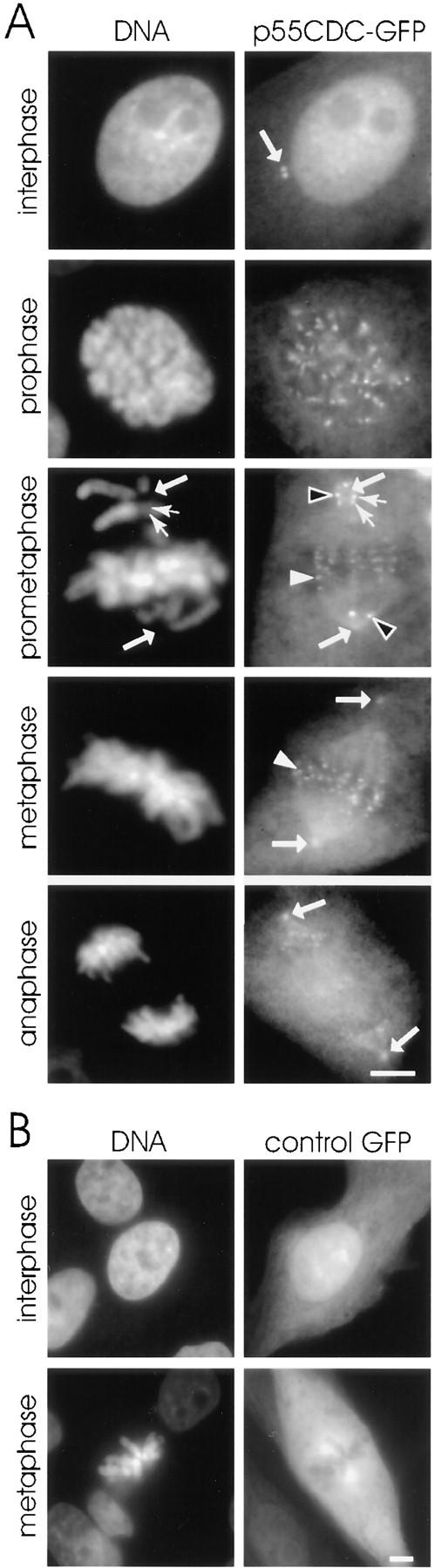
(A) Fixed LLC-PK1 cells expressing chimeric GFP-p55CDC. In interphase cells, a pair of dots at the centrosome (arrow) is brightly labeled throughout the cell cycle. In mitotic cells, the p55CDC-GFP concentrates at the kinetochores from late prophase to late anaphase. A pool of the protein also remains diffusely distributed. In addition, the spindle poles (arrows) and the kinetochore microtubules show concentrations of the p55CDC-GFP. During prometaphase, the intensity of the kinetochore p55CDC-GFP signal is more intense at the kinetochores of unaligned chromosomes located near the spindle poles (black arrowheads) compared with the kinetochores of chromosomes aligned at metaphase plate (white arrowheads). No apparent difference is seen in intensity of the p55CDC-GFP signal between the leading and the trailing kinetochore of individual unaligned chromosome (small arrows in prometaphase row). In anaphase cells, the spindle poles (big arrows) remain p55CDC-GFP-positive while kinetochore labeling diminishes. (B) An interphase and a mitotic LLC-PK1 cell expressing the GFP alone show no specific fluorescent kinetochore, spindle microtubule, or spindle pole signals. Bars, 5 μm.
p55CDC Binds Mad2 and the Anaphase-promoting Complex in Mitotic Cell Extracts
Anti-p55CDC immunoprecipitates made from mitotic HeLa cell extracts (at least 70% mitotic cells) contained Mad2, a checkpoint pathway protein, and Cdc27, a component of the APC (Fig. 4 A). In contrast, extracts prepared from unsynchronized HeLa cell cultures (less than 5% mitotic cells) showed little association between p55CDC and Mad2 or Cdc27.
Figure 4.
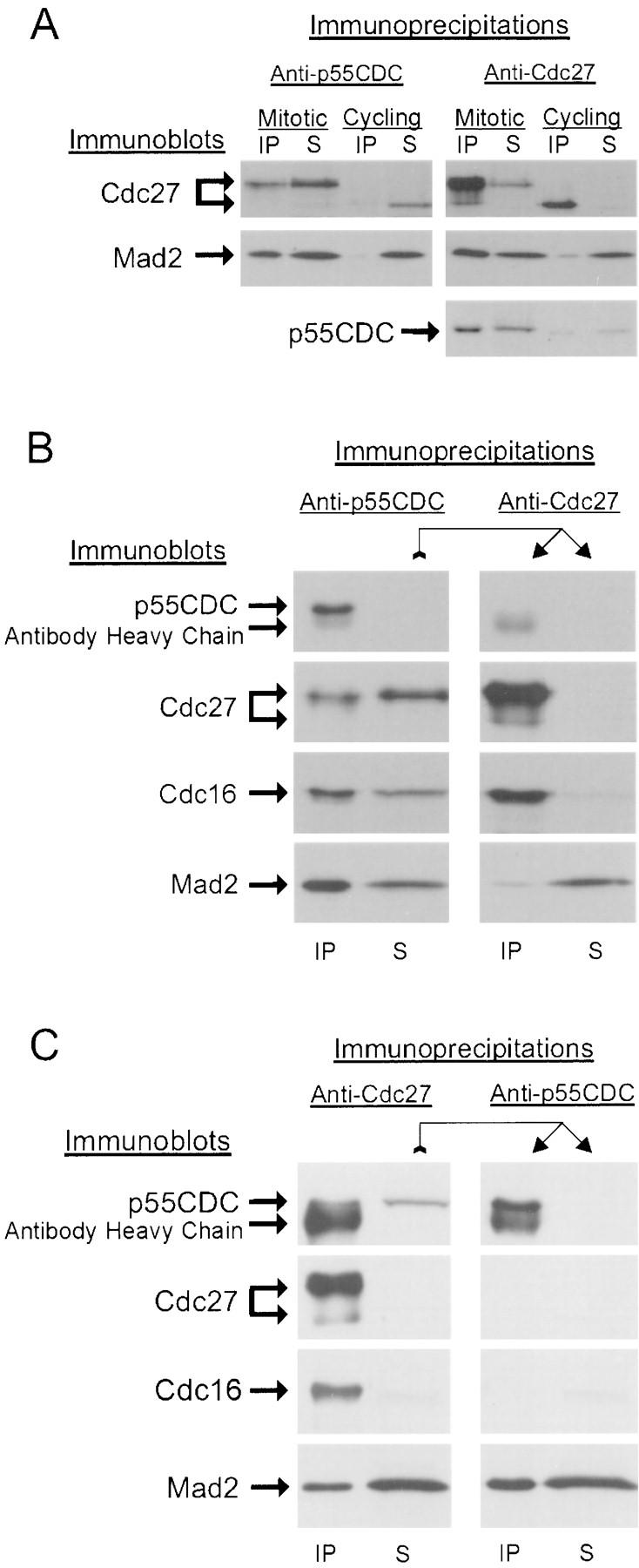
p55CDC binds to Mad2 protein and to the APC. (A) Cdc27 and Mad2 proteins are detected in anti-p55CDC immunoprecipitates from mitotic, but not interphase HeLa cell extracts (Cdc27 protein is indicated in two locations since it undergoes a phosphorylation shift in M phase). Mad2 and p55CDC proteins are detected in anti-Cdc27 immunoprecipitates from mitotic, but not interphase HeLa cell extracts. (B) A sequential immunoprecipitation experiment showing that p55CDC is necessary for Mad2 binding to the APC. A mitotic extract of HeLa cells was first immunodepleted of p55CDC protein (left). Some of the APC remains in the supernatant. The p55CDC-depleted extract was then used in a second immunoprecipitation with anti-Cdc27 antibody. In the absence of p55CDC, almost none of the remaining Mad2 coprecipitates with the Cdc27 protein. (C) Sequential immunoprecipitation experiment demonstrating that p55CDC and Mad2 form complexes independent of the APC. The mitotic HeLa cell extract was first immunodepleted of APC components by immunoprecipitation with anti-Cdc27 protein. The APC-depleted extract was then used for a second immunoprecipitation with anti-p55CDC. In the absence of APC components, a substantial portion of the Mad2 protein is complexed with p55CDC.
In mitotic HeLa cell extracts immunodepleted of all detectable p55CDC, substantial amounts of Mad2 protein and Cdc27/Cdc16 (APC component proteins) remained. To test if Mad2 was bound to the APC independently of p55CDC, we performed sequential immunoprecipitation experiments. We first immunoprecipitated the p55CDC protein from the mitotic extracts. As expected, a portion of Mad2 and Cdc27/Cdc16 coimmunoprecipitated, but much remained in the extract. We then used anti-Cdc27 antibody to immunoprecipitate the residual APC in the extract. We found only trace amounts of Mad2 bound to this residual APC (Fig. 4 B). Thus, Mad2, when not complexed with p55CDC, does not bind the APC.
To determine if p55CDC and Mad2 are complexed with each other in the absence of the APC, we first immunodepleted the APC from the mitotic extracts with anti-Cdc27 antibody (Fig. 4 C). A large amount of Mad2 remained in the supernatant. From this supernatant we again immunoprecipitated, this time with anti-p55CDC, and then immunoblotted with anti-Mad2 antibody. p55CDC was completely removed by the immunoprecipitation, and a substantial amount of Mad2 protein was coimmunoprecipitated. However, some Mad2 also remained in the second supernatant (Fig. 4 C). Therefore, in mitotic HeLa cell extracts, Mad2 complexes with p55CDC in the absence of the APC and some Mad2 is not associated with either p55CDC or APC.
In summary, the immunoprecipitation experiments indicate that Mad2 is present in two pools: one associated with p55CDC and one not. Only in the presence of p55CDC does Mad2 also associate with the APC. Since p55CDC associates with Mad2 in the absence of the APC, the simplest interpretation is that p55CDC and Mad2 must be present in a complex in order to bind to the APC. However, it is likely that other proteins are involved in the binding of Mad2, p55CDC, and the APC.
Microinjection of Mitotic HeLa Cells with Anti-p55CDC Induces a Delay at Metaphase
To examine the function of p55CDC in living mammalian cells, we microinjected anti-p55CDC antibody or control rabbit IgG into prometaphase HeLa cells. In control cells (n = 10) the average duration of metaphase was 15.2 ± 4.6 min (range 9–22 min), and the average length of anaphase was 3.2 ± 0.9 min (range 2–6 min; Table I).
Table I.
Cell Cycle Progression in HeLa and PtK1 Cells Injected with Control Rabbit IgG or Anti-p55CD
| Injection/cell phase injected | n | No. of cells arrested at metaphase | Duration of metaphase in min (mean ± SD) | Duration of anaphase in min (mean ± SD) | Specific notes | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| HeLa | ||||||||||
| control rabbit IgG‡ | 10 | 0 | 15.2 ± 4.6 | 3.2 ± 0.9 | ||||||
| early/late prometaphase | ||||||||||
| anti-p55CDC‡ | 19 | 0 | 35.0 ± 30.7‖ | 5.7 ± 3.0 | ||||||
| early/late prometaphase | ||||||||||
| PtK1 | ||||||||||
| control rabbit IgG | 32 | 0 | 10.8 ± 4.7 | 9.3 ± 4.0 | ||||||
| late prophase/early metaphase | 3 arrested at early anaphase | |||||||||
| anti-p55CDC§ | 31 | 10 | 38.0 ± 20.7‖ | 23.3 ± 13.1‖ | 4 arrested at late anaphase | |||||
| late prophase/early metaphase | 5 without cytokinesis |
8.0 mg/ml in KCl-buffer.
2.0 mg/ml in KCl-buffer.
1.0-2.5 mg/ml in KCl- or Tris-buffer.
P < 0.01.
Microinjection of prometaphase HeLa cells (n = 19) with anti-p55CDC (micropipette concentration 2.0 mg/ml) caused a transient metaphase arrest (Fig. 5 A). The cells delayed onset of anaphase significantly (35.0 ± 30.7 min; range: 7–109 min) when compared with control IgG-injected cells (P < 0.01, Table I). In addition, the duration of anaphase was slightly longer in the anti-p55CDC injected HeLa cells (5.7 ± 3.0 min; range: 3–13 min), although the difference from controls was not statistically significant (P < 0.1).
Figure 5.
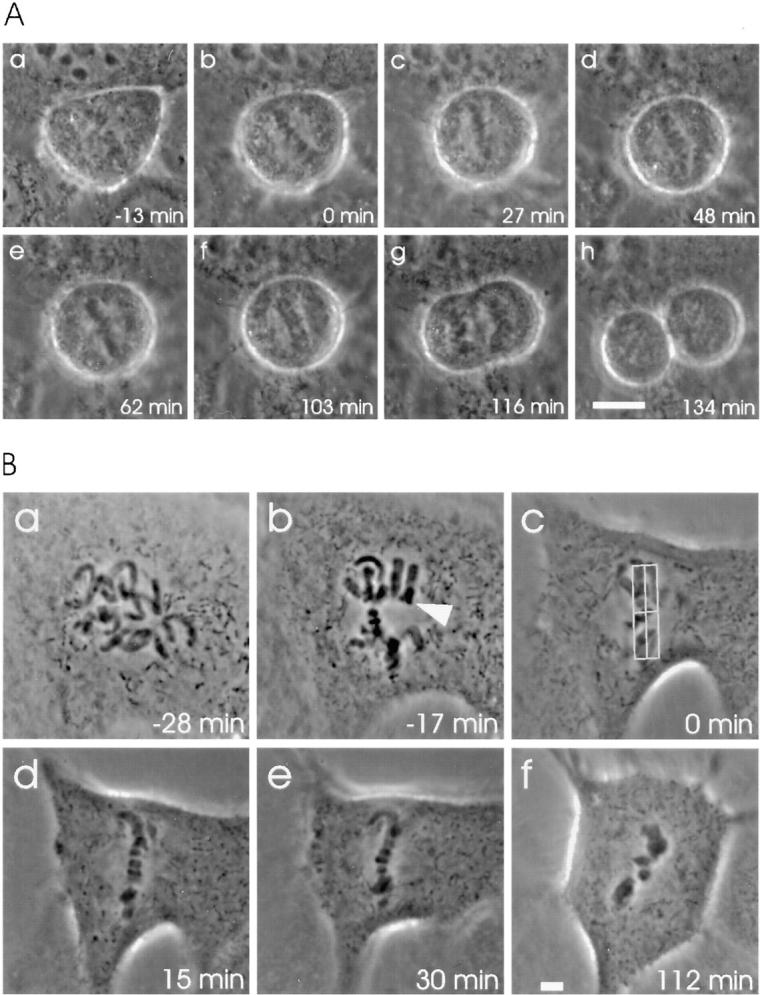
Microinjection with antibodies against p55CDC hampers the metaphase-to-anaphase transition. (A) Anti-p55CDC microinjection induces a transient metaphase arrest in HeLa cells. The antibody (2.0 mg/ml in the micropipette) was injected at prometaphase 18 min before the metaphase plate formed (time 0 = metaphase). (a) The first image was taken 5 min after injection. (b–f) During the metaphase arrest, the chromosomes underwent occasional stretching, but were rapidly pulled back to form a tight metaphase plate. (f) The onset of anaphase occurred 103 min after the tight metaphase plate formed for the first time. (f–g) Separation and movement of the sister chromatids required 13 min, which is slightly longer than the average of control cells. (g–h) Cytokinesis and exit from mitosis appeared normal. (B) Anti-p55CDC microinjection induces metaphase arrest in PtK1 cells. The cell was injected with anti-p55CDC (2.0 mg/ml) at late prophase a few minutes before breakdown of the nuclear envelope. (a) The first image was taken 5 min later, when the cell was at early prometaphase. (b) The last chromosome (arrowhead) became bioriented 17 min before the metaphase plate was formed. Progression through prometaphase appeared normal. (c) The last chromosome aligned at the spindle equator, and a metaphase plate was formed. The box (3 μm on either side of the spindle equator) used to define the start of metaphase is shown. (c–f) The cell did not initiate anaphase during the time it was monitored (total time = 168 min after the beginning of metaphase). Bars, 5 μm.
Microinjection of PtK1 Cells with Anti-p55CDC Causes Mitotic Defects
To study the effects of anti-p55CDC antibody injection in more detail, we injected prophase-to-early metaphase PtK1 cells either with anti-p55CDC antibody (n = 31, micropipette concentration 1.0–2.5 mg/ml) or with nonspecific rabbit IgG antibodies (n = 32, 8.0 mg/ml). In control PtK1 cells, the average duration of metaphase was 10.8 ± 4.7 min (range 4–23 min), and the average length of anaphase was 9.3 ± 4.0 min (range 3–16 min; Table I).
Cells injected with anti-p55CDC at late metaphase (8 min after the last chromosome had arrived to the metaphase plate; n = 2) or at early anaphase (n = 3) showed no apparent effects. Duration of anaphase in these cells was 6.6 ± 0.9 min. Cells injected between late prophase and early metaphase exhibited several mitotic defects when compared with control injected cells (Fig. 6). The frequency of specific defects did not correlate with the phase of mitosis in which the antibody was introduced. In prophase cells, identical effects were observed whether the cells were injected in the nucleus or the cytoplasm. Mitotic PtK1 cells microinjected with anti-p55CDC antibody before late metaphase showed three main responses (Table I): (a) metaphase arrest/delay in anaphase onset; (b) slow anaphase; and (c) failure to exit mitosis. In all, 29 of 31 cells (94%) injected before late metaphase showed at least one type of deficiency. Many cells showed a combination of responses, of which the most frequent was a delay in the onset of anaphase followed by slow poleward chromatid movement. The other two injected cells (6%) progressed through mitosis normally (Fig. 6).
Figure 6.

Cell cycle progression in PtK1 cells injected with nonspecific rabbit IgG (8.0 mg/ml) or anti-p55CDC (1.0–2.5 mg/ml). The numbers above the bars denote the proportion of cells in each category. Control IgG or anti-p55CDC injected cells were considered to exhibit a delay at metaphase if the duration of metaphase was more than double the mean duration of metaphase in the control cells. Cells were considered to exhibit an aberrant anaphase if they arrested during anaphase or if the duration of anaphase was more than double the mean duration of anaphase in control cells.
As with HeLa cells, the most frequent outcome of anti-p55CDC microinjection into PtK1 cells was a delay in the onset of anaphase which varied in duration among individual cells. Ten out of 31 cells (32%) injected with anti-p55CDC antibody before late metaphase failed to initiate anaphase within 90 minutes after the last chromosome reached the metaphase plate. As a rule, after 90 min in metaphase, observation of a cell was suspended, and it was categorized as permanently arrested at metaphase (Table I, Fig. 5 B, Fig. 6). Some of these cells were cultured overnight to determine if they would remain arrested. The cells were still arrested at M phase with condensed chromosomes the following morning.
In addition to the permanently arrested cells, a number of PtK1 cells microinjected with anti-p55CDC antibody exhibited a transient metaphase arrest (Table I, Fig. 7 A) with an average duration of 38.0 ± 20.7 min (range, 8–75 min). The duration of metaphase exhibited by these transiently arrested cells is significantly greater (P < 0.01) than that of control cells.
Figure 7.

Microinjection of mitotic PtK1 cells with anti-p55CDC antibodies impairs normal progression of late mitotic events. (A) Slow separation of sister chromatids and impeded exit from mitosis by anti-p55CDC microinjection. The cell was microinjected with anti-p55CDC antibodies (1.5 mg/ ml in the micropipette) at early prometaphase (6 min after NEB). (a) The first image was taken 13 min later when the cell was at late prometaphase. (b) The chromosomes reached the metaphase plate. (c) Anaphase initiated after a short delay of ∼26 min. (c–g) Separation of sister chromatids was very slow, requiring over 50 min before the sister chromatids reached the spindle poles. The cell failed to undergo cytokinesis, and exited mitosis as a binucleate cell (arrowheads in h). To the right of the microinjected cell, an untreated cell progressed through a normal mitosis. The asterisk in h denotes one of the progeny of this normal division. (B) Inhibition of sister chromatid separation by anti-p55CDC microinjection. The cell was injected at late prometaphase with anti-p55CDC antibodies (1.5 mg/ml). (a) The first evidence of chromatid segregation was seen 5 min after the start of metaphase (time point = 0 min). (b–h) Despite pulling forces evident from stretching of the kinetochore regions (e, arrows), full separation of sister chromatid arms was never achieved. Bars, 5 μm.
Microinjection of Mitotic PtK1 Cells with Anti-p55CDC Impairs Sister Chromatid Separation at Anaphase, Hampers Cytokinesis, and Exit from M phase
Most of the PtK1 cells that eventually proceeded to anaphase after anti-p55CDC microinjection showed atypical features, including failure to separate sister chromatids or impaired sister chromatid movement after anaphase onset. The most common type of deficiency was a very slow poleward movement of the chromatids during anaphase, even after the chromatids had separated (Fig. 7 A). The average duration of anaphase in those anti-p55CDC microinjected PtK1 cells that initiated anaphase either after a normal or prolonged metaphase was 23.3 ± 13.1 min (range: 6–48 min), a time significantly greater (P < 0.01) than that of cells injected with control IgG (Table I).
Three of the anti-p55CDC microinjected cells arrested in early anaphase, failing to separate their chromatids completely (Fig. 7 B). Anaphase onset was evident in these cells. The kinetochore regions of the chromosomes stretched and pulled apart, but the chromosome arms did not fully separate. These cells remained arrested at this early anaphase configuration for hours.
A third type of deficiency observed in the cells microinjected with anti-p55CDC antibody was the failure to decondense the chromosomes and exit mitosis. 4 out of 21 anti-p55CDC microinjected cells (19%) that eventually progressed into anaphase did not exit M phase. Instead, the cells were arrested at late anaphase-like configuration with condensed chromatids at the poles. 5 of 21 cells (24%) failed to initiate cytokinesis, but did decondense their chromosomes, reformed nuclear envelopes, and progressed into the next cell cycle as binucleate cells (Fig. 7 A).
To investigate whether the anti-p55CDC injection affects cell cycle progression at prometaphase, we monitored chromosome movements from nuclear envelope breakdown (NEB) to metaphase in cells that were injected at late prophase either with anti-p55CDC antibody or nonspecific rabbit IgG. The duration of prometaphase in anti-p55CDC–injected cells was not significantly different from uninjected or control-injected cells from similar cultures (Table II). However, again onset of anaphase was significantly delayed by anti-p55CDC antibody injection, and cells were observed to arrest at metaphase (Table II).
Table II.
Duration of Prometaphase in PtK1 Cells in Minutes
| Injection (pipette concentration) | No. of cells analyzed | NEB-metaphase | Lb-metaphase* | Lb-anaphase‡ | No. of cells arrested at metaphase | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| mean ± SD | mean ± SD | mean ± SD | ||||||||
| None | 10 | 18.0 ± 5.3 | 7.3 ± 4.5 | 14.8 ± 4.4 | 0 | |||||
| Rabbit IgG (8.0 mg/ml) | 10 | 16.8 ± 3.8 | 8.9 ± 2.8 | 17.4 ± 6.3 | 0 | |||||
| Anti-p55CDC (1.0 mg/ml) | 10 | 19.0 ± 4.1 | 8.4 ± 3.2 | 35.0 ± 8.7§ | 3 |
A fresh isolate of PtK1 cells was used. Data are from three independent experiments.
Last chromosome to biorientate (Lb) to formation of metaphase plate.
Last chromosome to biorientate (Lb) to onset of anaphase.
Significantly different from other values; P < 0.01.
At various times after microinjection, cells were fixed and labeled with a fluorescent anti-rabbit secondary antibody to track the fate of the microinjected anti-p55CDC antibody. The anti-p55CDC antibody bound to kinetochores within a few seconds after injection (data not shown). Interestingly, in microinjected cells that did complete anaphase and exit M phase, the antibody remained bound at the kinetochore region of the late telophase/ early G1 nuclei as demonstrated by colabeling with Crest autoimmune sera (Fig. 8). This result is in contrast to the results obtained with conventional immunofluorescence, where labeling of kinetochores fades as cells reach telophase (Fig. 2).
Figure 8.

Tracking of anti-p55CDC antibody injected into living cells. Cells were microinjected with anti-p55CDC, observed for a time, and then fixed for immunofluorescence to determine the localization of the injected antibody. Cells were colabeled with human autoimmune antibodies to localize kinetochores. (a) PtK1 cell arrested at metaphase after microinjection with anti-p55CDC at early prometaphase. (b) An anti-p55CDC–microinjected PtK1 cell at late anaphase. The cell was microinjected at late prometaphase. (c) A anti-p55CDC–microinjected PtK1 cell at late telophase. The antibodies were introduced at late prometaphase. Note that the microinjected anti-p55CDC antibody remained bound to kinetochores even at late telophase. Bar, 10 μm.
Microinjection with Anti-p55CDC Does Not Perturb Expression of the 3F3/2 Phosphoepitope at Kinetochores
We have postulated that the kinetochore phosphoprotein recognized by the 3F3/2 anti-phosphoepitope is a component of the spindle checkpoint. To investigate the possibility that the metaphase arrest induced by anti-p55CDC microinjections could result from activation of the spindle checkpoint upstream of the 3F3/2 phosphoepitope, cells microinjected with anti-p55CDC were probed with 3F3/2 antibody. 3F3/2 phosphoepitope expression was not altered in cells microinjected with anti-p55CDC (Fig. 9 A). The 3F3/2 signal at the kinetochores was lost by metaphase as the chromosomes aligned at the spindle equator. If cells were microinjected with anti-p55CDC, allowed to proceed into metaphase, and subsequently treated with the microtubule poison nocodazole, the 3F3/2 phosphoepitope reappeared at all kinetochores (Fig. 9 B). As we had shown previously, the intensity of 3F3/2 phosphoepitope expression varied among the kinetochores of prometaphase cells, diminishing as the chromosomes align at the metaphase plate. We found that labeling with anti-p55CDC paralleled the changes seen with the 3F3/2 antibody. As chromosomes became aligned, their labeling with the anti-3F3/2 antibody diminished (Fig. 9 A). However, unlike the 3F3/2 antibody labeling, labeling with anti-p55CDC antibody persisted in metaphase and, albeit more weakly, in anaphase (Fig. 9 A; Fig. 2).
Figure 9.
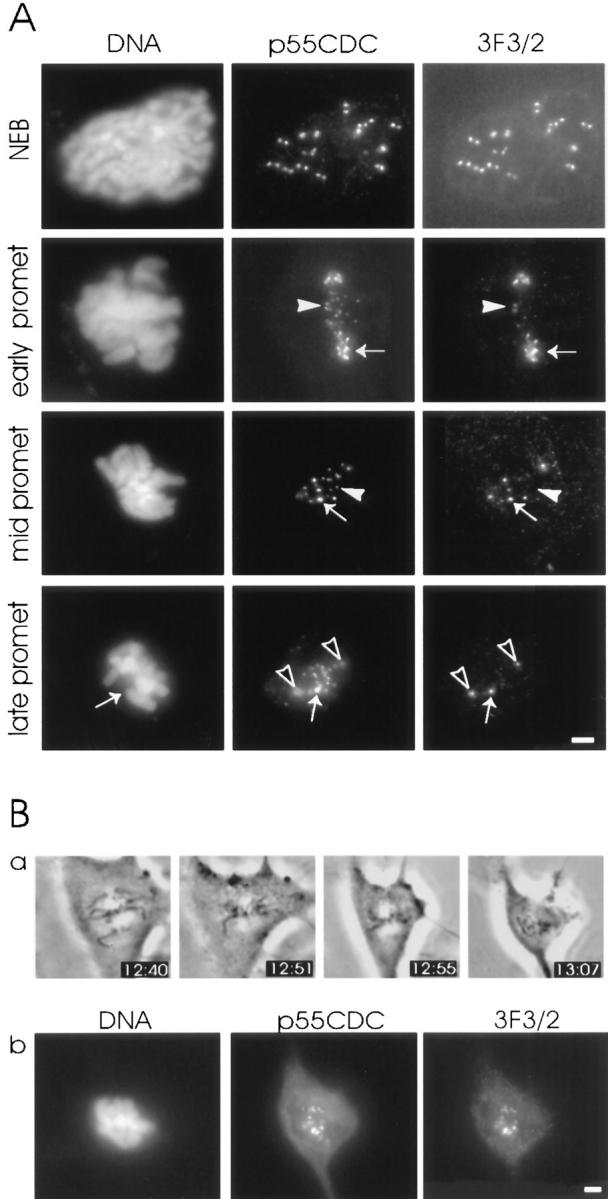
Expression of 3F3/2 phosphoepitope at the kinetochores of anti-p55CDC microinjected PtK1 cells. (A) Similar to p55CDC signal, the intensity of the fluorescent label of 3F3/2 epitope varies among chromosomes with different positions along the spindle. The kinetochores of chromosomes closer to spindle poles possess brighter 3F3/2 and p55CDC signals (arrows in early prometaphase and mid prometaphase rows) compared with kinetochores of aligned chromosomes (arrowheads in early prometaphase and mid prometaphase rows). At late prometaphase and metaphase the 3F3/2 signal disappears from the kinetochores of aligned chromosomes, while p55CDC is still present at all kinetochores. One misaligned chromosome having a bright p55CDC signal also shows strong labeling with the 3F3/2 antibody (arrows in late prometaphase row). The spindle poles that are labeled by both p55CDC and 3F3/2 are denoted with open arrowheads. (B) The 3F3/2 phosphoepitope expression reappears at the kinetochores of anti-p55CDC injected cells at metaphase if the spindle is destroyed with nocodazole. (a) At 12:35 the early prometaphase cell was injected with antibodies against p55CDC (1.0 mg/ml). At 12:55, all the chromosomes were aligned at the spindle equator. Nocodazole (5 μg/ml) was added at 12:57. (b) The cell was fixed 15 min later, and was immunolabeled to detect injected anti-p55CDC and the 3F3/2 phosphoepitope. Bars, 5 μm.
Discussion
We have investigated the M phase role of p55CDC, a mammalian protein related to Cdc20 and Hct1/Cdh1 in S. cerevisiae, and to fizzy and fizzy-related gene products in Drosophila. By immunofluorescence and by expression of a p55CDC-GFP fusion protein, we determined that p55CDC is present diffusely and also concentrated at kinetochores and spindle poles in M phase cells. In extracts of M phase cells, we found that p55CDC associates with Mad2, an element of the spindle checkpoint pathway, and with the APC, the 20S ubiquitin ligase complex. Moreover, we discovered that p55CDC was necessary for the interaction between Mad2 and the APC. To test the function of p55CDC in M phase, we injected anti-p55CDC antibody into M phase cells. Anti-p55CDC caused several alterations in mitosis, including metaphase arrest, delayed anaphase onset, aberrant chromatid movement in anaphase, and inhibition of exit from M-phase.
p55CDC Associates with Kinetochores of Chromosomes in M Phase
Kinetochores play multiple roles in M phase. Classically they were found to be essential for equal segregation of chromosomes in meiosis and mitosis. A number of microtubule motor proteins reside at the kinetochore (Rudner and Murray, 1996; Yen and Schaar, 1996), and in vitro experiments demonstrate that chromosomes, presumably by action of their kinetochores, are capable of translocation on microtubules (Hyman and Mitchison, 1991; McIntosh, 1991). Recently it was recognized that kinetochores play an additional critical role as signaling organelles in cell cycle regulation. Most cells delay anaphase onset if one or more chromosomes are misaligned. Genetic manipulations in yeast (Spencer and Hieter, 1992; Wang and Burke, 1995) and laser ablation experiments in mammalian cells (Rieder et al., 1995) demonstrate that the kinetochores of misaligned chromosomes generate an M phase checkpoint signal that inhibits anaphase onset until chromosomes are properly attached to the spindle microtubules.
Labeling with anti-p55CDC antibodies and expression of GFP-p55CDC fusion proteins showed that p55CDC is present diffusely and also concentrated at kinetochores and other spindle structures in cultured mitotic cells. In interphase, p55CDC appears to be present at reduced levels diffusely in the nucleus and cytoplasm with a concentration in two dots, perhaps centrioles, within the centrosome.
During M phase, immunofluorescence and p55CDC-GFP localization show that the concentration of p55CDC at the kinetochores changes with progressive stages of mitosis. Up to and including prophase, the majority of the protein is diffuse with some concentration in the nucleus. At late mitotic prophase, the protein begins to be found associated with kinetochores. It remains there until late telophase, when the p55CDC protein wanes in abundance. During chromosome congregation in prometaphase, the anti-p55CDC immunolabeling and the p55CDC-GFP fluorescence are most intense on kinetochores of chromosomes near the poles, and diminishes as chromosomes move closer to metaphase plate. These changes suggest that p55CDC concentration progressively diminishes at kinetochores as chromosomes establish stable attachments to the spindle. These dynamics in kinetochore association are similar to changes with other kinetochore elements, including the 3F3/2 phosphoepitope, the Mad2 protein, and the Bub1 protein, all implicated as components of the spindle checkpoint (Campbell and Gorbsky, 1995; Chen et al., 1996; Li and Benezra, 1996; Taylor and McKeon, 1997).
p55CDC is Associated with Mad2 and the APC in Mitosis
The spindle checkpoint signal appears to function by inhibiting the M phase ubiquitin ligase called the APC. Recent studies show that Mad2 protein does not bind the APC in interphase, but does in M phase, and inhibits APC activity (Li et al., 1997). The concentration of Mad2 protein does not appear to vary with the cell cycle (our unpublished observation) and, in yeast, Mad2 protein does not appear to be phosphorylated (Hartwick and Murray, 1995). Thus, a likely mechanism for cell cycle regulation of Mad2 activity is through complex formation with other cell cycle–regulated proteins. Our immunoprecipitation and immunoblotting experiments revealed that in extracts prepared from M phase, but not interphase cells, p55CDC is complexed with Mad2. Similarly, we found that p55CDC was also associated with the APC in M phase, but not interphase extracts. Recently, Cdc20 of budding yeast and Slp1 of fission yeast were shown to complex with Mad2, although the association in these organisms was not reported to be cell cycle–dependent (Hwang et al., 1998; Kim et al., 1998). Lim et al. (1998) also recently reported that tagged forms of Cdc20 and Cdc23, a component of the yeast APC, when overexpressed in S. cerevisiae, can be coimmunoprecipitated, but again no cell cycle dependence of this association was demonstrated in this organism.
The presence of both p55CDC and Mad2 complexed with the APC prompted us to ask whether Mad2 binding to the APC might depend on p55CDC. We first determined that p55CDC and Mad2 were complexed in the absence of APC. Second, quantitative immunoprecipitation of the p55CDC protein coprecipitated some of the APC and Mad2 from the cell extracts. However, the supernatant remaining after p55CDC immunoprecipitation contained significant amounts of both APC and Mad2. This residual APC and Mad2 were not complexed. One explanation for this result is that Mad2 association with the APC is mediated by p55CDC.
Microinjection of Antibody to p55CDC Interferes with Normal Mitotic Timing
Several related mitotic events—anaphase onset, anaphase chromatid movement, and exit from mitosis—are blocked or delayed by microinjection of anti-p55CDC antibody. The multiple effects detected after injection of anti-p55CDC antibody suggest that the protein is important in several pathways, or in a single pathway that regulates others. In cells injected with anti-p55CDC antibodies, we did not detect alterations in the movement of chromosomes during prometaphase. Moreover, the chromosome oscillations and stretching at the spindle equator at metaphase appeared typical, even in injected cells that showed significant delays in the onset of anaphase.
Many mitotic cells arrested transiently at metaphase after anti-p55CDC antibody injection, suggesting that p55CDC function is involved in the metaphase-to-anaphase transition. In PtK1 cells this transient metaphase arrest was often followed by an aberrant progression of anaphase. Sister chromatid movements were slow, and cells sometimes failed to complete cytokinesis or decondense their chromosomes. Thus, p55CDC-dependent events are also apparently essential in late mitosis. However, cells microinjected at late metaphase or anaphase completed division without notable effects. This result suggests that by late metaphase p55CDC has completed its functions, or that it becomes immune to antibody perturbation.
The precise mechanisms by which antibody injection affects mitotic progression is uncertain. For the effects of the antibody in delaying the metaphase-to-anaphase transition, our favored hypothesis is that the anti-p55CDC antibody interferes with the requisite ubiquitination of anaphase inhibitor proteins by the APC. In budding yeast, the Cdc20, a protein related to p55CDC, is required for timely degradation of the anaphase inhibitor protein Pds1 (Visintin et al., 1997; Lim et al., 1998) and mitotic cyclin Clb2 (Lim et al., 1998). Thus, anti-p55CDC antibody injections may also delay onset of anaphase by interfering with the APC-dependent destruction of mammalian cyclin A. In Drosophila, the presence of nondegradable forms of cyclin A have been shown to delay onset of anaphase (Sigrist et al., 1995). Moreover, mutations in the Drosophila fizzy gene product, another protein related to p55CDC, blocks destruction of mitotic cyclins including cyclin A, and induces cell cycle arrest at metaphase (Sigrist et al., 1995).
To our knowledge, the very slow anaphase movements of chromatids seen in the injected PtK1 cells that enter anaphase are without precedents in antibody microinjection experiments. Qualitatively similar effects in mammalian cells are seen after treatment with low concentrations of the microtubule-stabilizing agent taxol (Rieder et al., 1994; Waters et al., 1996). Thus, p55CDC may be important in regulating microtubule dynamics in anaphase. Disruption of microtubule dynamics at metaphase normally activates the 3F3/2 phosphoepitope at kinetochores. However, in anti-p55CDC-injected cells that have progressed to metaphase, the 3F3/2 phosphoepitope is extinguished, suggesting that microtubule dynamics before anaphase are sufficiently normal to turn off the spindle checkpoint. Thus, it appears that the metaphase delay induced by anti-p55CDC antibody injection is independent of effects on microtubule dynamics, and occurs downstream of the spindle checkpoint. Anti-p55CDC antibody could block or delay APC-mediated degradation of proteins whose destruction is necessary for normal anaphase spindle function. One such protein identified in budding yeast is Ase1. This protein is required for spindle elongation and separation of the spindle poles during anaphase B (Pellman et al., 1995). The gene product is an APC substrate, and is degradated as the cells undergo cytokinesis (Juang et al., 1997). If protein destruction is prevented by expressing a nondegradable Ase1, the spindle disassembly is delayed and the cells exhibit a slow anaphase (Juang et al., 1997).
The third general result from cell injection with anti-p55CDC antibody is failure to exit mitosis. Once more we hypothesize that this effect may be attributable to the failure of the APC to target appropriate substrates for ubiquitination. In this instance, the likely candidate substrates include isoforms of cyclin B. Cyclin B degradation and the subsequent inactivation of Cdk1 has been shown to be required for decondensation of mitotic chromosomes, nuclear envelope reassembly, and exit from mitosis (Holloway et al., 1993; Irniger et al., 1995). Microinjecting mRNA coding for nondegradable cyclin B in mammalian cells results in late anaphase arrest, but anaphase onset and chromatid movement occur normally (Wheatley et al., 1997). Similarly, a nondegradable form of cyclin B3 induces cell cycle arrest at late anaphase in Drosophila (Sigrist et al., 1995). Recently, new members of the Cdc20 protein family were identified in S. cerevisiae. Hct1 and Cdh1 were both shown to be required for yeast mitotic cyclin degradation in an APC-dependent manner (Schwab et al., 1997; Visentin et al., 1997). Together these results suggest that the failure to degrade different cyclin B isoforms, and maintenance of high levels of Cdk1 activity may account for the observed difficulty to exit mitosis.
The mechanisms by which p55CDC itself and its cognate proteins in other species function in regulating cell cycle events are only beginning to be explored. Moreover, we cannot at present formally distinguish whether the anti-p55CDC antibody inhibits or enhances the function of its target protein in living cells. Our presumption is that the anti-p55CDC antibody interferes with the ability of p55CDC to carry out its function, perhaps in mediating interactions between the APC and its substrates. However, an alternative explanation for the antibody microinjection results stems from biochemical data indicating that a large proportion of the p55CDC protein is normally degraded about the time of M phase exit (Weinstein, 1997). Conventional immunofluorescence and p55CDC-GFP studies suggests that p55CDC disappears from kinetochores after late anaphase. In our studies, microinjected anti-p55CDC antibody remains bound to the kinetochore regions of chromosomes even into early G1 phase for cells that succeed in exiting M phase. Thus, instead of inhibiting p55CDC function in living cells, the presence of the antibody may preserve p55CDC beyond the time at which it should normally be degraded, thus interfering with the normal progression of mitosis.
Our results suggest that mammalian p55CDC is used at metaphase and anaphase to target several substrates. This result is in agreement with the latest genetic studies from yeast, demonstrating that a single targeting protein such as Cdc20 may be involved in the ubiquitination of several specific substrates by the APC at appropriate stages of cell division. Other Cdc20-like proteins such as Hct1/Cdh1 and Fizzy-related appear to be required at G1 when no Cdc20 is present. However, at present we cannot formally eliminate the possibility that anti-p55CDC antibody injection into PtK1 cells also affects related members of the mammalian family of Cdc20 proteins.
Models of p55CDC Function and the Spindle Checkpoint
Based on the results presented here and on genetic studies of p55CDC homologs in other systems, we hypothesize that p55CDC serves as an essential mitotic targeting component for the APC (Fig. 10). We propose that a complex composed of p55CDC, Mad2, and likely other yet unidentified proteins, is formed during prometaphase in normal mitosis, and in cells arrested with antimicrotubule drugs. This complex binds the APC. We postulate that this p55CDC/Mad2 complex contains elements that prime or preactivate the APC and others that simultaneously inhibit its ubiquitin ligase activity. We speculate that unattached or unstably attached kinetochores serve as staging areas for catalytic assembly of these p55CDC/Mad2 complexes, which are released to bind the APC throughout the cell. We suggest that the inhibitory capability of these complexes spontaneously decays, but in the presence of unattached kinetochores they continue to be produced. Thus, in a normal cell the checkpoint is maintained until the last chromosome achieves bipolar attachment to the spindle. The presence of a microtubule poison will artificially produce unattached kinetochores, and thus will perpetuate inhibitor complex production.
Figure 10.
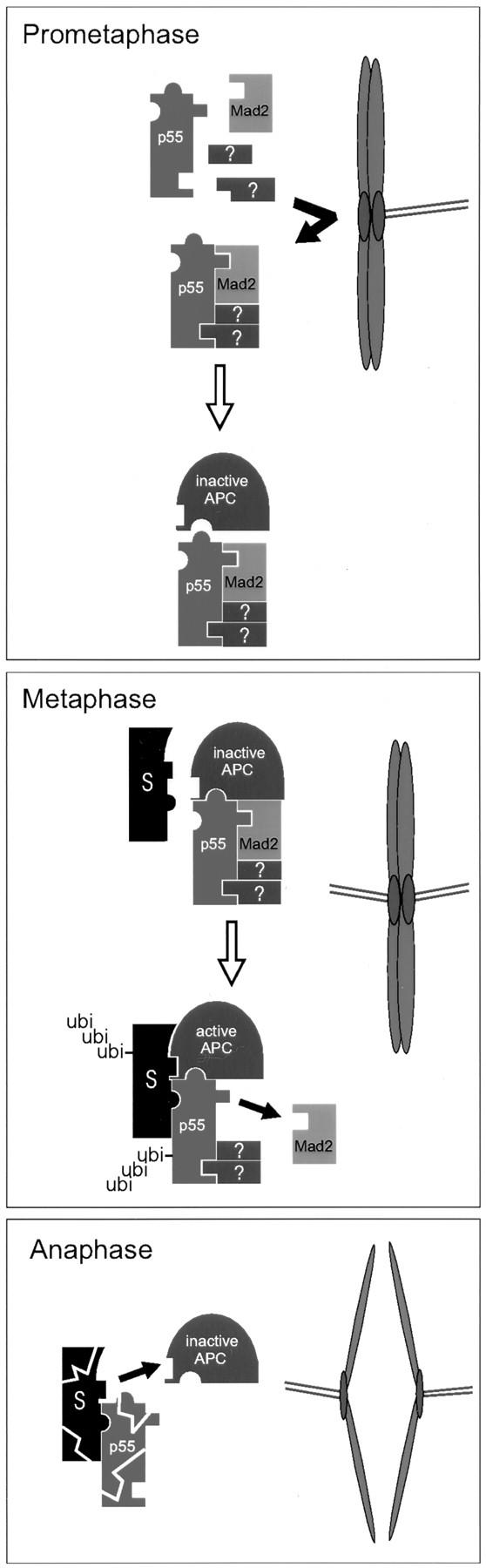
Model for p55CDC function. p55CDC and Mad2 proteins associate with unattached kinetochores in prometaphase cells (Figs. 2 and 3; Chen et al., 1996) where p55CDC and Mad2 complexes are formed (Fig. 5 A). p55CDC/Mad2 complex formation does not require the presence of APC (Fig. 5 C). The complex may also contain other yet uncharacterized elements (question marks). p55CDC/Mad2 complex production ceases when all chromosomes are properly oriented in the spindle. The p55CDC/ Mad2 complex then binds the APC (Fig. 5 B). The APC activity is inhibited by Mad2 (Li et al., 1997). p55CDC participates in targeting the APC to proteolytic substrates (S). Substrate binding may occur before or after activation of the APC (binding to inactive APC/p55CDC/Mad2 complex shown). The APC is activated upon loss of Mad2. This activation leads to ubiquitination of mitotic target proteins and p55CDC by the APC. At anaphase onset, the polyubiquitinated substrate proteins and p55CDC are proteolysed by the proteosome. During late mitotic events, APC may associate with other p55CDC-like protein/substrate complexes and be reused for their destruction.
Upon attachment of the final chromosome, inhibitor production ceases. We suggest that, at a certain threshold of decay of the inhibitory complex, an autocatalytic mechanism is initiated to assure cell-wide spontaneous activation of the APC. At the molecular level we speculate that activation involves loss of the inhibitory components (e.g., Mad2) with retention of the essential targeting components (e.g., p55CDC until it too is degraded by APC-mediated ubiquitination). While highly speculative, this model does propose testable predictions. For example, it predicts that p55CDC and Mad2 should be transient components of kinetochores in prometaphase cells. Second, it predicts that inhibitory components such as Mad2, but not targeting components such as p55CDC, should be released from the APC after activation of the ubiquitin ligase activity.
In summary, we have presented evidence that p55CDC participates in the mammalian checkpoint pathway that regulates various processes of M phase, and is involved in the APC-mediated protein proteolysis.
Acknowledgments
This study was supported by grants to G.J. Gorbsky and D.J. Burke from the National Institute of General Medical Sciences, and to M. Kallio from the Academy of Finland and Cultural Foundation of Finland. The authors wish to thank Drs. R.-H. Chen, A. Murray, S. Tugendreich, P. Hieter, and J.B. Rattner for providing antibodies.
Abbreviations used in this paper
- APC
anaphase-promoting complex
- Ase
anaphase spindle elongation
- NEB
nuclear envelope breakdown
- Pds1
precocious division of sister chromatids
Footnotes
Address all correspondence to Gary J. Gorbsky, Department of Cell Biology, Health Sciences Center, Box 439, University of Virginia, Charlottesville, VA 22908. Tel.: 804-982-1654. Fax: 804-982-3912; E-mail: gjg5y@Virginia.edu
References
- Campbell MS, Gorbsky GJ. Microinjection of mitotic cells with 3F3/2 anti-phosphoepitope antibody delays the onset of anaphase. J Cell Biol. 1995;129:1195–1204. doi: 10.1083/jcb.129.5.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R-H, Waters JC, Salmon ED, Murray AW. Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science. 1996;274:242–246. doi: 10.1126/science.274.5285.242. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Peters J-M, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996;10:3081–3093. doi: 10.1101/gad.10.24.3081. [DOI] [PubMed] [Google Scholar]
- Cyert MS, Scherson T, Kirschner MW. Monoclonal antibodies specific for thiophosphorylated proteins recognize XenopusMPF. Dev Biol. 1988;129:209–216. doi: 10.1016/0012-1606(88)90175-3. [DOI] [PubMed] [Google Scholar]
- Dawson IA, Roth S, Akam M, Artavanis-Tsakonas S. Mutations of the fizzy locus cause metaphase arrest in Drosophila melanogasterembryos. Development. 1993;117:359–376. doi: 10.1242/dev.117.1.359. [DOI] [PubMed] [Google Scholar]
- Dawson IA, Roth S, Artavanis-Tsakonas S. The Drosophila cell cycle gene fizzy is required for normal degradation of cyclins A and B during mitosis and has homology to the Cdc20 gene of Saccharomyces cerevisiae. . J Cell Biol. 1995;129:725–737. doi: 10.1083/jcb.129.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funabiki H, Yamano H, Kumada K, Nagao K, Hunt T, Yanagida M. Cut2 proteolysis is required for sister-chromatid separation in fission yeast. Nature. 1996a;381:85–97. doi: 10.1038/381438a0. [DOI] [PubMed] [Google Scholar]
- Funabiki H, Kumada K, Yanagida M. Fission yeast Cut1 and Cut2 are essential for sister chromatid separation, concentrate along the metaphase spindle and form large complexes. EMBO (Eur Mol Biol Organ) J. 1996b;15:6617–6628. [PMC free article] [PubMed] [Google Scholar]
- Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- Gorbsky GJ. Cell cycle checkpoints: arresting progress in mitosis. Bioessays. 1997;19:193–197. doi: 10.1002/bies.950190303. [DOI] [PubMed] [Google Scholar]
- Gorbsky GJ, Ricketts WA. Differential expression of a phosphoepitope at the kinetochores of moving chromosomes. J Cell Biol. 1993;122:1311–1321. doi: 10.1083/jcb.122.6.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guacci V, Koshland D, Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. . Cell. 1997;91:47–57. doi: 10.1016/s0092-8674(01)80008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Smith D. Altered fidelity of mitotic chromosome transmission in cell cycle mutants of S. cerevisiae. . Genetics. 1985;110:381–395. doi: 10.1093/genetics/110.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KG. The spindle checkpoint. Trends Genet. 1998;14:1–4. doi: 10.1016/S0168-9525(97)01340-1. [DOI] [PubMed] [Google Scholar]
- Hartwick KG, Murray AW. Mad1p, a phosphoprotein component of the spindle assembly checkpoint in budding yeast. J Cell Biol. 1995;131:709–720. doi: 10.1083/jcb.131.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway SL, Glotzer M, King RW, Murray AW. Anaphase is initiated by proteolysis rather than by the inactivation of MPF. Cell. 1993;73:1393–1402. doi: 10.1016/0092-8674(93)90364-v. [DOI] [PubMed] [Google Scholar]
- Hwang AH, Lau LF, Smith DL, Mistrot CA, Hardwick KG, Hwang ES, Amon A, Murray AW. Budding yeast Cdc20: a target of the spindle checkpoint. Science. 1998;279:1041–1044. doi: 10.1126/science.279.5353.1041. [DOI] [PubMed] [Google Scholar]
- Hyman AA, Mitchison TJ. Regulation of the direction of chromosome movement. Cold Spring Harbor Symp Quant Biol. 1991;56:745–750. doi: 10.1101/sqb.1991.056.01.083. [DOI] [PubMed] [Google Scholar]
- Irniger S, Piatti S, Michaelis C, Nasmyth K. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell. 1995;81:269–278. doi: 10.1016/0092-8674(95)90337-2. [DOI] [PubMed] [Google Scholar]
- Juang Y-L, Huang J, Peters J-M, McLaughlin ME, Tai C-Y, Pellman D. APC-mediated proteolysis of Ase1 and the morphogenesis of the mitotic spindle. Science. 1997;275:1311–1314. doi: 10.1126/science.275.5304.1311. [DOI] [PubMed] [Google Scholar]
- Kim SH, Lin DP, Matsumoto S, Kitazono A, Matsumoto T. Fission yeast Slp1: an effector of the Mad2-dependent spindle checkpoint. Science. 1998;279:1045–1047. doi: 10.1126/science.279.5353.1045. [DOI] [PubMed] [Google Scholar]
- King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- Li Y, Benezra R. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 1996;274:246–248. doi: 10.1126/science.274.5285.246. [DOI] [PubMed] [Google Scholar]
- Li Y, Corbea C, Mahaffey D, Rechsteiner M, Benezra R. MAD2 associates with the cyclosome/anaphase-promoting complex and inhibits its activity. Proc Natl Acad Sci USA. 1997;94:12431–12436. doi: 10.1073/pnas.94.23.12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Nicklas RB. Mitotic forces control a cell-cycle checkpoint. Nature. 1995;373:630–632. doi: 10.1038/373630a0. [DOI] [PubMed] [Google Scholar]
- Lim HH, Goh P-Y, Surana U. Cdc20 is essential for the cyclosome-mediated proteolysis of both Pds1 and Clb2 during M phase in budding yeast. Curr Biol. 1998;8:231–234. doi: 10.1016/s0960-9822(98)70088-0. [DOI] [PubMed] [Google Scholar]
- Matsumoto T. A fission yeast homolog of Cdc20/p55CDC/Fizzy is required for recovery from DNA damage and genetically interacts with p34cdc2 . Mol Cell Biol. 1997;17:742–750. doi: 10.1128/mcb.17.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JR. Structural and mechanical control of mitotic progression. Cold Spring Harbor Symp Quant Biol. 1991;56:613–619. doi: 10.1101/sqb.1991.056.01.070. [DOI] [PubMed] [Google Scholar]
- Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91:35–45. doi: 10.1016/s0092-8674(01)80007-6. [DOI] [PubMed] [Google Scholar]
- Nicklas RB. How cells get the right chromosomes. Science. 1997;275:632–637. doi: 10.1126/science.275.5300.632. [DOI] [PubMed] [Google Scholar]
- Nicklas RB, Ward S, Gorbsky GJ. Kinetochore chemistry is sensitive to tension and may link mitotic forces to a cell cycle checkpoint. J Cell Biol. 1995;130:929–939. doi: 10.1083/jcb.130.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellman D, Bagget M, Tu Y-H, Fink GR. Two microtubule-associated proteins required for anaphase spindle movement in Saccharomyces cerevisiae. . J Cell Biol. 1995;130:1373–1385. doi: 10.1083/jcb.130.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzi L, Gersch MS, Campbell MS, Wu L, Osmani SA, Gorbsky GJ. MPM-2 antibody-reactive phosphorylations can be created in detergent-extracted cells by kinetochore-bound and soluble kinases. J Cell Sci. 1997;110:2013–2025. doi: 10.1242/jcs.110.17.2013. [DOI] [PubMed] [Google Scholar]
- Rieder CL, Schultz A, Cole R, Sluder G. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J Cell Biol. 1994;127:1301–1310. doi: 10.1083/jcb.127.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Cole RW, Khodjakov A, Sluder G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol. 1995;130:941–948. doi: 10.1083/jcb.130.4.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner AD, Murray AW. The spindle assembly checkpoint. Curr Opin Cell Biol. 1996;8:773–780. doi: 10.1016/s0955-0674(96)80077-9. [DOI] [PubMed] [Google Scholar]
- Schwab M, Schulze A, Lutum, Seufert W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 1997;90:683–693. doi: 10.1016/s0092-8674(00)80529-2. [DOI] [PubMed] [Google Scholar]
- Sigrist S, Jacobs H, Stratmann R, Lehner CF. Exit from mitosis is regulated by Drosophila Fizzyand sequential destruction of cyclins A, B and B3. EMBO (Eur Mol Biol Organ) J. 1995;14:4827–4838. doi: 10.1002/j.1460-2075.1995.tb00164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist S, Lehner CF. Drosophila fizzy-relateddown-regulates mitotic cyclins. Cell. 1997;90:671–681. doi: 10.1016/s0092-8674(00)80528-0. [DOI] [PubMed] [Google Scholar]
- Spencer F, Hieter P. Centromere DNA mutations induce a mitotic delay in Saccharomyces cerevisiae. . Proc Natl Acad Sci USA. 1992;89:8908–8912. doi: 10.1073/pnas.89.19.8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Ganoth D, Dahan A, Heller H, Hersko J, Luca F, Ruderman JV, Hersko A. The cyclosome, a large complex containing cyclin-selective ubiquitination ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell. 1995;6:185–198. doi: 10.1091/mbc.6.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S, McKeon F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 1997;89:727–735. doi: 10.1016/s0092-8674(00)80255-x. [DOI] [PubMed] [Google Scholar]
- Tugendreich S, Tomkiel J, Earnshaw W, Hieter P. CDC27Hs colocalizes with CDC16Hs to the centrosome and mitotic spindle and is essential for the metaphase to anaphase transition. Cell. 1995;81:261–268. doi: 10.1016/0092-8674(95)90336-4. [DOI] [PubMed] [Google Scholar]
- Visintin R, Prinz S, Amon A. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 1997;278:460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- Wang Y, Burke DJ. Checkpoint genes required to delay cell division in response to nocodazole respond to impaired kinetochore function in the yeast Saccharomyces cerevisiae. . Mol Cell Biol. 1995;15:6838–6844. doi: 10.1128/mcb.15.12.6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters JC, Mitchison TJ, Reider CL, Salmon ED. The kinetochore microtubule minus-end disassembly associated with poleward flux produces a force that can do work. Mol Biol Cell. 1996;7:1547–1558. doi: 10.1091/mbc.7.10.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J, Jacobsen FW, Hsu-Chen J, Wu T, Baum LG. A novel mammalian protein, p55CDC, present in dividing cells is associated with protein kinase activity and has homology to the Saccharomyces cerevisiae cell division cycle proteins Cdc20 and Cdc4. Mol Cell Biol. 1994;14:3350–3363. doi: 10.1128/mcb.14.5.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J. Cell cycle-regulated expression, phosphorylation and degradation of p55CDC: a mammalian homolog of CDC20/Fizzy/slp1. J Biol Chem. 1997;272:28501–28511. doi: 10.1074/jbc.272.45.28501. [DOI] [PubMed] [Google Scholar]
- Wheatley SP, Hinchcliffe EH, Glotzer M, Hyman AA, Sluder G, Wang YL. CDK1 inactivation regulates anaphase spindle dynamics and cytokinesis in vivo. J Cell Biol. 1997;138:385–393. doi: 10.1083/jcb.138.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto YA, Guacci V, Koshland D. Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s) J Cell Biol. 1996;133:99–110. doi: 10.1083/jcb.133.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen TJ, Schaar BT. Kinetochore function: molecular motors, switches and gates. Curr Opin Cell Biol. 1996;8:381–388. doi: 10.1016/s0955-0674(96)80014-7. [DOI] [PubMed] [Google Scholar]